Key Points
Automated analysis of megakaryocyte features can discriminate MPN samples from reactive mimics and facilitate disease classification.
Simplified visual representation of machine-learned features is a new tool for assessing patient samples and monitoring disease progression.

Abstract
Accurate diagnosis and classification of myeloproliferative neoplasms (MPNs) requires integration of clinical, morphological, and genetic findings. Despite major advances in our understanding of the molecular and genetic basis of MPNs, the morphological assessment of bone marrow trephines (BMT) is critical in differentiating MPN subtypes and their reactive mimics. However, morphological assessment is heavily constrained by a reliance on subjective, qualitative, and poorly reproducible criteria. To improve the morphological assessment of MPNs, we have developed a machine learning approach for the automated identification, quantitative analysis, and abstract representation of megakaryocyte features using reactive/nonneoplastic BMT samples (n = 43) and those from patients with established diagnoses of essential thrombocythemia (n = 45), polycythemia vera (n = 18), or myelofibrosis (n = 25). We describe the application of an automated workflow for the identification and delineation of relevant histological features from routinely prepared BMTs. Subsequent analysis enabled the tissue diagnosis of MPN with a high predictive accuracy (area under the curve = 0.95) and revealed clear evidence of the potential to discriminate between important MPN subtypes. Our method of visually representing abstracted megakaryocyte features in the context of analyzed patient cohorts facilitates the interpretation and monitoring of samples in a manner that is beyond conventional approaches. The automated BMT phenotyping approach described here has significant potential as an adjunct to standard genetic and molecular testing in established or suspected MPN patients, either as part of the routine diagnostic pathway or in the assessment of disease progression/response to treatment.
Introduction
Philadelphia-negative myeloproliferative neoplasms (MPNs) are a group of disorders in which acquired mutations in hematopoietic stem cells affecting the MPL-JAK-STAT signaling pathway drive excessive proliferation of ≥1 blood lineages.1,2 The 3 most common Philadelphia-negative MPNs (essential thrombocythemia [ET], polycythemia vera [PV], and primary myelofibrosis [PMF]) have overlapping clinical and laboratory features that can make their distinction challenging, particularly at early disease time points.3,4 In >90% of MPNs, mutations are detected in 1 of 3 genes: JAK2, CALR, and MPL. Mutations in JAK2, typically JAK2V617F, are detected in almost all cases of PV and >60% of ET and PMF, while mutations in CALR and MPL occur in ET and PMF. Approximately 5% to 10% of patients with ET and PMF do not have a detectable “driver” mutation.5-7
Currently, subjective histological features form an important component of the classification scheme central to the diagnosis of MPN and are integral to the diagnosis of various other human cancers.4,8 Recent advances in computational image analysis have the potential to transform the conventional morphological assessment of human tissues.9,10 In particular, quantification of specific cell populations and systematic description of tissue architecture can replace or augment the categorical classification systems that are currently employed in the diagnosis of cancer.11-17 Moreover, translation of advanced tissue and single-cell–based genomic and proteomic technologies into clinical strategies will require sophisticated approaches to the assessment of complex pathological tissues that are beyond the scope of routine histopathology.
In MPNs, accurate classification is crucial for optimal management, as treatment targets and the risks of progression differ among the disorders.18 In cases of ET and PMF that lack a mutation in 1 of the 3 main MPN driver genes (so-called triple negative [TN]), their distinction from a “reactive” process, for example due to chronic inflammation, remains particularly challenging. This is reflected in the revised 2016 World Health Organization (WHO) classification scheme of myeloid malignancies, in which particular emphasis is placed upon the integration of clinical, genetic, and histological features for the diagnosis of MPNs.4,8 Central to the histological interpretation of bone marrow trephines (BMTs) from suspected MPN patients is the assessment of megakaryocytes using long-established but highly subjective descriptions of their cytological and topographic features. These include variations in cell size, atypia (eg, nuclear lobulation/complexity), and cell clustering that may be subconsciously based on assessment of only a subset of the megakaryocytes in the tissue section. Despite their importance in the 2016 WHO classification system, there is controversy about the relative significance and reliability of these subjective cytological descriptions.18-22 An improved quantitative approach to the description, analysis, and classification of the complex cytological and topographical features of BMT megakaryocytes has the potential to significantly enhance the histological component of integrated MPN diagnosis.
Materials and methods
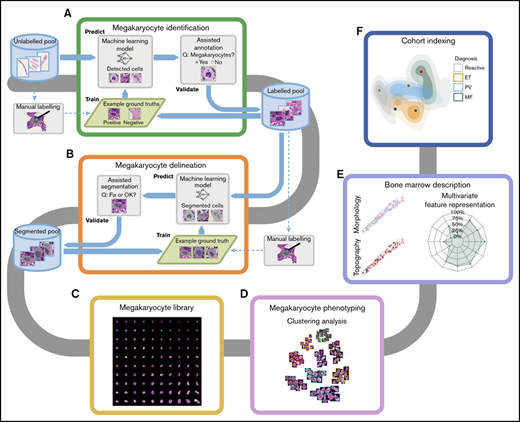
An overview of the analytical pipeline detailed below is given as Figure 1.
Overview of the computational pipeline for the disease cohort analysis of MPNs. We characterized the megakaryocyte morphology and topography of each BMT sample. To effectively build an annotated library of megakaryocytes, assisted annotation tools for identification (A) and delineation (B) have been developed. (C) A library of 62 479 annotated megakaryocytes from reactive and MPN samples was generated, of which 37 284 have been validated by a hematopathologist. (D) Clustering analysis performed on the library of megakaryocytes identified candidate phenotypes. (E) The phenotypic and topographical profile of megakaryocytes was extracted and used to create abstract representations of each trephine sample. (F) Based on these abstract representations, the analyzed samples can be represented in 2-dimensional space with new samples indexed to annotated disease cohorts.
Overview of the computational pipeline for the disease cohort analysis of MPNs. We characterized the megakaryocyte morphology and topography of each BMT sample. To effectively build an annotated library of megakaryocytes, assisted annotation tools for identification (A) and delineation (B) have been developed. (C) A library of 62 479 annotated megakaryocytes from reactive and MPN samples was generated, of which 37 284 have been validated by a hematopathologist. (D) Clustering analysis performed on the library of megakaryocytes identified candidate phenotypes. (E) The phenotypic and topographical profile of megakaryocytes was extracted and used to create abstract representations of each trephine sample. (F) Based on these abstract representations, the analyzed samples can be represented in 2-dimensional space with new samples indexed to annotated disease cohorts.
Clinical samples
BMT samples were obtained from the archive of Oxford University Hospitals NHS Foundation Trust. All specimens were of sufficient technical quality (staining and section thickness) for use in conventional histological reporting and contained ≥5 intact intertrabecular spaces. Samples were fixed in 10% neutral buffered formalin for 24 hours prior to decalcification in 10% EDTA for 48 hours. Whole-slide scanned images (Hamamatsu NanoZoomer 2.0HT/40X/NDPI files) were prepared from 4 μm hematoxylin and eosin–stained sections cut from formalin-fixed paraffin-embedded blocks. The data set comprised 131 samples (45 ET, 18 PV, 25 myelofibrosis [MF], and 43 reactive/nonneoplastic) with reactive/nonneoplastic samples sourced from patients in whom there was no evidence of bone marrow malignancy or underlying myeloid disorder. ET, PV, and MF (PMF or secondary myelofibrosis) cases represent patients in whom this was either an established or new diagnosis, satisfying the diagnostic criteria of the latest WHO classification (2016), and were designated following review by a myeloid multidisciplinary meeting. BMTs were identified from the laboratory reporting system or myeloid multidisciplinary meeting records. This work was conducted as part of the INForMeD study (INvestigating the genetic and cellular basis of sporadic and Familial Myeloid Disorders; Integrated Research Approval System ID 199833; Research Ethics Committee reference 16/LO/1376; principal investigator, A.J.M.). A summary of the key patient characteristics is provided in supplemental Table 1. The distribution of BMT sample sizes across diagnostic categories is provided in supplemental Figure 1.
Automated identification of megakaryocytes
Accurate detection and delineation of megakaryocytes was fully automated using deep-learning–based approaches. The detection task required predicting the locations of megakaryocytes on a sample (Figure 2A) using a deep neural network called Single Shot Multibox Detector that demonstrates good tradeoffs among accuracy, computational complexity, and speed when compared with other algorithms in the same class.23,24 This method generates bounding boxes and scores for the presence of megakaryocytes within each box (supplemental Methods: Detection). Following detection, image segmentation was required to partition the images into different regions to locate the boundaries of objects of interest (in this case megakaryocyte cells) (supplemental Figure 2A). This segmentation task was performed using U-Net, a method that has provided consistently good results in several medical image analysis tasks.25,26 U-Net delineates the boundaries of megakaryocytes, segmenting the cell area of interest from the background microenvironment (supplemental Methods: Segmentation).
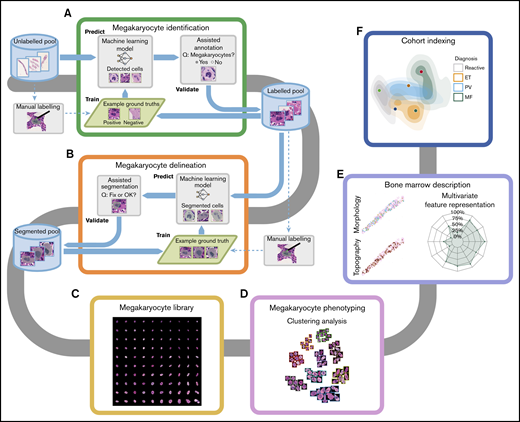
Efficiency of the assisted tool for megakaryocyte identification. Our Web-based interface for assisted annotation. (A) Candidate megakaryocytes are automatically identified by the AI algorithm, allowing hematopathologists to quickly review and confirm these as megakaryocytes (green boxes) or nonmegakaryocytes (red boxes). The human-in-the-loop assisted annotation achieved a high level of accuracy as measured by the mean average precision (mAP) within a few training iterations (B), and the annotation time was significantly reduced (C) (Wilcoxon rank-sum test, significance level 0.05).
Efficiency of the assisted tool for megakaryocyte identification. Our Web-based interface for assisted annotation. (A) Candidate megakaryocytes are automatically identified by the AI algorithm, allowing hematopathologists to quickly review and confirm these as megakaryocytes (green boxes) or nonmegakaryocytes (red boxes). The human-in-the-loop assisted annotation achieved a high level of accuracy as measured by the mean average precision (mAP) within a few training iterations (B), and the annotation time was significantly reduced (C) (Wilcoxon rank-sum test, significance level 0.05).
Megakaryocyte library
From an initial training set of 2427 manually annotated megakaryocytes taken from 14 reactive and 6 ET samples, we curated a total of 62 479 delineated megakaryocytes from the whole data set. This library was used for the identification of a set of discrete cytomorphological subtypes (Figure 1C-D) forming the basis of our quantitative approach (Figure 1E-F). To ensure accurate capture of cytomorphological information, a significant portion (60% [37 284]) of the total megakaryocytes in the library were reviewed by a hematopathologist. This was achieved using an iterative approach in which custom-built computer-assisted annotation tools highlighted individual cells within a bounding box and prompted either acceptance or rejection of the enclosed feature as a megakaryocyte (Figures 1A-B and 2A). All user interactions were logged to support an audit of this data if required (supplemental Methods: Iterative Pipeline).
Identification of cytomorphological subtypes
We learned feature representation (a numerical vector encoding the megakaryocyte cytomorphology) using a type of neural network (autoencoder) that can be trained in an unsupervised manner to learn efficient data encodings (supplemental Figures 4A and 5).24 To train the autoencoder, we used validated megakaryocytes from 43 reactive and 30 ET samples. This training was then successfully generalized to the remaining PV and MF samples (supplemental Figure 4B). Validation involved review by hematopathologists who indicated that a detected megakaryocyte was a true-positive detection, an unclear result, or a false-positive.
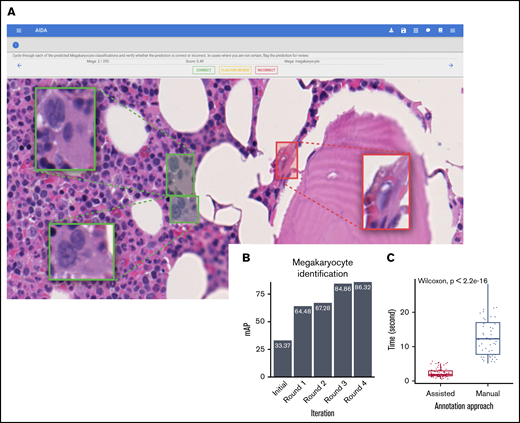
Following feature extraction, we performed a clustering analysis to group cytomorphologically similar megakaryocytes. We trained a self-organizing map on the learned latent representation vectors from samples that were reactive (n = 43 cases [7336 cells]), ET (n = 45 cases [17 535 cells]), and PV (n = 18 cases [12 413 cells]) to produce a 10-by-10 grid with 100 megakaryocyte subtypes or groups (Figure 1C).27 To further reduce the subtypes, we applied Markov clustering on the self-organizing groups with the graph structure determined by the grid configuration.28 We chose the Markov clustering result that maximizes the modularity (ie, how well nodes on the graph structure are separated into modules [clusters]) (supplemental Methods: Identification of Phenotype). This resulted in 9 distinct cytomorphological subtypes to which each megakaryocyte was assigned (Figures 1D and 3A).
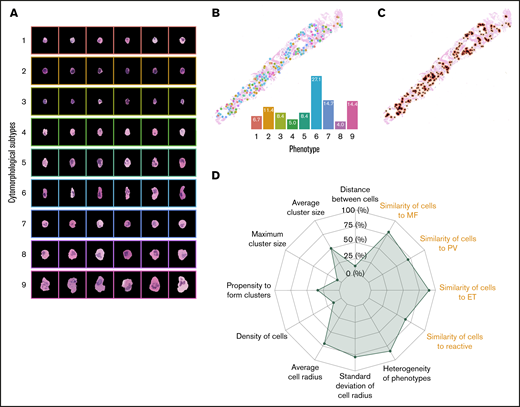
Abstract representation of BMT samples. (A) A total of 9 megakaryocytic phenotypes were automatically discovered in the unsupervised clustering analysis. Morphological representation characterized by proportions and heterogeneity of the megakaryocytic phenotypes (B) and topographical distribution (C) can be used to describe a BMT sample in the form of a radar plot (D). In total, we analyzed 17 features, 9 of which were related to the proportions of the megakaryocyte phenotypes and visualized in the form of morphological similarities to megakaryocytes in different subgroups of MPNs or reactive cases (highlighted in orange on the radar plot).
Abstract representation of BMT samples. (A) A total of 9 megakaryocytic phenotypes were automatically discovered in the unsupervised clustering analysis. Morphological representation characterized by proportions and heterogeneity of the megakaryocytic phenotypes (B) and topographical distribution (C) can be used to describe a BMT sample in the form of a radar plot (D). In total, we analyzed 17 features, 9 of which were related to the proportions of the megakaryocyte phenotypes and visualized in the form of morphological similarities to megakaryocytes in different subgroups of MPNs or reactive cases (highlighted in orange on the radar plot).
Combining megakaryocyte subtype classification and topographical features into a sample megakaryocyte “fingerprint”
In addition to determining megakaryocyte cytomorphological subtypes, we established their topographic distribution throughout the marrow space (Figures 1E and 3B-C). These features were then represented by a set of numerical values using machine learning methods to quantitatively describe the megakaryocytes in each sample (supplemental Methods: Abstract Representation of BMT Samples). These quantitative data were visually represented in the form of a radar plot, allowing samples to be directly compared (cohort indexing) (Figures 1E, 3D, and 6).
Principal-component analysis (PCA) was used to further enhance the visualization of samples in the reference cohort in an abstract 2-dimensional space (Figure 4A). This allowed a single sample to be indexed into a reference cohort using the learned PCA space. A confidence score of each specific disease subtype was determined through a Gaussian kernel density estimation conditional on each subtype in the PCA space. Where sequential patient samples were available, changes in megakaryocyte phenotype (cell subtype and topography) were readily appreciated as a shift in the location of features on the multivariable plots (Figure 6A-B). Of note, the features presented on the radar plot do not map consistently onto changes in sample location on the PCA plot; a unit change on one radar plot axis may result in a greater or smaller change on the PCA plot. This reflects the different relative weights of each feature and the dimensionality reduction when compressed onto the PCA.
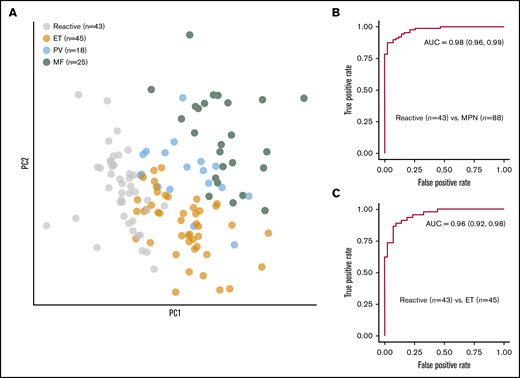
Discriminating reactive and MPN samples. (A) PCA of the abstract representations of samples from an annotated cohort showing clusters of reactive cases and MPN disease subtypes. The random forest classifier reached an AUC of 0.98 for discriminating reactive and MPN samples (B) and 0.96 for discriminating reactive and ET samples (C). PC, principal component.
Discriminating reactive and MPN samples. (A) PCA of the abstract representations of samples from an annotated cohort showing clusters of reactive cases and MPN disease subtypes. The random forest classifier reached an AUC of 0.98 for discriminating reactive and MPN samples (B) and 0.96 for discriminating reactive and ET samples (C). PC, principal component.
Results
Assisted megakaryocyte library construction
We employed a human-in-the-loop methodology to efficiently build a large library of annotated megakaryocytes (62 729 cells). Figure 2A shows the interface of our Web-based annotation tool for megakaryocyte identification. The identification tool detected candidate megakaryocytes for which the delineation tool suggested segmentation between the boundaries of the cell cytoplasm and adjacent/background structures (Figure 1A-B; supplemental Figure 2A). To ensure accuracy and quality, these predicted results were reviewed and edited by hematopathologists and fed into the artificial intelligence (AI) models for further training to iteratively improve the model performance (Figure 2B; supplemental Figure 2B-C). The tools achieved high levels of accuracy for identification (mean average precision = 0.86) and delineation (intersection over union = 0.93) within 4 training iterations. The time spent on annotating megakaryocytes with our assisted AI tools was significantly less than manual annotation (median detection time: 1.86s vs 12.29s, P = 2.2e-16; median segmentation time: 5.41s vs 8.62s, P = 3.2e-11) (Figure 2C; supplemental Figure 2D). The number of megakaryocytes identified within MPN samples was significantly greater than reactive samples, with cell numbers in PV and MF being significantly greater than ET (supplemental Figure 3A-B).
Megakaryocyte cytomorphology and topography and their diagnostic associations
An autoencoder neural network was used to identify a feature set that best captured the megakaryocyte cytomorphology. A total of 9 cytomorphological subtypes were identified through clustering analysis performed on these learned features. Several of the 9 identified subtypes have distinct cellular characteristics (Figure 3A; supplemental Figure 6). For example, subtypes 8 and 9 are large cells with atypical, polylobated nuclei. By contrast, cells of subtypes 2 and 3 are small with a high nuclear-to-cytoplasmic ratio. However, other megakaryocyte subtypes are not easily distinguished by hematopathologists, emphasizing the benefits of automated over conventional subjective assessment.
As expected, when compared with reactive samples, the MPN BMTs contained significantly more megakaryocytes, with greater average cell size and heterogeneity in cytological features (supplemental Figure 7B-E). MPN megakaryocytes were also significantly more clustered (defined as ≥2 cells in direct contact) when compared with reactive samples, as determined by the proportion of megakaryocytes within clusters, their density, and relative cluster size (supplemental Figure 7F-I).
We used the proportion of megakaryocyte subtypes (Figure 3B) and their topographical features (Figure 3D) to characterize the samples. PCA demonstrated clear separation of reactive and MPN samples, as well as separation of MPN subtypes (Figure 4A). Based on these feature representations, we trained a random forest classifier to distinguish (1) reactive (n = 43) and MPN (n = 88) samples and (2) reactive (n = 43) and ET (n = 45) samples. In fivefold cross validation (used to estimate the performance of a model by which data are split into 5 groups of approximately equal size) the classifier reached an area under the curve (AUC) of 0.98 for discriminating the reactive and MPN samples, and an AUC of 0.96 for discriminating the reactive and ET samples (Figure 4B-C; supplemental Table 9).
To determine the risk of tangentially sampled megakaryocytes introducing sampling bias in our analysis and subsequent classification confidence, each sample was divided into roughly equal parts with the abstracted features of both parts compared with the sample as a whole on the PCA plot (supplemental Figure 10). This confirmed that the calculated classification confidence of the majority of samples was not significantly influenced by selective megakaryocyte sampling.
Morphomolecular association analysis
Next, we explored whether there was an association between the megakaryocyte cytological subtypes and the underlying driver mutation status (including TN and the 2 most common driver mutations [JAK2V617F and CALR] in our cohort). Statistically significant associations were observed for 8 of the 9 megakaryocyte subtypes (Figure 5A-I). Among the associations seen, megakaryocyte subtypes 1 and 4 were significantly underrepresented in CALR mutation–bearing samples when compared with both TN- and JAK2-mutated cases, while subtype 7 was significantly increased in TN cases when compared with JAK2- and CALR-mutated samples.
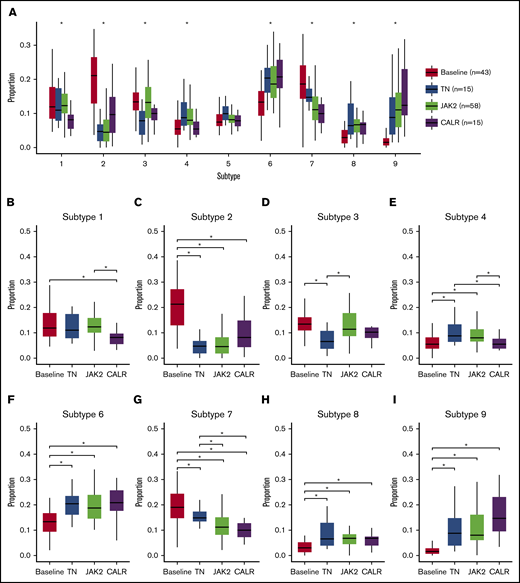
Relationship between megakaryocyte subtypes and mutational status. Distribution of megakaryocytic phenotypes with respect to underlying mutational status. Difference between groups was tested using a Kruskal-Wallis test adjusted for false discovery rate using the Benjamini-Hochberg procedure. (A) Results with P < .05 were considered statistically significant and are indicated by an asterisk. Pairwise difference between mutational groups was tested using a Wilcoxon rank-sum test adjusted for false discovery rate using the Benjamini-Hochberg procedure. (B-I) Results with P < .05 were considered statistically significant.
Relationship between megakaryocyte subtypes and mutational status. Distribution of megakaryocytic phenotypes with respect to underlying mutational status. Difference between groups was tested using a Kruskal-Wallis test adjusted for false discovery rate using the Benjamini-Hochberg procedure. (A) Results with P < .05 were considered statistically significant and are indicated by an asterisk. Pairwise difference between mutational groups was tested using a Wilcoxon rank-sum test adjusted for false discovery rate using the Benjamini-Hochberg procedure. (B-I) Results with P < .05 were considered statistically significant.
Cohort indexing
To determine the utility of cohort indexing sample cases (ie, visualizing single samples with reference to cohorts of previously analyzed cases), we analyzed sequential BMTs from 4 patients in whom clinical and laboratory follow-up data were available. This included 2 patients with evidence of stable disease: a patient with ET for whom 2 samples had been taken at an interval of 2 years, and a patient with PV who had undergone 5 bone marrow biopsies over a period of 6 years. In both cases, the sequential samples closely aggregated on the cohort PCA plot, indicating relative stability of the megakaryocyte features (Figure 6A). By contrast, analysis of serial samples from 2 MPN patients who progressed to post-ET and post-PV myelofibrosis demonstrated a marked shift in their location on the PCA plot (Figure 6B), consistent with the histological findings (supplemental Figure 8).
![Examples of disease cohort indexing. Multivariable representations of individual patient samples in the context of the analyzed disease cohorts. (A) Two patients with stable disease (ET [patient 1] and refractory PV [patient 2]) in whom sequential samples were available showed close clustering of the analyzed samples on the PCA plot. (B) Two patients with transformed ET (patient 3) and PV (patient 4) showed a marked shift in the multivariable representations of megakaryocytes between the chronic MPN samples and the transformed samples (post-ET myelofibrosis and post-PV myelofibrosis, respectively) on the PCA plot. The confidence level indicates the likelihood of samples belonging to a particular MPN classification.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/14/10.1182_bloodadvances.2020002230/1/m_advancesadv2020002230f6.png?Expires=1768289228&Signature=QQSjHiGC7Ga7iGhkKSGqZiLOiXrO0bO56rFAyuPFqeMLnjjYFeVJyb33EUW66FUrWzppPZ8YsLGlPVWvpL7gmdcdooRwFEIf4VuUmhGrQ-MIOMbK8l6HhcohEQIx9Xg6OOZ2ULlqxZPQ4XDVFZ6EPpM88VupfVvcoZO47rkimmkWJeUHvU-XP~3oJYr1rLLYaWmwYaFCUfejhzR4xMQZTEGUCa-Tv7BfzBOu5IdF5sujRgEKwlgmTHOz8N8LYUE4RooOeMgtuEqY398AKie78TZApXaSAv-C8kqZSG1y6Incxbu2tUyug0BfkcG3392f14nplIZtREB3ME4259yF1w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Examples of disease cohort indexing. Multivariable representations of individual patient samples in the context of the analyzed disease cohorts. (A) Two patients with stable disease (ET [patient 1] and refractory PV [patient 2]) in whom sequential samples were available showed close clustering of the analyzed samples on the PCA plot. (B) Two patients with transformed ET (patient 3) and PV (patient 4) showed a marked shift in the multivariable representations of megakaryocytes between the chronic MPN samples and the transformed samples (post-ET myelofibrosis and post-PV myelofibrosis, respectively) on the PCA plot. The confidence level indicates the likelihood of samples belonging to a particular MPN classification.
Examples of disease cohort indexing. Multivariable representations of individual patient samples in the context of the analyzed disease cohorts. (A) Two patients with stable disease (ET [patient 1] and refractory PV [patient 2]) in whom sequential samples were available showed close clustering of the analyzed samples on the PCA plot. (B) Two patients with transformed ET (patient 3) and PV (patient 4) showed a marked shift in the multivariable representations of megakaryocytes between the chronic MPN samples and the transformed samples (post-ET myelofibrosis and post-PV myelofibrosis, respectively) on the PCA plot. The confidence level indicates the likelihood of samples belonging to a particular MPN classification.
Discussion
In this study, we demonstrate that the complex morphological features of megakaryocytes can be captured on digital images of routinely prepared hematoxylin and eosin–stained sections and used to develop improved disease diagnosis, classification, and monitoring tools through image analysis and AI technologies. We present a novel computational method for the systematic analysis of tissue megakaryocytes and describe a set of machine learning approaches capable of associating the extracted features with an underlying diagnosis of MPN or a reactive/nonneoplastic mimic. This incorporates a platform that combines manual annotation tools with support from AI models trained on small data sets that can be used to assist hematopathologists in the efficient identification of megakaryocytes from routinely prepared BMT slides. For the first time, it is possible to perform quantitative assessment of megakaryocytic distributions, with clustering based on deeply learned features deployed to identify distinct megakaryocyte features within the marrow of normal and MPN samples. Our analysis accurately refines the association between a diagnosis of MPN (including subtype) and the distribution frequency of particular megakaryocyte cytomorphological subtypes, along with their propensity to form clusters and the cluster characteristics. Importantly, in addition to facilitating a more comprehensive analysis of an established or suspected MPN, our approaches have the potential to monitor or track the morphological and topographical changes in megakaryocytes that occur as part of normal disease progression or resulting from therapy.
The statistical description of bone marrow features is a significant departure from conventional histological approaches. Automated identification and quantitation of 9 distinct megakaryocyte cell subtypes facilitates an unbiased and accurate description of the entire megakaryocyte population within a given BMT sample. When combined with topographical data summarizing the distribution of each cell subtype throughout the marrow space, it is possible to visualize complex megakaryocyte morphological features in a manner readily appreciable by non–expert pathologists and hematologists inexperienced in trephine reporting.
A data-driven phenotyping approach is ideally suited to detailed comparisons between specific patient cohorts, such as annotated trial cohorts, patients with different mutation status (including key MPN driver mutations), and other common laboratory or clinical features. Indeed, 8 megakaryocyte subtypes were significantly associated with an MPN diagnosis. While some of these phenotypic groups (eg, large atypical cells [subtypes 8 and 9] forming clusters) are key pathological hallmarks of current classification schemes, new and more subtle morphological features were identified using this platform that would not have been recognized by conventional approaches. Further studies are required to determine the biological significance of identified megakaryocyte subtypes, for example by single-cell analyses or laser microdissection. This is supported by the finding that 8 of the 9 identified megakaryocyte subtypes were significantly associated with the underlying MPN driver mutation status, consistent with observations of mutational-phenotypic associations in ET.29 However, establishing the relationships between particular cell subtypes and the underlying mutation is complicated by the observation that each of the megakaryocyte subtypes identified in our analysis can be observed in both reactive and TN MPN samples.
The ability to review an index case in the context of a larger annotated tissue cohort is of particular value, as it allows the interpretation of megakaryocyte populations with respect to library reactive and MPN samples. Further, it is possible to visualize changes in the megakaryocyte population in serial samples over the course of disease progression, as demonstrated by clear evidence of a shift in the location of samples on the PCA plot from 2 patients who developed myelofibrosis-like appearances following progression of ET and PV. This highlights the potential of this platform in routine monitoring and assessment of patients receiving conventional or novel therapeutic interventions, particularly where hematopathologist expertise is not available. Of note, there is clear overlap of various regions describing the reactive and MPN sample subgroups on the PCA plot, consistent with the diagnostic ambiguity of a significant proportion of MPN patients when classified using current WHO criteria.
The machine learning approaches presented here have several major benefits over conventional histological analysis. Firstly, a fully automated pipeline has the potential to provide a fast and reliable initial diagnostic assessment of specimens in advance of formal pathology reporting, likely to be of value when access to hematopathology expertise is restricted (for example in low-resource health care systems). This compliments other novel AI strategies aimed at utilizing mutational and peripheral blood data for more accurate MPN diagnosis,30 along with improved interpretation of peripheral blood/bone marrow smear preparations.31-34 Secondly, a comprehensive and easily interpreted summary of the megakaryocytic population will enable the pathologist to concentrate on the “higher-level” process of integrating the broader pathological features with the clinical and laboratory findings. Thirdly, this approach is ideally suited for more accurate assessment of sequential specimens from patients undergoing treatment and/or repeated investigation, in whom quantitative morphological correlates of disease response are currently unavailable.
Although the morphological assessment of megakaryocytes is central to the histological assessment of MPNs, other features such as marrow cellularity, lineage maturation, degree of fibrosis, and blast cell estimation are required for the current WHO classification system of MPNs. It is therefore apparent that the optimal clinical application of the machine learning approaches presented here will require structured integration into a more comprehensive quantitative description of the bone marrow microenvironment. Variants of the machine learning approaches and strategies presented here are ideally suited to incorporating the accurate and objective assessment of these diverse histological features. These approaches are also readily applicable to other related hematological malignancies and a diverse range of human cancers in which quantitative histological assessment is currently unavailable.
The data sets generated and/or analyzed during the study are available from the corresponding authors upon request.
Acknowledgments
J.R. is adjunct professor of the Ludwig Oxford Branch.
The research was funded by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre. J.R. is supported through the Engineering and Physical Sciences Research Council–funded Seebibyte program (EP/M013774/1). Computation was supported by Oxford Biomedical Research Computing, a joint development between the Wellcome Centre for Human Genetics and the Big Data Institute supported by Health Data Research UK and the NIHR Oxford Biomedical Research Centre. H.T. was supported by funding from the Engineering and Physical Sciences Research Council and Medical Research Council (grant EP/L016052/1).
The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR, or the Department of Health. The funding sources had no role in the study design, collection or interpretation of data, writing of this manuscript, or the decision to submit this paper for publication. The corresponding authors (D.R. and J.R.) had full access to the data and are responsible for the decision to submit this article for publication.
Authorship
Contribution: D.R. and J.R. conceived and supervised the study with input from B.P. and A.J.M.; K.S., A.A., D.R., and J.R. designed the study; A.A. and K.S. designed the human-in-the-loop protocol for annotation; A.A. implemented Web annotation tools and collected and summarized expert annotations; K.S. implemented machine learning algorithms; D.R., G.D.H.T., and G.R. verified AI predictions for megakaryocyte identification; K.S. and H.T. reviewed AI predictions for megakaryocyte delineation; K.S. and H.T. performed statistical analyses; N.S., B.P., and A.J.M. identified samples and collected/provided access to clinical and genetic data; and K.S., A.A., H.T., J.R., and D.R. drafted the manuscript; and all authors read and have given approval of the final manuscript.
Conflict-of-interest disclosure: K.S., A.A., and J.R. are cofounders and equity holders of Ground Truth Labs. Both University of Oxford and Ground Truth Labs have intellectual property interests relevant to the work that is the subject of this paper. The remaining authors declare no competing financial interests.
Correspondence: Daniel Royston, Nuffield Division of Clinical Laboratory Sciences, University of Oxford, John Radcliffe Hospital, Oxford OX3 9DU, United Kingdom; e-mail: daniel.royston@ndcls.ox.ac.uk; and Jens Rittscher, Big Data Institute, University of Oxford, Li Ka Shing Centre for Health Information and Discovery, Old Road Campus, Oxford OX3 7LF, United Kingdom; e-mail: jens.rittscher@eng.ox.ac.uk.