Key Points
Heterozygous pathogenic germ line CSF3R variants increase risk for developing both myeloid and lymphoid malignancies.
Detection of invariant variants on tumor-based sequencing panels can be used to identify potential germ line variants.

Abstract
Colony-stimulating factor 3 receptor (CSF3R) encodes the receptor for granulocyte colony-stimulating factor (G-CSF), a cytokine vital for granulocyte proliferation and differentiation. Acquired activating heterozygous variants in CSF3R are the main cause of chronic neutrophilic leukemia, a hyperproliferative disorder. In contrast, biallelic germ line hypomorphic variants in CSF3R are a rare cause of severe congenital neutropenia, a hypoproliferative condition. The impact of heterozygous germ line CSF3R variants, however, is unknown. We identified CSF3R as a new germ line hematologic malignancy predisposition gene through analysis of 832 next-generation sequencing tests conducted in 632 patients with hematologic malignancies. Among germ line CSF3R variants, 3 were abnormal in functional testing, indicating their deleterious nature. p.Trp547* was identified in 2 unrelated men with myelodysplastic syndromes diagnosed at 76 and 33 years of age, respectively. p.Trp547* is a loss-of-function nonsense variant in the extracellular domain that results in decreased CSF3R messenger RNA expression and abrogation of CSF3R surface expression and proliferative responses to G-CSF. p.Ala119Thr is a missense variant found in 2 patients with multiple myeloma and acute lymphoblastic leukemia, respectively. This variant is located between the extracellular immunoglobulin-like and cytokine receptor homology domains and results in decreased G-CSF sensitivity. p.Pro784Thr was identified in a 67-year-old man with multiple myeloma. p.Pro784Thr is a missense variant in the cytoplasmic domain that inhibits CSF3R internalization, producing a gain-of-function phenotype and G-CSF hypersensitivity. Our findings identify germ line heterozygous CSF3R variants as risk factors for development of myeloid and lymphoid malignancies.
Introduction
With the advent of more accessible and affordable next-generation sequencing (NGS) technology, routine sequencing of hematologic malignancies is becoming commonplace. Although designed for the purpose of somatic variant detection, tumor-based NGS testing can identify germ line variants, because tumor cells contain germ line DNA, as do all tissues.1,2 For example, variants that remain at a consistent variant allele fraction (VAF) of about 50% across sequential testing and disease status time points are more likely to be of germ line origin than those with substantial fluctuation. We will henceforth refer to such variants as invariant variants. In this study, we identified unique invariant variants within the colony-stimulating factor 3 receptor (CSF3R) gene using results from tumor-based NGS panels performed in patients with hematologic malignancies.
CSF3R is a type 1 cytokine receptor that binds granulocyte colony-stimulating factor (G-CSF), a cytokine vital for granulocyte proliferation and differentiation.3 Acquired nonsense and frameshift truncation variants in the cytoplasmic domain and activating missense variants in the membrane-proximal region of CSF3R are found in a majority of cases of chronic neutrophilic leukemia (CNL).4-7 Similar variants have also been found in a minority of cases of atypical chronic myeloid leukemia, chronic myelomonocytic leukemia, and de novo acute myeloid leukemia (AML).6,8-11
Germ line homozygous and compound heterozygous nonsense and frameshift truncating variants in the extracellular domain of CSF3R are a rare cause of G-CSF–resistant severe congenital neutropenia (SCN).12,13 Other cases of SCN, resulting from germ line variants in genes such as ELANE and HAX1, frequently acquire cytoplasmic truncating CSF3R variants, akin to those found in CNL, and have a high incidence of transformation to myelodysplastic syndrome (MDS) and AML.14,15 These acquired variants are believed to occur early and drive leukemogenesis.16
Two genome-wide association studies have found an association between CSF3R and white blood cell count.17,18 Interestingly, in 1 of these studies, CSF3R variants were associated with both myeloid and lymphoid cell counts.18 Deleterious germ line heterozygous CSF3R variants that predispose patients to myeloid and/or lymphoid malignancies have not yet been described.
Materials and methods
Candidate germ line predisposition gene identification strategy
We analyzed the results from the custom OncoPlus NGS panel test19 conducted on blood and/or bone marrow collected from patients with hematologic malignancies at the University of Chicago between June 2017 and October 2019. OncoPlus comprises ∼1200 genes, from which the results of ∼150 genes are reported clinically based on their recognized importance in oncogenesis (supplemental Table 1). Details on the methods used to prioritize gene variants for further study are provided in the supplemental Data.
Testing for germ line status
Germ line DNA was extracted from cultured skin fibroblasts and/or bone marrow mesenchymal stromal cells. Genomic regions encompassing the CSF3R variants were amplified using optimized polymerase chain reaction conditions (supplemental Table 2). Additional details are provided in the supplemental Data. All tissues were collected from patients who consented to institutional review board–approved research protocols at the University of Chicago.
Generation and characterization of CSF3R-expressing cell lines
CSF3R mutants were generated in the pcDNA40-CSF3R expression vector containing the wild-type (WT) human CSF3R coding region using the GeneArt Site Directed Mutagenesis System (ThermoFisher) as previously described.20 Additional details on the characterization of CSF3R-expressing cell lines are provided in the supplemental Data. Sequences of the mutagenic oligonucleotides used are shown in supplemental Table 3.
See the supplemental Data for additional methods.
Results
Identification of CSF3R as a candidate germ line susceptibility gene
Analysis of 832 OncoPlus tests conducted in 632 patients (median age, 65 years; range, 1 month to 95 years) with hematologic malignancies revealed 990 unique variants within 33 genes. Among these patients, 128 (20.3%) had an invariant variant, seen over at least 2 time points, and 117 had invariant variants with a VAF ≥0.4 (Figure 1A).
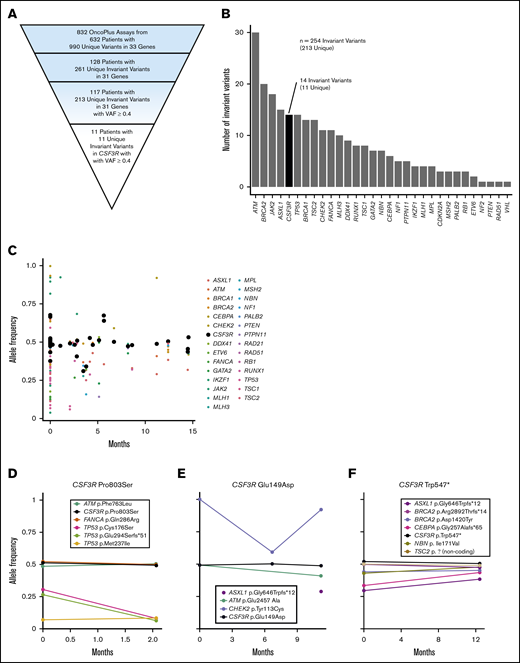
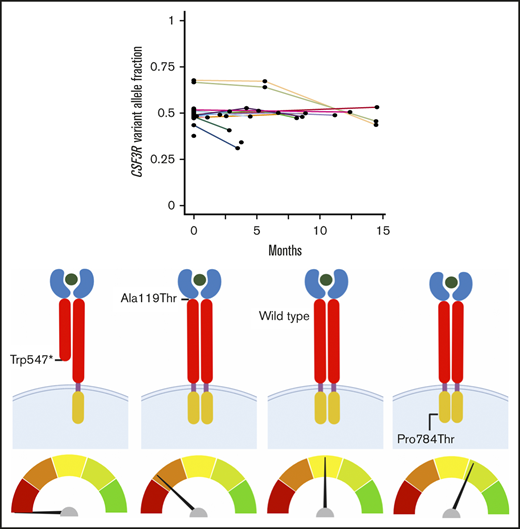
Discovery of CSF3R as a candidate germ line predisposition gene through the analysis of invariant variants on tumor-based NGS panels. (A) Pyramid chart showing the algorithm used to identify CSF3R as a candidate germ line hematologic malignancy predisposition gene. OncoPlus is a custom NGS panel of ∼1200 genes important in oncogenesis. Invariant variants are defined as variants present at consistent VAFs of about 50% in ≥2 OncoPlus tests conducted at different times from the same patient. (B) Histogram of the 31 genes found to have invariant variants with a VAF ≥0.4 in the 832 OncoPlus testing cohort displaying the number of such variants present within each of these genes. (C) Change in VAF across different testing time points for 33 genes found to be mutated among 632 patients who underwent OncoPlus testing. Time points were normalized to the date of each patient’s first test, set as time point 0 months. Variants in CSF3R remained at approximately the same VAF across multiple different testing time points. (D-F) Examples of variation in VAF across multiple testing time points for individual patients found to have variants in multiple different genes, including CSF3R. The CSF3R variants remained at a VAF of ∼0.5, whereas the VAFs of variants in most other genes fluctuated over time. p.Ala119Thr and p.Pro784Thr are not shown because the patients harboring these variants underwent a single OncoPlus test.
Discovery of CSF3R as a candidate germ line predisposition gene through the analysis of invariant variants on tumor-based NGS panels. (A) Pyramid chart showing the algorithm used to identify CSF3R as a candidate germ line hematologic malignancy predisposition gene. OncoPlus is a custom NGS panel of ∼1200 genes important in oncogenesis. Invariant variants are defined as variants present at consistent VAFs of about 50% in ≥2 OncoPlus tests conducted at different times from the same patient. (B) Histogram of the 31 genes found to have invariant variants with a VAF ≥0.4 in the 832 OncoPlus testing cohort displaying the number of such variants present within each of these genes. (C) Change in VAF across different testing time points for 33 genes found to be mutated among 632 patients who underwent OncoPlus testing. Time points were normalized to the date of each patient’s first test, set as time point 0 months. Variants in CSF3R remained at approximately the same VAF across multiple different testing time points. (D-F) Examples of variation in VAF across multiple testing time points for individual patients found to have variants in multiple different genes, including CSF3R. The CSF3R variants remained at a VAF of ∼0.5, whereas the VAFs of variants in most other genes fluctuated over time. p.Ala119Thr and p.Pro784Thr are not shown because the patients harboring these variants underwent a single OncoPlus test.
We ranked the genes containing invariant variants by the frequency of these variants without controlling for gene length (Figure 1B). Notably, many known germ line predisposition genes were among those ranked at the top, including ATM, BRCA2, TP53, BRCA1, and DDX41, suggesting that this approach can identify cancer susceptibility loci. CSF3R was the fifth ranked gene, although it had never been recognized as an autosomal dominant germ line cancer susceptibility gene. Eleven (9.4%) of the 117 patients found to have invariant variants with a VAF ≥0.4 had variants in CSF3R, accounting for 1.7% of patients within the 632-patient OncoPlus testing cohort (Figure 1A). This 1.7% represents all variants in CSF3R and therefore includes both somatic and germ line variants. Most CSF3R variants, however, remained at a VAF of ∼0.5 across sequential time points, suggestive of germ line origin (Figure 1C). Furthermore, numerous individual patients had CSF3R variants that persisted at a VAF of ∼0.5, whereas the VAFs of variants in other genes tended to fluctuate (Figure 1D-F). Given the high frequency of invariant variants found in CSF3R and the association of the gene with CNL and SCN, we evaluated its potential as a candidate germ line predisposition gene.
Identification of novel pathogenic germ line CSF3R variants
To assess CSF3R variants comprehensively, we reviewed all CSF3R variants identified in patients with hematopoietic malignancies, including those obtained from a single OncoPlus test. After CSF3R was selected for further evaluation, we identified and included an additional patient with a CSF3R p.Trp547* variant who was not included in the OncoPlus test cohort, because the patient underwent molecular testing at an outside institution. This patient was identified through review of molecular profiling results of all cases under study in the research laboratory of author L.A.G. A total of 85 patients with 91 total and 28 unique CSF3R variants were identified. Among these 28 unique CSF3R variants, 14 (50%) were confirmed to be germ line, 6 (21.4%) were determined to be somatic, and 8 (28.6%) were unconfirmed because of a lack of available germ line tissue (Figure 2; Table 1). Table 1 lists all 28 CSF3R variants identified, including those from cases outside the original OncoPlus testing cohort. Disease phenotypes for patients found to have a germ line or possible germ line CSF3R VUS, pathogenic variant, or likely pathogenic variant are listed in supplemental Table 4.
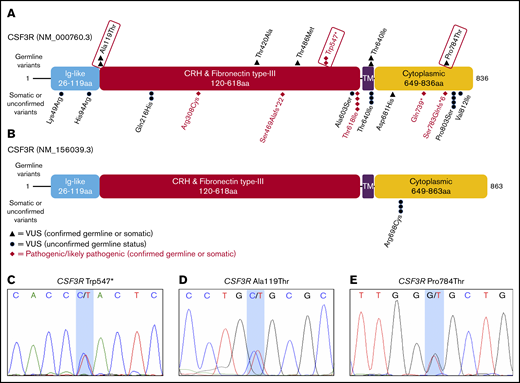
Germ line status of identified CSF3R variants. Protein schematics of CSF3R NM_000760.3 (A) and NM_156039.3 transcripts (B) showing the locations of known protein domains and 19 unique pathogenic variants, likely pathogenic variants, or variants of uncertain significance (VUSs) identified among 84 patients with CSF3R variants found on OncoPlus testing. Variants selected for further study of oncogenic potential are boxed in red. Transcript NM_156039.3 contains an additional 27 amino acid residues in the cytoplasmic domain that the primary transcript, NM_000760.3, lacks and includes the p.Arg698Cys variant. Sanger sequencing of genomic DNA extracted from cultured mesenchymal stromal cells confirmed the germ line origin of the CSF3R p.Trp547* variant in a patient with therapy-related MDS (C), the p.Ala119Thr variant in a patient with multiple myeloma (D), and the p.Pro784Thr variant in a patient with multiple myeloma (E). CRH, cytokine receptor homology; Ig, immunoglobulin; TM, transmembrane.
Germ line status of identified CSF3R variants. Protein schematics of CSF3R NM_000760.3 (A) and NM_156039.3 transcripts (B) showing the locations of known protein domains and 19 unique pathogenic variants, likely pathogenic variants, or variants of uncertain significance (VUSs) identified among 84 patients with CSF3R variants found on OncoPlus testing. Variants selected for further study of oncogenic potential are boxed in red. Transcript NM_156039.3 contains an additional 27 amino acid residues in the cytoplasmic domain that the primary transcript, NM_000760.3, lacks and includes the p.Arg698Cys variant. Sanger sequencing of genomic DNA extracted from cultured mesenchymal stromal cells confirmed the germ line origin of the CSF3R p.Trp547* variant in a patient with therapy-related MDS (C), the p.Ala119Thr variant in a patient with multiple myeloma (D), and the p.Pro784Thr variant in a patient with multiple myeloma (E). CRH, cytokine receptor homology; Ig, immunoglobulin; TM, transmembrane.
Among the 14 confirmed germ line variants, p.Trp547* was classified as pathogenic based on American College of Medical Genetics and Genomics/Association for Molecular Pathology criteria (PVS1, PM3, and PP3), 5 were classified as VUSs (p.Ala119Thr [PP3], p.Thr420Ala [PM2 and BP4], p.Thr486Met [BP4], p.Thr640Ile [PM5 and BP4], and p.Pro784Thr [PM2 and PP3]), 6 were classified as likely benign, and 2 were classified as benign (Table 1). Along with p.Trp547*, 2 of the 5 VUSs, p.Pro784Thr and p.Ala119Thr, met preset criteria for the selection of VUSs most likely to be pathogenic (supplemental Data; Table 1) and were therefore further evaluated in functional assays.
p.Trp547*, a nonsense variant in the extracellular domain of CSF3R, was found in the heterozygous state in 2 unrelated men, each with MDS. The first was a white man who had undergone surgery and chemotherapy for bladder cancer 3 years earlier at age 74 years. He was diagnosed with a therapy-related myeloid neoplasm with 9% bone marrow blasts at age 76 years. Molecular profiling revealed a CSF3R p.Trp547* variant at a VAF of 0.51 (Figure 1D-F) as well as pathogenic variants in BRCA2 (c.8673_8674del, p.Arg2892Thrfs*14), ASXL1 (c.1934dup, p.Gly646Trpfs*12), CEBPA (c.766_769dup, p.Gly257Alafs*65), SRSF2 (c.284C>A, p.Pro95His), STAG2 (c.1997del, p.Asn666Thrfs*26), and TET2 (c.1968dup, p.Ser657Leufs*24) as well as several VUSs (supplemental Table 5). The patient had a normal neutrophil count at the time of MDS diagnosis. He was treated with multiple cycles of 5-azacitidine, and after 1 year, a bone marrow biopsy showed stable disease, with 8% to 9% blasts. There were no significant changes in the molecular profile of the patient’s MDS 1 year after diagnosis, despite treatment with 5-azacitidine. The BRCA2 frameshift variant was present in both bone marrow samples at a VAF of ∼0.5 and was confirmed to be germ line. The second patient, a 33-year-old man, was initially diagnosed with aplastic anemia and was treated unsuccessfully with cyclosporine, antithymocyte globulin, and methylprednisolone. Reevaluation led to a revision in the diagnosis to hypoplastic MDS with single-lineage dysplasia. At the time of MDS diagnosis, the patient was neutropenic, with an absolute neutrophil count of 0.83 × 103/μL. The patient was subsequently treated with 5-azacitidine followed by allogeneic stem cell transplantation. The p.Trp547* variant was present in both mesenchymal stromal cells and hematopoietic cells collected from this patient before allogeneic stem cell transplantation, confirming its germ line origin. Molecular profiling was performed at an outside institution, and no other pathogenic or likely pathogenic variants were identified among 160 other genes evaluated as part of an inherited bone marrow failure panel (supplemental Table 6).
p.Ala119Thr, a missense variant positioned at the junction between the extracellular immunoglobulin-like and cytokine receptor homology domains of CSF3R, was detected in 2 unrelated white men with lymphoid malignancies: a patient with a history of melanoma who was diagnosed with immunoglobulin G κ multiple myeloma at age 57 years and another who was diagnosed with Philadelphia chromosome–negative B-cell acute lymphoblastic leukemia (ALL) at age 58 years. Both patients had a normal neutrophil count at diagnosis.
p.Pro784Thr, a rare missense variant located in the cytoplasmic domain of CSF3R, was found in a White man diagnosed with λ light chain multiple myeloma at age 67 years. The patient had a normal neutrophil count at diagnosis. After diagnosis, the patient received 4 cycles of carfilzomib, lenalidomide, and dexamethasone, followed by autologous stem cell transplantation; he remains on carfilzomib, lenalidomide, and dexamethasone for consolidation, with achievement of a stringent complete remission. Molecular profiling of the patient’s multiple myeloma also revealed a pathogenic KRAS variant (c.183A>C, p.Gln61His) at a VAF of 0.18 and VUSs in ATR, DDX41, FOXL2, PALB2, PDGFRA, and POLE (supplemental Table 5). The PALB2 (c.13C>T, p.Pro5Ser) and POLE (c.2683G>A, p.Ala895Thr) VUSs were confirmed to be germ line.
Notably, none of these patients reported a family history of hematologic malignancies. Additional clinical data, including complete blood cell counts at diagnosis, treatment history, and family history, can be found in supplemental Table 5. In total, 4 (0.6%) of the 632 patients in our OncoPlus testing cohort were found to have germ line variants in CSF3R that were deleterious.
Functional analysis of CSF3R germ line variants
To investigate the functional properties of the p.Trp547*, p.Ala119Thr, and p.Pro784Thr germ line variants, we expressed WT CSF3R or the p.Trp547*, p.Ala119Thr, or p.Pro784Thr CSF3R variants in Ba/F3 cells. Immunoblot analysis confirmed expression of all 4 CSF3R proteins (Figure 3A). Notably, Trp547* migrated as a lower molecular weight band, consistent with the location of the nonsense variant in CSF3R. Ba/F3 cells expressing the different CSF3R forms displayed varying proliferative responses to G-CSF (Figure 3B), and none conferred cytokine independence when cultured in the absence of interleukin-3 or G-CSF (data not shown). Compared with WT transfectants, p.Trp547*-transfected cells exhibited a G-CSF proliferation profile consistent with a loss-of-function phenotype, with no response to G-CSF, even at concentrations as high as 100 ng/mL. p.Ala119Thr-transfected cells exhibited a hypoproliferative phenotype, with markedly diminished proliferation observed at G-CSF concentrations <1 ng/mL. In contrast, p.Pro784Thr-transfected cells displayed a hypersensitive phenotype, with increased sensitivity to G-CSF as demonstrated by the left shift in the G-CSF dose-response curve. Examination of G-CSF responsiveness of 2 other rare germ line CSF3R variants identified in our cohort, p.Arg583His and p.Thr640Ile, which are likely benign and a VUS per American College of Medical Genetics and Genomics/Association for Molecular Pathology criteria, respectively, demonstrated no change in 50% effective concentration for G-CSF (supplemental Figure 1). p.Thr640Ile has been reported to be nontransforming in Ba/F3 cells.21
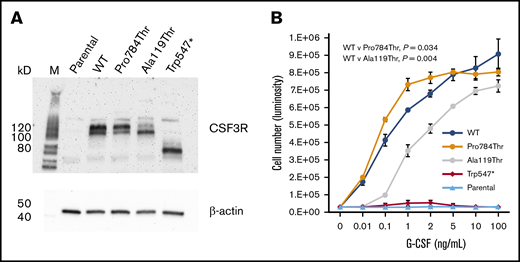
Expression and proliferative capacity of the p.Pro784Thr, p.Ala119Thr, and p.Trp547* CSF3R variants. (A) Immunoblot analysis of receptor expression in untransfected (parental) Ba/F3 cells and Ba/F3 cells transfected with WT, p.Pro784Thr, p.Ala119Thr, and p.Trp547* CSF3R. (B) Proliferative responses of cells in panel A in response to varying concentrations of G-CSF. Cell numbers were quantified using a luminescence-based assay at 48 hours after culture in the indicated concentrations of G-CSF. EC50 values calculated by dynamic curve fitting of the data for WT, p.Pro784Thr, and p.Ala119Thr were 0.25 ± 0.11, 0.05 ± 0.01, and 1.16 ± 0.08, respectively, with P values of .034 and .004 for p.Pro784Thr and p.Ala119Thr relative to WT, respectively (determined by 1-way analysis of variance). Data shown are representative of 4 independent experiments.
Expression and proliferative capacity of the p.Pro784Thr, p.Ala119Thr, and p.Trp547* CSF3R variants. (A) Immunoblot analysis of receptor expression in untransfected (parental) Ba/F3 cells and Ba/F3 cells transfected with WT, p.Pro784Thr, p.Ala119Thr, and p.Trp547* CSF3R. (B) Proliferative responses of cells in panel A in response to varying concentrations of G-CSF. Cell numbers were quantified using a luminescence-based assay at 48 hours after culture in the indicated concentrations of G-CSF. EC50 values calculated by dynamic curve fitting of the data for WT, p.Pro784Thr, and p.Ala119Thr were 0.25 ± 0.11, 0.05 ± 0.01, and 1.16 ± 0.08, respectively, with P values of .034 and .004 for p.Pro784Thr and p.Ala119Thr relative to WT, respectively (determined by 1-way analysis of variance). Data shown are representative of 4 independent experiments.
p.Trp547* CSF3R is secreted and absent from the cell surface
We investigated whether a loss of surface expression of the truncated p.Trp547* protein, resulting from deletion of the entire CSF3R transmembrane and cytoplasmic domains, could account for its observed insensitivity to G-CSF. Flow cytometry of multiple single clones generated by limiting dilution demonstrated the absence of p.Trp547* expression on the surface of transfected cells (Figure 4A). Analysis of these single clones confirmed their lack of proliferative responsiveness to G-CSF (supplemental Figure 2). To determine whether p.Trp547* CSF3R, which could be detected in whole-cell lysates when expressed as a complementary DNA transgene in Ba/F3 cells (Figure 4B lane 1), was secreted, immunoblot analysis of conditioned media harvested from p.Trp547*-expressing cells was performed and confirmed the presence of the truncated p.Trp547* CSF3R in the media (Figure 4B lane 5). There was no evidence that the WT (Figure 4B lanes 2 and 7) or p.Pro784Thr protein (Figure 4B lanes 3 and 9) was secreted.
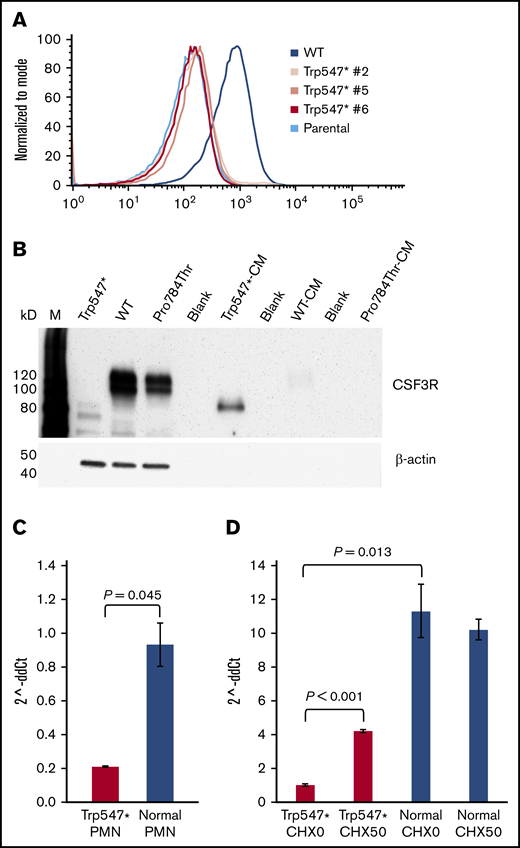
The p.Trp547* CSF3R variant abrogates receptor surface expression, induces receptor secretion, and decreases CSF3R messenger RNA (mRNA) expression. (A) Flow cytometric analysis of CSF3R (CD114) surface expression on parental Ba/F3 cells, 3 Ba/F3 clones grown from single cells expressing p.Trp547* CSF3R (Trp547* #2, Trp547* #5, and Trp547* #6), and Ba/F3 cells expressing WT CSF3R. (B) Immunoblot analysis of whole-cell lysates and conditioned media harvested from cells expressing p.Trp547* CSF3R, WT CSF3R, or p.Pro784Thr CSF3R. Lanes indicate the relevant CSF3R forms, and M denotes molecular weight markers. Lanes 1 to 3 are whole-cell lysates, and lanes 5, 7, and 9 are conditioned media (CM). Lanes 4, 6, and 8 were intentionally left empty. (C) Quantitation of CSF3R mRNA transcripts by TaqMan quantitative reverse transcription polymerase chain reaction (qRT-PCR) in neutrophils (PMNs) isolated from 3 different healthy donors (blue) and the elderly patient with the heterozygous p.Trp547* CSF3R variant (red). CSF3R mRNA expression (expressed as 2^-ddCt) in PMNs isolated from the patient with the p.Trp547* allele was 0.207 ± 0.003, compared with 0.9 ± 0.1 for PMNs isolated from 3 healthy donors. A Q test to determine if the p.Trp547* expression value was outside the range of normal values yielded a P value of .045. Error bars show standard error. (D) Analysis of nonsense-mediated decay (NMD) of CSF3R mRNA transcripts in monocytes from the same patient in panel C with a heterozygous p.Trp547* CSF3R variant. Monocytes from the patient (red) and a normal donor (blue) were incubated in vitro in the absence (CHX0) or presence of 50 μg/mL of cycloheximide (CHX50). Total CSF3R mRNA transcript levels from triplicate samples were quantified using TaqMan qRT-PCR. Basal CSF3R mRNA expression was significantly decreased in the p.Trp547* monocytes compared with normal monocytes (P = .013). After CHX treatment, a significant change in CSF3R mRNA transcripts was detected in p.Trp547* monocytes (P < .001), but not in monocytes from the normal donor (P = .570). Error bars show the standard error.
The p.Trp547* CSF3R variant abrogates receptor surface expression, induces receptor secretion, and decreases CSF3R messenger RNA (mRNA) expression. (A) Flow cytometric analysis of CSF3R (CD114) surface expression on parental Ba/F3 cells, 3 Ba/F3 clones grown from single cells expressing p.Trp547* CSF3R (Trp547* #2, Trp547* #5, and Trp547* #6), and Ba/F3 cells expressing WT CSF3R. (B) Immunoblot analysis of whole-cell lysates and conditioned media harvested from cells expressing p.Trp547* CSF3R, WT CSF3R, or p.Pro784Thr CSF3R. Lanes indicate the relevant CSF3R forms, and M denotes molecular weight markers. Lanes 1 to 3 are whole-cell lysates, and lanes 5, 7, and 9 are conditioned media (CM). Lanes 4, 6, and 8 were intentionally left empty. (C) Quantitation of CSF3R mRNA transcripts by TaqMan quantitative reverse transcription polymerase chain reaction (qRT-PCR) in neutrophils (PMNs) isolated from 3 different healthy donors (blue) and the elderly patient with the heterozygous p.Trp547* CSF3R variant (red). CSF3R mRNA expression (expressed as 2^-ddCt) in PMNs isolated from the patient with the p.Trp547* allele was 0.207 ± 0.003, compared with 0.9 ± 0.1 for PMNs isolated from 3 healthy donors. A Q test to determine if the p.Trp547* expression value was outside the range of normal values yielded a P value of .045. Error bars show standard error. (D) Analysis of nonsense-mediated decay (NMD) of CSF3R mRNA transcripts in monocytes from the same patient in panel C with a heterozygous p.Trp547* CSF3R variant. Monocytes from the patient (red) and a normal donor (blue) were incubated in vitro in the absence (CHX0) or presence of 50 μg/mL of cycloheximide (CHX50). Total CSF3R mRNA transcript levels from triplicate samples were quantified using TaqMan qRT-PCR. Basal CSF3R mRNA expression was significantly decreased in the p.Trp547* monocytes compared with normal monocytes (P = .013). After CHX treatment, a significant change in CSF3R mRNA transcripts was detected in p.Trp547* monocytes (P < .001), but not in monocytes from the normal donor (P = .570). Error bars show the standard error.
To determine whether the presence of the premature stop codon in the p.Trp547* CSF3R gene in primary cells from the elderly patient with the heterozygous p.Trp547* variant resulted in a decrease in p.Trp547* CSF3R mRNA transcription as a result of NMD, CSF3R mRNA transcription was analyzed by quantitative reverse transcription polymerase chain reaction. CSF3R mRNA expression was found to be reduced in both neutrophils and monocytes from the patient compared with healthy (normal) donor cells (Figure 4C-D). Consistent with these results, decreased CSF3R protein expression was also detected in neutrophils isolated from the same patient with the p.Trp547* allele compared with neutrophils from 2 normal donors (supplemental Figure 3). As shown in Figure 4D, incubation of monocytes from the patient with the p.Trp547* allele with cycloheximide, a potent inhibitor of NMD,22,23 resulted in a more than fourfold increase in CSF3R mRNA expression compared with untreated monocytes from the patient (P = .00001), whereas there was no change in CSF3R mRNA expression in normal donor monocytes after cycloheximide treatment. Thus, either by NMD and/or secretion of the truncated protein, the p.Trp547* variant results in a dysfunctional allele.
Aberrant signaling by p.Ala119Thr and p.Pro784Thr CSF3R proteins
G-CSF–induced phosphorylation of STAT3 and STAT5 was examined in Ba/F3 cells expressing the p.Ala119Thr or p.Pro784Thr CSF3R protein. Because p.Trp547* variants induce a loss-of-function phenotype, these studies were not performed in cells expressing p.Trp547* CSF3R. Phosphorylation of both STAT3 and STAT5 was reduced in p.Ala119Thr transfectants compared with cells expressing WT CSF3R (Figure 5A-B), indicating that the previously observed decrease in proliferation of p.Ala119Thr-transfected cells was due to defective ligand-induced receptor activation. In contrast, robust activation of both STAT5 and STAT3 in response to G-CSF was observed in cells expressing the p.Pro784Thr mutant (Figure 5C-D). Densitometric analyses of the immunoblots exhibited a delay in peak activation of STAT5 (Figure 5C) and slightly increased pSTAT3 levels (Figure 5D) in Pro784Thr cells. Increased STAT3 phosphorylation was most evident at 30 minutes, when the cells were stimulated with lower concentrations of G-CSF (supplemental Figure 4). Additionally, analysis of STAT activation as a function of G-CSF concentration demonstrated that p.Pro784Thr CSF3R produced an increase in STAT3 phosphorylation at G-CSF concentrations <2 ng/mL (Figure 6B), consistent with the hypersensitivity observed for G-CSF–induced cell proliferation (Figure 3B). Analysis of STAT dephosphorylation shown in Figure 7 demonstrated an initial delay in extinction of STAT5 phosphorylation, seen immediately after removal from 10 ng/mL of G-CSF (Figure 7A-B time 0) and STAT3 phosphorylation (Figure 7C-D) in p.Pro784Thr cells. Although the observed trends in delayed STAT5 and STAT3 dephosphorylation did not reach statistical significance, these data are consistent with the sustained STAT3 activation shown in Figure 5 and supplemental Figure 4. Delays in peak CSF3R signaling and prolonged STAT activation have been reported for other mutant CSF3R forms defective in internalization.24-26 Notably, p.Pro784 lies within a region in CSF3R previously shown to mediate receptor internalization.27 Moreover, defective receptor downregulation and persistence of cell surface binding have also been shown to induce G-CSF hyperresponsiveness,24 prompting us to examine internalization of p.Pro784Thr CSF3R (Figure 7E). Although we observed no differences in the basal level of CSF3R on the cell surface (P = .7), G-CSF induced rapid downregulation of WT CSF3R, with a 50% reduction at 30 minutes. In contrast, a 35% reduction in cell surface receptor was detected in p.Pro784Thr-expressing cells at the same time point, indicative of delayed receptor internalization.
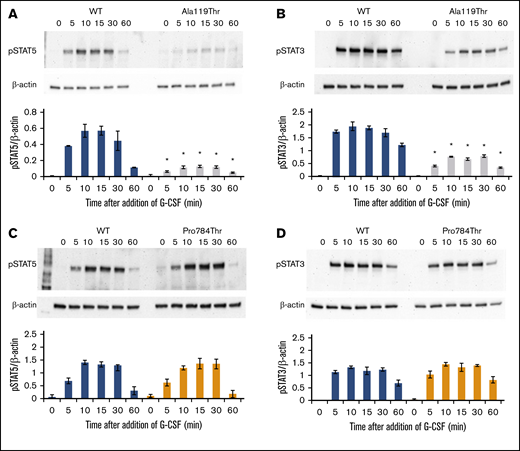
p.Ala119Thr CSF3R variants result in defective STAT activation, whereas p.Pro784Thr CSF3R variants show a trend toward increased STAT activation. Cells expressing p.Ala119Thr CSF3R (A-B), p.Pro784Thr CSF3R (C-D), or WT CSF3R (A-D) were serum and cytokine deprived for 2 hours and then stimulated with 10 ng/mL of G-CSF for the indicated times in minutes. Whole-cell lysates were immunoblotted with antibodies to phosphorylated STAT5 (pSTAT5), pSTAT3, and β-actin. Representative blots of 4 independent experiments for each CSF3R variant are shown. Densitometric analyses of the immunoblots were performed using ImageJ software. The data are presented as the ratios of pSTAT/β-actin. Error bars show the standard error. *P < .05.
p.Ala119Thr CSF3R variants result in defective STAT activation, whereas p.Pro784Thr CSF3R variants show a trend toward increased STAT activation. Cells expressing p.Ala119Thr CSF3R (A-B), p.Pro784Thr CSF3R (C-D), or WT CSF3R (A-D) were serum and cytokine deprived for 2 hours and then stimulated with 10 ng/mL of G-CSF for the indicated times in minutes. Whole-cell lysates were immunoblotted with antibodies to phosphorylated STAT5 (pSTAT5), pSTAT3, and β-actin. Representative blots of 4 independent experiments for each CSF3R variant are shown. Densitometric analyses of the immunoblots were performed using ImageJ software. The data are presented as the ratios of pSTAT/β-actin. Error bars show the standard error. *P < .05.
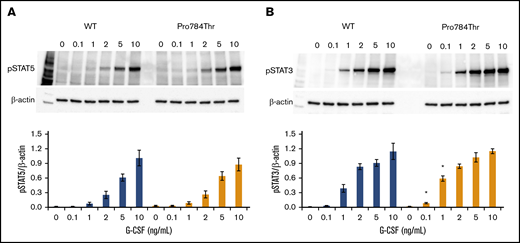
G-CSF hypersensitivity of the p.Pro784Thr CSF3R variant correlates with STAT3 phosphorylation responses. Cells expressing p.Pro784Thr CSF3R or WT CSF3R were serum and cytokine deprived for 2 hours and then stimulated with the indicated concentrations of G-CSF for 30 minutes. (A-B) Whole-cell lysates were immunoblotted with antibodies to phosphorylated STAT5 (pSTAT5), pSTAT3, and β-actin. Representative blots of 3 independent experiments are shown. Densitometric analyses of the immunoblots were performed using ImageJ software. The data are presented as the ratios of pSTAT/β-actin. Error bars show the standard error. *P < .05.
G-CSF hypersensitivity of the p.Pro784Thr CSF3R variant correlates with STAT3 phosphorylation responses. Cells expressing p.Pro784Thr CSF3R or WT CSF3R were serum and cytokine deprived for 2 hours and then stimulated with the indicated concentrations of G-CSF for 30 minutes. (A-B) Whole-cell lysates were immunoblotted with antibodies to phosphorylated STAT5 (pSTAT5), pSTAT3, and β-actin. Representative blots of 3 independent experiments are shown. Densitometric analyses of the immunoblots were performed using ImageJ software. The data are presented as the ratios of pSTAT/β-actin. Error bars show the standard error. *P < .05.
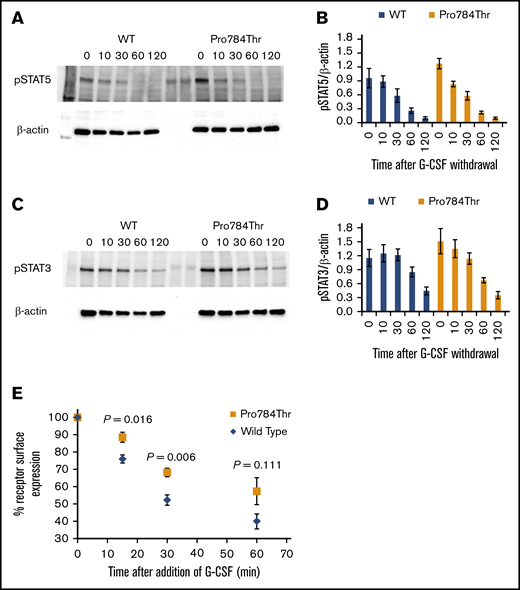
STAT dephosphorylation and delayed internalization of the p.Pro784Thr CSF3R variant. (A,C) Cells expressing p.Pro784Thr CSF3R or WT CSF3R were serum and cytokine deprived for 2 hours, stimulated with 10 ng/mL of G-CSF for 10 minutes, removed from G-CSF–containing media, and centrifuged and resuspended in lysis buffer at the indicated times in minutes. Whole-cell lysates were immunoblotted with antibodies to phosphorylated STAT5 (pSTAT5), pSTAT3, and β-actin. Representative blots of 3 independent experiments are shown (A,C), and densitometric analyses of the immunoblots were performed using ImageJ software (B,D). The data are presented as the ratios of pSTAT/β-actin. Error bars show the standard error. P values were calculated using Student t test. Statistical significance (P < .05) was not met for any of the time points; however, there was a trend toward increased pSTAT5/β-actin at 0 minutes (P = .104). (E) Receptor internalization kinetic data are shown from flow cytometric analyses of cells expressing WT or p.Pro784Thr CSF3R. Data are expressed as a percentage relative to the quantity of surface-bound CSF3R before the addition of G-CSF. WT and p.Pro784Thr receptor surface expressions were compared, and P values calculated using Student t test were .016, .006, and .111 at 15, 30, and 60 minutes, respectively.
STAT dephosphorylation and delayed internalization of the p.Pro784Thr CSF3R variant. (A,C) Cells expressing p.Pro784Thr CSF3R or WT CSF3R were serum and cytokine deprived for 2 hours, stimulated with 10 ng/mL of G-CSF for 10 minutes, removed from G-CSF–containing media, and centrifuged and resuspended in lysis buffer at the indicated times in minutes. Whole-cell lysates were immunoblotted with antibodies to phosphorylated STAT5 (pSTAT5), pSTAT3, and β-actin. Representative blots of 3 independent experiments are shown (A,C), and densitometric analyses of the immunoblots were performed using ImageJ software (B,D). The data are presented as the ratios of pSTAT/β-actin. Error bars show the standard error. P values were calculated using Student t test. Statistical significance (P < .05) was not met for any of the time points; however, there was a trend toward increased pSTAT5/β-actin at 0 minutes (P = .104). (E) Receptor internalization kinetic data are shown from flow cytometric analyses of cells expressing WT or p.Pro784Thr CSF3R. Data are expressed as a percentage relative to the quantity of surface-bound CSF3R before the addition of G-CSF. WT and p.Pro784Thr receptor surface expressions were compared, and P values calculated using Student t test were .016, .006, and .111 at 15, 30, and 60 minutes, respectively.
Discussion
We have identified germ line CSF3R variants as rare risk alleles for the development of hematopoietic malignancies through screening invariant variants identified in patients with hematologic malignancies. In addition to identifying germ line susceptibility loci, this approach can also detect genes for which somatic invariant variants are common and persistent if no disease-directed treatment is administered or if treatment is ineffective, such as JAK2 and ASXL1 variants.
We used functional testing to identify 3 heterozygous germ line CSF3R variants that encode proteins with altered function. In 2 cases, the CSF3R variant was hypomorphic, and in 1 case, the variant showed gain of function. Other germ line predisposition syndromes exhibit the phenomenon of either hypo- or hypermorphic alleles conferring risk to hematopoietic malignancies of either myeloid or lymphoid lineage. For example, germ line RUNX1 mutations are most often hypomorphs,28 but some variants show increased activity when tested in vitro,29 and significant phenotypic heterogeneity has been observed in families with germ line RUNX1 variants.30
We identified 3 deleterious CSF3R variants. First, p.Trp547*, a nonsense variant, was found in 2 unrelated patients with MDS. This variant resulted in diminished mRNA expression in patient neutrophils and monocytes and a loss-of-function phenotype with absent CSF3R cell surface expression in transfected Ba/F3 cells. This is the first study confirming the loss-of-function phenotype of p.Trp547* and the first describing its association with hematologic malignancies when present in the heterozygous state. p.Trp547* was previously found to be pathogenic in a compound heterozygous state with c.998-2A>T in an infant with SCN, and as we observed in our elderly patient, this infant also lacked CSF3R expression on the surface of neutrophils and monocytes.12 Despite the p.Trp547* variant CSF3R allele, our elderly patient was not neutropenic. Consistent with this observation, coexpression of p.Trp547* with WT CSF3R did not alter proliferative responsiveness to G-CSF (supplemental Figure 5). The absence of neutropenia and advanced age at onset of MDS in this case suggest that heterozygosity of the p.Trp547* allele and CSF3R haploinsufficiency are insufficient to induce MDS and that additional acquired pathogenic variants are required for malignant transformation. In this case, we suspect that the germ line BRCA2 variant along with acquisition of pathogenic variants in ASXL1, CEBPA, SRSF2, STAG2, and/or TET2 contributed to the development of malignancy. The hypothesis that additional pathogenic variants are required for development of malignancy is consistent with evidence from the SCN literature, where acquisition of CSF3R variants precedes nearly all cases of SCN-associated MDS/AML.15 In cases of SCN-associated myeloid neoplasms, latency periods vary, and acquisition of additional variants in genes such as ASXL1 and RUNX1 occurs before leukemic evolution.15,31 Our data support the postulate that CSF3R-mutated hematopoietic stem cell clones are preleukemic, with a high propensity for secondary hits and subsequent leukemic transformation.15
Second, p.Ala119Thr, a missense variant, was identified in 2 patients, 1 with multiple myeloma and 1 with ALL, resulting in hyposensitivity to G-CSF. This mutation localizes to the previously identified ligand binding domain of CSF3R.32,33 The observed reduction in proliferative response and defective STAT activation detected in p.Ala119Thr transfectants are consistent with prior reports of other CSF3R variants within the same extracellular region that demonstrated an increase in the 50% effective concentration for G-CSF and decreased ligand binding capacity.32 The p.Arg308Pro variant, which was identified in a family with SCN, is another germ line missense variant in the extracellular domain of CSF3R that results in decreased ligand-induced JAK/STAT activation.13
Third, p.Pro784Thr was identified as a novel germ line heterozygous gain-of-function missense variant in the cytoplasmic domain of CSF3R in a patient with multiple myeloma. Missense activating germ line CSF3R variants have previously been reported. p.Thr640Asn, located within the transmembrane domain, was identified in a family with chronic neutrophilia, including 1 family member who was diagnosed with MDS.34 The membrane-proximal p.Asn610His and p.Asn610Ser variants were identified in 2 patients with myeloproliferative neoplasms and shown to confer ligand-independent signaling.35 p.Thr618Ile has been identified as a germ line heterozygous variant in patients with CNL, including a child with a de novo mutation who had had persistent leukocytosis since birth36 and 3 individuals with CNL from a 2-generation pedigree.37
Gain-of-function somatic cytoplasmic CSF3R variants producing G-CSF hypersensitivity and impaired receptor internalization, like the p.Pro784Thr variant that we have identified, have been reported and associated with progression to AML in patients with SCN.38-40 However, Pro784Thr is unique in that it alters an identified receptor internalization domain without producing a truncation, thus leaving the cytoplasmic domain and the associated regulatory regions intact, potentially contributing to the observed phenotype.27,41 To our knowledge, ours is the first description of a germ line gain-of-function missense variant located within the cytoplasmic domain of CSF3R and also the first description of a germ line CSF3R variant associated with a lymphoid lineage malignancy. CSF3R is typically not thought to be expressed on the surface of lymphoid cells; however, CSF3R variants have been detected in lymphoid malignancies, and patients with SCN are at risk of developing lymphoid as well as myeloid malignancies.14,16,42,43 Aberrant expression of CSF3R on lymphoblasts has also been described.14,42,44 Collectively, these reports support our findings and assertion that germ line heterozygous CSF3R pathogenic variants predispose to hematologic malignancies of both myeloid and lymphoid lineages.
The association of pathogenic CSF3R variants with both myeloid and lymphoid malignancies is not unique. This dual association has also been described for many other germ line susceptibility genes, such as RUNX1, DDX41, and ETV6. In addition to myeloid malignancies, germ line RUNX1 variants predispose to T-cell ALL and less frequently to B-cell malignancies.30,45 Germ line DDX41 variants have been found to predispose to MDS/AML as well as a wide array of lymphoid malignancies, including non-Hodgkin lymphoma, Hodgkin lymphoma, and multiple myeloma.46,47 ETV6 predisposes to B-cell ALL most commonly and less frequently to myeloid malignancies.48
A previous study described 2 families with SCN resulting from inherited biallelic CSF3R variants.13 The parents of 1 family, who had 3 children with SCN resulting from homozygous p.Arg308Cys variants, were a healthy consanguineous couple with normal neutrophil counts at the time of sampling. The parents of the second family, who had 1 child with SCN who was compound heterozygous for p.Gly316fsTer322 and p.Gly415fsTer432 variants, were healthy and also had normal neutrophil counts at the time of sampling. It should be noted that both sets of parents were likely young at the time their neutrophil counts were recorded, because the oldest child in this series was 5 years of age. It is unknown whether the parents subsequently developed a hematologic malignancy, and like many established germ line predisposition genes, the penetrance of germ line heterozygous CSF3R variants is unknown. Although heterozygous CSF3R variants alone may be insufficient to trigger malignant evolution, they may act as a genetic risk factor that predisposes to hematologic malignancies through a propensity toward and cooperation with secondary hits. Additional studies will be required to fully understand the mechanisms by which heterozygous CSF3R variants can predispose to both myeloid and lymphoid malignancies.
One of our patients harboring the p.Trp547* CSF3R variant was also found to have a germ line pathogenic BRCA2 variant, and the patient with the p.Pro784Thr variant had a germ line PALB2 VUS (supplemental Table 5). PALB2 is vital for the function of BRCA2.49 Pathogenic germ line variants in both BRCA2 and PALB2 are known to confer an increased risk of breast cancer, and pathogenic variants in BRCA2, which is a Fanconi anemia gene also known as FANCD1, are associated with leukemia.50,51 The contribution of these variants to the patients’ hematologic malignancies and the significance of their cooccurrence with germ line CSF3R variants are unclear. Because of a lack of available data, it is unknown whether the other patient with a p.Trp547* CSF3R variant had additional similar germ line variants.
It is interesting that all 5 patients with germ line deleterious CSF3R variants were men. Some germ line predisposition genes, such as DDX41, are known to exhibit higher penetrance in men.52 However, because of the small number of germ line CSF3R variants identified in our cohort, we cannot determine the relevance of this observation.
In conclusion, we have identified germ line CSF3R variants as a novel predisposition for the development of both myeloid and lymphoid malignancies. Specifically, in vitro functional testing demonstrated deleterious loss-of-function effects of p.Trp547* and p.Ala119Thr and gain-of-function effects of p.Pro784Thr CSF3R variants. Additional studies should help to elucidate the precise mechanisms by which these variants predispose to hematologic malignancies. We show that detection of invariant variants on tumor-based NGS panels can be used to identify potential germ line variants to aid in new germ line syndrome discovery.
For data sharing requests, e-mail the corresponding author, Lucy A. Godley (lgodley@medicine.bsd.uchicago.edu).
Acknowledgments
This work was supported by a Helios Scholarship Fund (A.M.T.), the Kerry and Simone Vickar Family Foundation (B.R.A.), the Leon Levine Foundation (B.R.A.), and Swim Across America (B.R.A.).
Authorship
Contribution: I.L.K. and L.A.G. analyzed next-generation sequencing data and initially identified CSF3R as a potential candidate predisposition gene; M.H., J.P.S., and S.D. performed and analyzed next-generation sequencing data; S.F. analyzed next-generation sequencing data and contributed a patient to the cohort; A.M.T., L.J.D., and A.L. performed experiments; A.M.T., L.J.D., I.L.K., A.L., B.R.A., and L.A.G. analyzed results; A.M.T., L.J.D., and I.L.K. created the figures; and A.M.T., L.J.D., I.L.K., A.L., B.R.A., and L.A.G. designed the research and wrote the paper.
Conflict-of-interest disclosure: B.R.A. receives royalties from a coauthored article on evaluation of neutropenia in BMJ Best Practice and is a member of an advisory committee for Juno Therapeutics. L.A.G. receives royalties from a coauthored article on inherited hematopoietic malignancies in UpToDate, Inc., and is on the scientific advisory board of Invitae, Inc. The remaining authors declare no competing financial interests.
The current affiliation for A.M.T. is Division of Hematology, QEII Health Sciences Centre, Dalhousie University, Halifax, NS, Canada.
The current affiliation for M.H. is Invitae, Inc., San Francisco, CA.
Correspondence: Belinda R. Avalos, Levine Cancer Institute, 1021 Morehead Medical Dr, Charlotte, NC 28204; e-mail: belinda.avalos@atriumhealth.org; or Lucy A. Godley, University of Chicago, 5841 S Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: lgodley@medicine.bsd.uchicago.edu.