

Key Points
Deletion of individual residues in the FLT-3 juxtamembrane domain, such as Q575Δ, can be activating similar to the classic ITD mutation.
A systematic analysis informed by NGS points to a novel class of mutation that can be targeted with FLT-3 inhibitors.

Abstract
The FMS-like tyrosine kinase 3 (FLT-3) is the most frequently mutated gene in acute myeloid leukemia (AML), a high-risk feature, and now the target of tyrosine kinase inhibitors (TKIs), which are approved and in development. The most common mutation is the internal tandem duplication (ITD). We present a novel mutation, FLT-3/Q575Δ, identified in a patient with AML through next-generation sequencing (NGS). This mutation is activating, drives downstream signaling comparable to FLT-3/ITD, and can be targeted using available FLT-3 TKIs. We present the results of a systematic analysis that identified Y572Δ, E573Δ, and S574Δ as similarly activating and targetable deletions located in the FLT-3 juxtamembrane domain (JMD). These mutations target key residues in the JMD involved in the interactions within FLT-3 that regulate its activation. Our results suggest a new class of FLT-3 mutations that may have an impact on patient care and highlight the increasing importance of a systematic understanding of FLT-3 mutations other than ITD. It is likely that, as NGS becomes more commonly used in the diagnosis of patients with AML, these and other activating mutations will be discovered with increasing frequency.
Introduction
FMS-like tyrosine kinase-3 (FLT-3) is the most frequently deleted gene in acute myeloid leukemia (AML), with pathogenic mutations occurring in up to 30% of pediatric and adult cases.1 Activating mutations lead to constitutive autophosphorylation and downstream signaling, with subsequent increases in proliferation, differentiation blockade, and decreased apoptosis. The internal tandem duplication (ITD) is the most clinically significant class of FLT-3 mutations, and its presence constitutes a high-risk feature in AML, associated with increased rates of resistance, induction failure, relapse, and decreased overall survival.2 The ITD comprises a class of mutations that occur within the juxtamembrane domain (JMD) and adjacent tyrosine kinase domain (TKD) and disrupt the normal secondary and tertiary structures of FLT-3, preventing interactions between the JMD and the activation loop and allowing for constitutive activation of the protein kinase domain.3,4
Despite the relative chemoresistance conferred by FLT-3/ITD, AML blasts that harbor this mutation remain sensitive to FLT-3 tyrosine kinase inhibitors (TKIs), and, as such, FLT-3 remains an attractive target for drug development,5 prompting clinical trials that led to the recent approvals of midostaurin (Rydapt; Novartis) and gilteritinib (Xospata; Astellas), with additional trials actively investigating other TKIs, including quizartinib (AC220), sorafenib, and crenolanib in upfront, relapsed/refractory, and posttransplant settings.6-9 Although research continues to develop more potent and effective TKIs with fewer side effects, the application of FLT-3 TKIs has already led to improved outcomes in the pretransplant setting, with promising trials in the posttransplant setting under way.
With the promise of FLT-3 TKI therapy becoming a practical reality and given the role of FLT-3 as one of the most risk-defining mutations in AML, the proper identification of targetable FLT-3 mutations is becoming increasingly critical. The traditional diagnostic method for detecting FLT-3/ITD mutations is polymerase chain reaction (PCR) amplification of the FLT-3 JMD.10 FLT-3/ITD mutations are identified by the presence of an additional PCR product longer than the wild-type FLT-3 fragment, with the relative abundance of that additional fragment allowing for direct measurement of the variant allele frequency. The recent advent of next-generation sequencing (NGS) has provided clinicians with a new way of examining the FLT-3 status of patients with AML. With these new technologies, it is now possible to examine FLT-3 and other genes with nucleotide level resolution and identify gene derangements that previous PCR-based techniques would not have detected. With this new level of precision, a need has arisen for more complete information regarding the biological and clinical significance to assist clinicians in interpreting this new wealth of potentially treatment-informing data. Although databases such as the Catalog of Somatic Mutations in Cancer (COSMIC) provide repositories for such information, a systematic approach has not been undertaken to couple these correlative data with validated testing.11
We describe a novel deletion of a single amino acid residue in the FLT-3 JMD that was identified in a patient through NGS. The biochemical and genetic studies described demonstrate that this mutation is an activator in a manner molecularly similar to ITD. We present a brief review of existing literature reports and conduct a survey of publicly available clinical data that show small mutations in this domain, including deletions, represent a previously unappreciated class of FLT-3 mutations. We have undertaken a systematic analysis of single-residue deletions in this region and have identified a cluster of residues with similarly activating deletions, defining a novel class of FLT-3 mutations with clinical importance and relevance for future trial designs, which will become more critical with the growing clinical use of NGS and other genetic tools.
Methods
Deletion constructs
Wild-type FLT-3 was cloned into the pBABE expression vector with selectable puromycin resistance, as previously described.12,13 pBABE/FLT-3 was subjected to site-directed mutagenesis with the QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s protocols, using primers tiled across the region containing single amino acid residue deletions (supplemental Table 1). Mutant FLT-3 sequences were confirmed by Sanger sequencing.
Cell culture
The interleukin-3 (IL-3)–dependent murine cell line, Ba/F3 (ATCC), was grown in RPMI-1640 medium supplemented with 10% fetal calf serum and antibiotics. Nontransformed cells were maintained in 1 ng/mL recombinant murine IL-3 (rm IL-3; R&D Systems). Two million cells were transfected with 2 µg of pBABE construct, using the Amaxa Nucleofector II (Lonza), selecting in 2 mg/mL puromycin for 1 week, and tapering to 1.5 mg/mL for an additional week. To create stable, FLT-3-dependent cell lines, the cells were washed, and replated in rm IL-3–free medium.
Analysis of FLT-3 activity
Ba/F3 cells stably expressing FLT-3 constructs were washed to remove rm IL-3. The cells were grown in cytokine-free medium for 4 hours and then harvested for cell lysis and phospho-western analysis. To test TKI inhibition, the cells were exposed to a range of concentrations of drug for 1 hour before harvest. Cells expressing wild-type FLT-3 were exposed to 10 ng/mL FLT-3 ligand (R&D Systems) for 10 minutes before harvest. Inhibition of cell proliferation was measured by culturing FLT-3–expressing Ba/F3 cells in a range of concentrations of TKIs for 48 hours in 96-well format (25 000 cells per well), after which the cells were exposed to 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) for 4 hours and then lysed overnight in 5% sodium dodecyl sulfate with 0.05 M hydrochloric acid. Optical density at 570 nm was measured with an iMark (Bio-Rad) plate reader and software. Analysis was performed using the Excel and CompuSyn (ComboSyn, Inc) platforms.
Deletion screening
After Ba/F3 cells were transduced with the pBABE/FLT-3 mutation constructs and puromycin selected, the cells were plated in a 96-well format in the presence or absence of rm IL-3. The cells were cultured for 5 days, and growth was measured every 24 hours with the MTT assay. For pooled studies, the pBABE/FLT-3 mutation constructs were pooled in equimolar concentrations and transfected into Ba/F3 cells. After puromycin selection and expansion in rm IL-3, the cells were allowed to grow for 6 days in the absence of rm IL-3. The cells were then collected, and genomic DNA was purified. The JMD was amplified via PCR using the AmpliTaq Gold (Roche) system according to the manufacturer’s protocol, with the following primers: forward, GCCCCTTCCCTTTCATCCAA, and reverse, CTTTCAGCATTTTGACGGCAACC. The products were cloned into the pCR3-TOPO vector with the TOPO-TA cloning kit (Invitrogen) and transformed into the chemically competent One Shot TOP10 (Invitrogen), and single colony transformants were selected, prepped, and sequenced using the M13R standard sequencing primer.
Mutation frequency analysis
The COSMIC database was queried for all reported mutations in FLT-3. Mutations were curated to ensure defined locations, changes, and effects on protein coding. Permutation analysis (1 × 106 randomizations) was used to generate expectation distributions of different mutation types. Analysis was performed on the R platform. To increase sensitivity, known mutational hotspots (D835 and I836) were excluded from the analysis.
Results
Case
The patient was a 46-year-old woman presenting with fevers and ecchymoses who was found to have anemia (5.5 g/dL), thrombocytopenia (<10 000/µL), and leukocytosis. She had a total white blood cell count of 94 × 103/μL leukocytes with 90% blasts. Central nervous system (CNS) status evaluation was negative. Flow cytometry demonstrated low CD45 and low side scatter; positive staining for CD13, CD33, CD117, and CD34; and partial loss of HLA-DR. Karyotype and fluorescence in situ hybridization were normal. She was enrolled in a phase 1B trial of standard cytarabine and daunorubicin (7+3) induction chemotherapy in combination with gilteritinib (ASP2215), beginning on day 4 for all patients with newly diagnosed AML, regardless of mutation status (registered on www.clinicaltrials.gov as #NCT02236013). Despite her significantly elevated counts at presentation, a common finding in FLT-3–mutated AML, and rapid clearance of blasts with the initiation of FLT-3–directed therapy (Figure 1A), standard rapid PCR-based FLT-3 testing was negative for an ITD mutation. However, by the second week of induction, while still on protocol-based FLT-3 TKIs, NGS sequencing demonstrated a 3-nucleotide deletion leading to an in-frame, single-residue loss of the glutamine at position 575 (FLT-3/Q575Δ), and an IDH2/R172K mutation, both with variant allele frequencies >40%. Query of the COSMIC database indicated that this was a unique FLT-3 mutation.
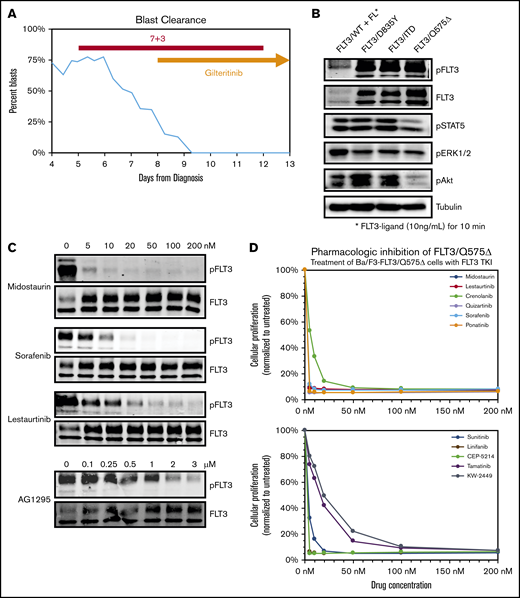
Deletion of Q575 leads to targetable activation of FLT-3 in a patient with AML. (A) Circulating blasts as a percentage of total leukocytes in a patient with AML found to harbor a novel FLT-3/Q575Δ mutation. Timing of induction chemotherapy (7+3) and gilteritinib is indicated. (B) Expression of different FLT-3 mutations, including Q575Δ, in Ba/F3 cells leads to autophosphorylation of FLT-3 and downstream activation of signaling pathways, as detected by phospho-western analysis of whole-cell lysates. For wild-type FLT-3, cells were simulated with FLT-3 ligand before lysis. (C) Cells expressing FLT-3/Q575Δ were treated with increasing concentrations of the indicated FLT-3 TKIs, and total and phosphorylated FLT-3 was detected by western blot analysis. (D) Cells were cocultured in increasing concentrations of the indicated TKIs, and growth was measured by colorimetric MTT assay after 48 hours.
Deletion of Q575 leads to targetable activation of FLT-3 in a patient with AML. (A) Circulating blasts as a percentage of total leukocytes in a patient with AML found to harbor a novel FLT-3/Q575Δ mutation. Timing of induction chemotherapy (7+3) and gilteritinib is indicated. (B) Expression of different FLT-3 mutations, including Q575Δ, in Ba/F3 cells leads to autophosphorylation of FLT-3 and downstream activation of signaling pathways, as detected by phospho-western analysis of whole-cell lysates. For wild-type FLT-3, cells were simulated with FLT-3 ligand before lysis. (C) Cells expressing FLT-3/Q575Δ were treated with increasing concentrations of the indicated FLT-3 TKIs, and total and phosphorylated FLT-3 was detected by western blot analysis. (D) Cells were cocultured in increasing concentrations of the indicated TKIs, and growth was measured by colorimetric MTT assay after 48 hours.
Deletion of Q575 activates FLT-3
To investigate the biological activity of this mutation, we used site-directed mutagenesis to generate it in a wtFLT-3 cDNA construct and transduced the IL-3–dependent, lymphoblastic murine cell line, Ba/F3, to express FLT-3/Q557Δ, as well as wild-type FLT-3, FLT-3/ITD, and the activating FLT-3 TKD mutation D835Y. Similar to the FLT-3/ITD and FLT-3/D835Y constructs, expression of FLT-3/Q575Δ rendered transduced Ba/F3 cells to IL-3–independent growth. Western blot analysis of whole-cell lysates from these cells demonstrated that expression of FLT-3/Q575Δ leads to activation of downstream signaling through STAT5, AKT, and ERK1/2 (Figure 1B), in a pattern similar to FLT-3/ITD. Of note, this downstream activation, similar to FLT-3/ITD and other activating mutations, leads to more robust signaling than stimulation of wild-type FLT-3 with ligand (B.N. and D.S., unpublished data, western analysis of downstream targets).
FLT-3/Q575Δ is sensitive to TKI inhibition
To confirm that Q575Δ is a targetable mutation, we exposed Ba/F3 cells expressing FLT-3/Q575Δ to increasing concentrations of clinically relevant TKIs (Figure 1C). The various TKIs inhibited FLT-3/Q575Δ autophosphorylation at concentrations comparable to those previously reported for FLT-3/ITD. Of particular note, FLT-3/Q575Δ is as sensitive, if not more so, to midostaurin (supplemental Figure 1B). Furthermore, when Ba/F3-FLT-3/Q575Δ cells are exposed to these different FLT-3 TKI, a dose-dependent cytotoxicity was noted (Figure 1D; supplemental Figure 1A). From these results, we concluded that FLT-3/Q575Δ is an activating mutation that can be targeted using conventional FLT-3 TKI, explaining the presentation and response in our patient.
The FLT-3 JMD is a hotspot for non-ITD mutations
We conducted a review of published case reports and series for mutations of the FLT-3 JMD. All classes of mutations (missense, deletion, insertions, and nonsense) have been identified (Table 1); however, there are limited functional data for the JMD mutations, especially the deletions.12,14-22 Existing functional data suggest that any changes in the wild-type JMD sequence are activating, similar to effects of the JMD-lengthening FLT-3/ITD mutations. Pharmacologic studies suggest that targeting of the other classes of JMD-altering mutations with small-molecule inhibitors may be possible, but the pattern of sensitivity to inhibition varies. Important to the case of this patient, no systematic approach has been used to examine the effects of single-residue deletions of the JMD.
To define the potential spectrum of mutations within the JMD that may have clinical relevance, we collected all FLT-3 mutations reported in the COSMIC database (supplemental Figure 2A). Examining the distribution of mutations throughout the gene, several patterns are noted: a tight, strong cluster of deletions and missense mutations at D835 and I836, correlating with well-established TKD mutations, and a broad cluster of insertions within the JMD, spanning much of its extent, correlating with the canonical ITD mutations. Interestingly, although not expected to have any effect on protein function, the silent mutation L561L was noted to occur frequently (supplemental Figure 2B). Permutation analysis, examining all possible silent mutations throughout the gene, suggests that this mutation is reported >3 times more frequently than would be expected based on chance (P < 1 × 10−6). This site may represent a mutational hotspot, either related to the presence of transcriptional regulatory domains or to a breakage hotspot, although there are no colocalizing regulatory domains reported within ENCODE, nor is this a putative DNAse hypersensitivity site.23,24
More importantly, when only deletions were examined within the FLT-3 gene, 2 patterns were again noted (Figure 2A). Deletions of the D835 and I836 were greatly overrepresented, again, consistent with activating TKD mutations. However, there was also a cluster of residues within the JMD, encompassing the Q575 residue, that were also frequently lost in reported FLT-3 mutations. Permutation analysis indicates that these residues are both deleted in particular and mutated in general at a twofold to threefold greater frequency than expected (Figure 2B; P ≤ 1 × 10−5). In this region, there are 2 areas of deletion enrichment: Q569-Q580 and D586-D600. We chose to focus on the first region, as it encompasses our patient’s deletion.
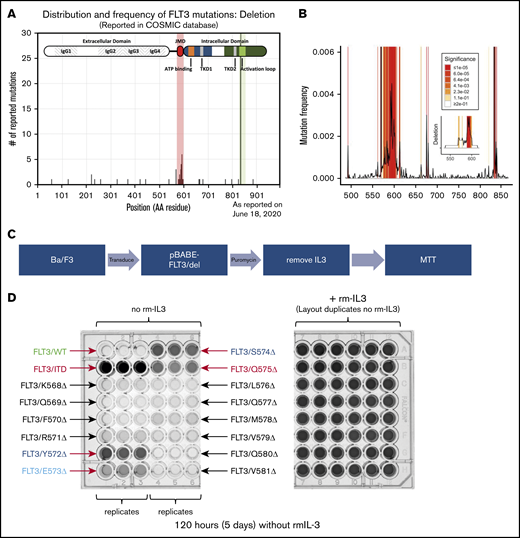
Clusters of activating deletions in the FLT-3 JMD. (A) Distribution of FLT-3 deletions as reported in the COSMIC database (query performed on 9 August 2017). The shaded regions indicate 2 enrichment clusters of deletions in the JMD and at the canonical TKD sites (D835/I836). (B) Permutation analysis of the distribution of mutated residues demonstrates significant enrichment of the expected ITD and TKD regions (both off scale). The frequency of mutation of each residue, as reported, is plotted. Shading indicates levels of significance compared with the predicted distributions (1 × 105 resamplings). The inset shows the same analysis but examines the deletions only. The entire CDS was analyzed, but only the JMD is plotted; the TKD was also enriched for deletions as expected. (C) Scheme for identifying activating mutations within the K568-V581 cluster. (D) Photographs of the MTT assay plates for cells expressing the different FLT-3 constructs in the presence (right) and absence (left) of rm IL-3, after 5 days. Relevant clones are indicated.
Clusters of activating deletions in the FLT-3 JMD. (A) Distribution of FLT-3 deletions as reported in the COSMIC database (query performed on 9 August 2017). The shaded regions indicate 2 enrichment clusters of deletions in the JMD and at the canonical TKD sites (D835/I836). (B) Permutation analysis of the distribution of mutated residues demonstrates significant enrichment of the expected ITD and TKD regions (both off scale). The frequency of mutation of each residue, as reported, is plotted. Shading indicates levels of significance compared with the predicted distributions (1 × 105 resamplings). The inset shows the same analysis but examines the deletions only. The entire CDS was analyzed, but only the JMD is plotted; the TKD was also enriched for deletions as expected. (C) Scheme for identifying activating mutations within the K568-V581 cluster. (D) Photographs of the MTT assay plates for cells expressing the different FLT-3 constructs in the presence (right) and absence (left) of rm IL-3, after 5 days. Relevant clones are indicated.
JMD deletions activate FLT-3 similar to ITD
Using a scanning mutagenesis approach, we created a panel of FLT-3 deletions tiling the entire region from K568 to V581. We again introduced these constructs into Ba/F3 cells and determined whether they were able to expand in the absence of rm IL-3 (Figure 2C). As expected, after 5 days the FLT-3/ITD control construct supported IL-3–independent growth, whereas the FLT-3/WT construct did not (Figure 2D; supplemental Figure 3). Although most of the FLT-3 deletion constructs did not support significant growth, 4 constructs (Y572Δ, E573Δ, S574Δ, and Q575Δ) demonstrated growth (Figure 2D). Indeed, when cells expressing these constructs were deprived of rm IL-3, they demonstrated reduced apoptosis levels compared with parental Ba/F3 cells, as measured by annexin V staining after 48 hours (Figure 3A; supplemental Figure 4). However, they did demonstrate less robust antiapoptotic protection than was conferred by the FLT-3/ITD construct. Furthermore, similar to the FLT-3/Q575Δ mutation, the cells expressing these constructs showed FLT-3 autophosphorylation accompanied by downstream signaling through the STAT5, AKT, and extracellular signal-regulated kinase-1/2 (ERK1/2) pathways when analyzed by western blot (Figure 3B).
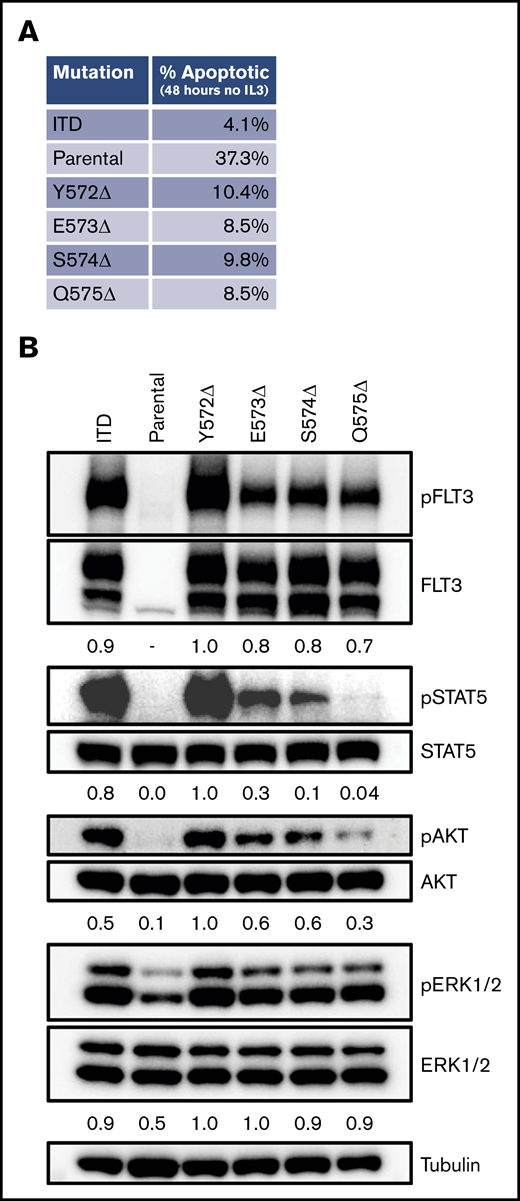
FLT-3 JMD deletions produce prosurvival signals. (A) Spontaneous apoptosis was measured in cells expressing the different constructs. After growing in rm IL-3 overnight, the cells were deprived of cytokines and cultured for 48 hours. Apoptosis was then measured by staining with annexin V, followed by flow cytometry. The flow plots from the assay are shown in supplemental Figure 4. (B) Signaling in cells expressing the 4 identified JMD deletions was measured by phospho-western analysis of whole-cell lysates. For comparison, lysates from cells expressing FLT-3/ITD and the parental cell line grown in the absence of rm IL-3 for 4 hours are included.
FLT-3 JMD deletions produce prosurvival signals. (A) Spontaneous apoptosis was measured in cells expressing the different constructs. After growing in rm IL-3 overnight, the cells were deprived of cytokines and cultured for 48 hours. Apoptosis was then measured by staining with annexin V, followed by flow cytometry. The flow plots from the assay are shown in supplemental Figure 4. (B) Signaling in cells expressing the 4 identified JMD deletions was measured by phospho-western analysis of whole-cell lysates. For comparison, lysates from cells expressing FLT-3/ITD and the parental cell line grown in the absence of rm IL-3 for 4 hours are included.
FLT-3 JMD deletions can be targeted by TKIs
We then tested the sensitivity of these 4 constructs to 6 FLT-3 TKIs in various stages of clinical testing and use (Figure 4). All 4 deletions demonstrated sensitivity to FLT-3 TKIs comparable to FLT-3/ITD sensitivity, in contrast to mutations such as D835Y, which have been shown to confer resistance to the drugs quizartinib (AC220), sorafenib (Nexavar), and ponatinib (Iclusig/AP24534).12,25 The deletion mutants were 5- to 10-fold more sensitive to the drugs midostaurin (Rydapt/PKC412) and lestaurtinib (CEP701) than to FLT-3/ITD. These compounds are both staurosporine derivatives, and it is possible that they have similar off-target effects here, suggesting that the deletion mutations are less protective against the general cytotoxicity of these drugs than FLT-3/ITD, consistent with the results seen in spontaneous apoptosis in the absence of IL-3 (Figure 3A). There does not appear to be a consistent pattern of response between what are felt to be type I TKIs that bind to the active conformation at the activation loop (midostaurin, lestaurtinib, and crenolanib) and type II TKIs that bind to the inactive conformation at the adenosine triphosphate–binding domain (quizartinib, sorafenib, and ponatinib).

FLT-3 JMD deletions can be targeted by FLT-3 TKIs. Cells expressing the FLT-3 JMD deletions or FLT-3/ITD mutations were exposed to increasing concentrations of the indicated FLT-3 TKIs for 48 hours. The colorimetric MTT assay was used to measure growth. Proliferation, normalized to untreated cells, was plotted as a function of TKI concentration.
FLT-3 JMD deletions can be targeted by FLT-3 TKIs. Cells expressing the FLT-3 JMD deletions or FLT-3/ITD mutations were exposed to increasing concentrations of the indicated FLT-3 TKIs for 48 hours. The colorimetric MTT assay was used to measure growth. Proliferation, normalized to untreated cells, was plotted as a function of TKI concentration.
Case follow-up
Our patient completed induction therapy with flow cytometric remission by day 14 and a day-28 marrow demonstrating complete remission (M1). She underwent high-dose cytarabine consolidation, and ultimately received a haploidentical peripheral blood stem cell transplant from her child with a prep consisting of fludarabine, cyclophosphamide, and total-body irradiation. After transplant, she received cyclophosphamide, mycophenolate, and sirolimus graft-versus-host disease prophylaxis. Posttransplant sorafenib maintenance was started after engraftment and count recovery. She suffered a CNS relapse 26 months after transplant. The disease did not have the original FLT-3/Q575Δ but rather a new, activating FLT-3 mutation near the ATP-binding site K663Q.26 It is not known if this mutation is sensitive to gilteritinib, although similar ATP-site mutations may not be, and as such could represent selection for drug resistance. She died of progressive CNS disease 3 years after the transplant (3.5 years after diagnosis).
Discussion
As the goals of targeted medicine and personalized health care become a reality through treatments such as small-molecule inhibitors and through rapid-turnaround, next-generation sequencing, clinicians will find themselves increasingly at the intersection of large data and multiple treatment options. Although singular mutations such as the FLT-3/ITD will continue to dominate the oncology landscape simply because of their ubiquity and outsized clinical significance, the remaining gains to be made in patient care will demand an understanding of less common disease features such as the FLT-3 Q575Δ mutation. Unfortunately, many of these are rare, if not unique, findings that can be acted upon only if systematic approaches are taken to understanding the effects of these changes within the context of a broader spectrum of disease-associated, frequent mutations.
We present a systematic analysis of a region of FLT-3 that can potentially mutate to cause constitutive kinase activation. Using unbiased approaches informed by the clinically significant data contained in repositories such as COSMIC, we have been able to confirm that this novel deletion is part of a larger collection of similar deletions and that the distribution of these deletions, far from random, is driven by the biology of FLT-3 autoinhibition. When these mutations are superimposed on the secondary structure of FLT-3, it can be seen that they cluster around the first β-pleated sheet (βJ1) of the JMD.3 Specifically, they are all found within the juxtamembrane binding motif, a highly conserved region of FLT-3 that shares considerable homology with other type III receptor tyrosine kinases, such as PDGFR, c-Fms, and c-KIT. Indeed, it is this motif that contacts the activation loop, normally holding FLT-3 in an inactive conformation until bound by a ligand, an interaction that is mediated specifically by the preferentially deleted Y572 residue.
Unsurprisingly, when the tyrosine at 572 is disturbed, the key contact between the JMD and the activation loop is lost, enabling the receptor to adopt the open, constitutively active conformation. However, this disturbance can take many forms beyond simple deletion. Being located at the end of the juxtamembrane binding motif associated with the rigid, amphipathic βJ1 sheet, this interaction is classically lost by the steric disruption caused by the larger insertion of the ITD mutations. Not surprisingly, the larger ITD mutations, pushing this interaction farther out of alignment, are associated with more active FLT-3. Yet, as seen in our series, even a small, single-residue disruption at a critical location can be sufficiently disruptive to activate FLT-3 and, in the case of our patient, cause disease. Indeed, it is telling that in examining the COSMIC data, we found that the JMD deletions form 2 very close, but separate clusters: Q569-Q580 and D586-D600. These clusters align closely with the 2 β-pleated sheets that make up the JMD (βJ1: Q575-T582 and βJ2: E588- Y591), with the gap between them aligning with the less rigid hinge domain linking the 2 sheets.
It should be noted that the secondary and tertiary structure of FLT-3 has been mapped through crystallographic, spectrographic, and genetic means.3 As such, it is possible to conceive of using this a priori knowledge to inform a candidate approach that would similarly identify activating and potentially clinically relevant mutations. However, with 953 different amino acid residues, at least 14 different domains and subdomains, and not less than 5 different critical interdomain contacts, such an undertaking would be prohibitive. This work demonstrates the power of coupling the large repositories of clinically derived, publicly available data with classic techniques to generate readily translatable knowledge to inform subsequent clinical study and even, with caution, patient care.
The importance of these mutations may extend beyond a target for TKI therapy. The FLT-3/ITD is an important risk-defining lesion for AML, and the treatment paradigm for this disease includes hematopoietic stem cell transplant for FLT-3/ITD and other poor-risk–prognostic indicators. With the application of TKI therapy and transplant, the outcomes of FLT-3/ITD are now approaching and even surpassing those of non-FLT-3–mutated disease. However, within the subset of supposedly lower risk non-FLT-3 mutant AML, there remains a subset of patients who will have poorer outcomes. Indeed, without NGS, our patient would have been included in the non-FLT-3 AML group. Although it is important to note that although we present evidence that FLT-3/Q575Δ is an activating, disease-causing mutation, it is not clear whether this mutation carries the same clinical significance as FLT-3/ITD, as evidenced by subtle differences in biological characteristics such as apoptosis, and such outcome data may never be possible with such sporadic mutations. Nonetheless, it is very provocative to think that at least part of the poorer outcome subset of patients comprises patients like our patient, whom traditional PCR-based diagnostic techniques would have overlooked. Ironically, with better supportive care, decreasing incidence of treatment-related morbidity and mortality, and an increasing acceptance of alternative-donor transplants, there is a growing desire to be able to identify those patients who may benefit from transplant as part of their upfront therapy. Yet, the more powerful, prospective clinical trials needed to define more completely that subset of patients can occur only if we appreciate the spectrum of genetic derangements that may contribute to AML. This work is only one step toward that appreciation.
Requests for protocols can be sent to the corresponding author (donsmall@jhmi.edu). This publication did not involve the generation of sequencing data sets.
Acknowledgments
This work was supported by National Institutes of Health, National Cancer Institute grants R01 CA090668 and P30 CA006973, a grant from Alex’s Lemonade Stand, and the Kyle Haydock Professorship (D.S.); and the Giant Food Pediatric Cancer Fund, the National Cancer Institute Laboratory-based Training in Pediatric Oncology/Hematology grant (T32 CA060441), and the Optimist Foundation Fellowship (D.J.Y.).
Authorship
Contribution: D.J.Y., A.S.D., and D.S. initiated the study; D.J.Y. was responsible for the design and conduct of the experiments and the data analysis and wrote the manuscript; B.N., R.Z., J.S., and L.L. designed and conducted the experiments; M.J.L., K.W.P., A.S.D., and D.S. were involved in experimental design and clinical studies; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: D.S. serves on the Safety Advisory Board of InSilico Medicine and receives research support and serves as a consultant for an unrelated project at Pharos I&BT Co, Ltd. D.J.Y. is now the principal investigator on an unrelated trial sponsored by Novartis, begun after his involvement in this work and does not receive compensation. K.W.P. receives research support from Millennium Pharmaceuticals (Takeda) and Agios Pharmaceuticals and serves as a consultant for Novartis. M.J.L. receives research support from and serves as a consultant for Novartis. The remaining authors declare no competing financial interests.
The current affiliation for D.J.Y is the National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD.
Correspondence: Donald Small, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, CRB1 Room 251, 1650 Orleans St, Baltimore, MD 21231; e-mail: donsmall@jhmi.edu.



