Key Points
Megadose radioimmunotherapy with 90Y-ibritumomab tiuxetan treats relapsed/refractory aggressive BCLs safely and successfully.
Radioimmunotherapy conditioning regimens warrant further exploration for curative-intent treatment in relapsed/refractory lymphomas.

Abstract
Allogeneic hematopoietic cell transplantation (allo-HCT) can be curative for relapsed or refractory B-cell lymphomas (BCLs), although outcomes are worse in aggressive disease, and most patients will still experience relapse. Radioimmunotherapy using 90Y-ibritumomab tiuxetan can induce disease control across lymphoma subtypes in a dose-dependent fashion. We hypothesized that megadoses of 90Y-ibritumomab tiuxetan with reduced-intensity conditioning could safely produce deeper remissions in aggressive BCL further maintained with the immunologic effect of allo-HCT. In this phase 2 study, CD20+ BCL patients received outpatient 90Y-ibritumomab tiuxetan (1.5 mCi/kg; maximum, 120 mCi), fludarabine, and then 2 Gy total body irradiation before HLA-matched allo-HCT. Twenty patients were enrolled after a median of 4.5 prior lines of therapy, including 14 with prior autologous transplant and 4 with prior anti-CD19 chimeric T-cellular therapy. A median 90Y-ibritumomab tiuxetan activity of 113.6 mCi (range, 71.2-129.2 mCi) was administered, delivering a median of 552 cGy to the liver (range, 499-2411 cGy). The estimated 1- and 5-year progression-free survival was 55% (95% confidence interval [CI], 31-73) and 50% (95% CI, 27-69) with a median progression-free survival of 1.57 years. The estimated 1- and 5-year overall survival was 80% (95% CI, 54-92) and 63% (95% CI, 38-81) with a median overall survival of 6.45 years. Sixteen patients (80%) experienced grade 3 or higher toxicities, although nonrelapse mortality was 10% at 1 year. No patients developed secondary acute myeloid leukemia/myelodysplastic syndrome. Megadose 90Y-ibritumomab tiuxetan, fludarabine, and low-dose total body irradiation followed by an HLA-matched allo-HCT was feasible, safe, and effective in treating aggressive BCL, exceeding the prespecified end point while producing nonhematologic toxicities comparable to those of standard reduced-intensity conditioning regimens.
Introduction
Despite improved survival, most individuals who experience relapsed or refractory (R/R) Hodgkin and non-Hodgkin lymphoma (NHL) will die of their disease. This is particularly true for those with aggressive B-cell lymphomas (BCLs), for which diffuse large BCL (DLBCL) comprises the majority of these patients.1 Following relapse, only a minority of those with chemosensitive disease derive long-term disease-free survival from high-dose therapy followed by autologous stem cell transplantation (ASCT); furthermore, ASCT is of limited utility in refractory disease.1-6 More recently, cellular, chimeric antigen receptor-modified T-cell (CAR-T) therapies offer potentially curative options. However, even with this approach, well over one-half of the patients who are able to receive this treatment will experience relapse.7-11 Allogeneic hematopoietic stem cell transplantation can induce long-term disease-free survival by taking advantage of the graft-versus-lymphoma (GVL) effect; however, this strategy is infrequently used due to unacceptable rates of treatment-related mortality and graft-versus-host disease (GVHD).12-14 To mitigate these toxicities, reduced-intensity conditioning (RIC) regimens were initially developed to facilitate allogeneic engraftment while diminishing treatment-related mortality rates, allowing for the immunologic GVL effect to eradicate disease, but earlier studies showed that rapidly progressive or bulky disease outpaced these beneficial effects.15,16 Echoed by the National Comprehensive Cancer Network guidelines, patients under consideration for allogeneic hematopoietic stem cell transplant due to mobilization failures, persistent bone marrow involvement, or lack of adequate response to second-line therapy should be in CR (complete response) or near-CR at the time of transplant.17
Radioimmunotherapy (RIT) has been studied as an approach for further cytoreduction of active disease before transplant, allowing sufficient time for the GVL effect to establish.18 Since its approval by the US Food and Drug Administration in the early 2000s for the treatment of patients with indolent B-cell follicular NHL, two CD20-targeting agents, 90Y-ibritumomab tiuxetan (Zevalin, Acrotech Biopharma) and 131I-tositumomab (Bexxar, GlaxoSmithKline), have been studied in front-line and R/R settings. Although 131I-tositumomab is no longer commercially available, 90Y-ibritumomab tiuxetan continues to be used clinically. The advantage of RIT includes the targeted delivery of tumoricidal radiation to multifocal lymphoma sites using monoclonal antibodies, thereby minimizing nonhematologic toxicity to the surrounding normal tissue. Our center and others have taken advantage of the exquisite radiosensitivity of lymphomas, reporting on standard and minimally escalated doses of 90Y-ibritumomab tiuxetan before non-myeloablative or RIC allogeneic transplantation for high-risk BCLs. These studies included a mixture of indolent and aggressive lymphomas and reported an estimated 2-year event-free survival and progression-free survival (PFS) rate of 25% to 43% and 31% to 40%, respectively.19-22 Our prior work showed that standard-dose RIT using 90Y-ibritumomab tiuxetan (anti-CD20) was found to be safe and effective at inducing early disease control across B-cell NHL subtypes before an RIC allograft; however, it did not yield sustained disease control in those with aggressive B-cell NHL. We therefore hypothesized that further escalation of RIT as part of RIC allogeneic transplantation would deepen and prolong remissions, allowing for additional time to establish a GVL effect and an improved PFS in aggressive BCL.
Herein we describe the results of a phase 2 study investigating the highest dose to date of 90Y-ibritumomab tiuxetan of 1.5 mCi/kg (maximum, 120 mCi) combined with fludarabine and low-dose total body irradiation (TBI), followed by HLA-matched allogeneic hematopoietic stem cell transplantation, for patients with R/R aggressive BCL.
Materials and methods
Study design and oversight
This single-center, phase 2 trial was reviewed and approved by the Institutional Review Board of the Fred Hutchinson Cancer Research Center and was registered at ClinicalTrials.gov as #NCT01434472. It complied with the Declaration of Helsinki and the Guidelines for Good Clinical Practice. All participating patients provided written informed consent and understood that study participation was voluntary. The study investigators assume responsibility for the accuracy and completeness of the data and analyses, as well as for the fidelity of the trial conduct and this report.
Selection of patients
Patients were eligible if they were aged ≥18 years with a histologically confirmed diagnosis of an aggressive BCL expressing the CD20 antigen. They must have had disease progression after at least one prior standard systemic therapy, have relapsed after high-dose therapy and autologous transplantation, or be ineligible for high-dose therapy and autologous transplantation. Patients with persistent disease >30 days after autologous transplant were also eligible. Patients had acceptable renal (creatinine <2.0 mg/dL) and hepatic (bilirubin < 1.5 mg/dL, and aspartate transaminase/alanine aminotransferase <3 × upper limit of normal) function, an expected survival without treatment of >60 days, and were free of major infections, including HIV. Patients had an HLA-identical related or HLA-matched unrelated donor. Patients were excluded if they received systemic anti-lymphoma therapy within the following intervals before the therapeutic 90Y-ibritumomab tiuxetan dose: (1) <30 days for IV administered cytotoxic chemotherapy and/or monoclonal antibodies; and (2) <5 half-lives for all other anticancer agents (eg, targeted therapies, corticosteroids, immunomodulatory agents). Other exclusions included the inability to understand or give informed consent; active central nervous system lymphoma; pregnancy or fertile men or women unwilling to use contraceptive methods during study treatment and for 12 months after treatment; Eastern Cooperative Oncology Group performance score ≥2; high-dose chemotherapy or external beam radiation therapy to the lung, liver, or kidneys >20 Gy within the previous 100 days before therapeutic 90Y-ibritumomab tiuxetan dose; or any medical condition that would contraindicate allogeneic transplantation as per standard practice guidelines (eg, impaired cardiopulmonary function, hepatitis).
Donor selection
HLA-identical related donor or HLA-matched unrelated donor met standard Seattle Cancer Care Alliance and/or National Marrow Donor Program criteria for peripheral blood stem cell (PBSC) donation. Participants with HLA-matched, related, non-sibling donors were treated according to the regimen for participants with HLA-matched unrelated donors. PBSC was the only permitted stem cell source.
Antibody infusions and conditioning regimen
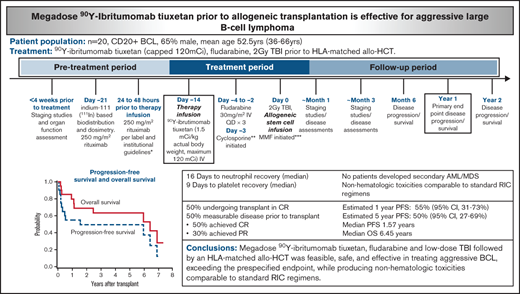
Staging studies and organ function assessment were assessed <4 weeks before the initiation of study therapy. The treatment plan was similar to that previously described.20 In brief, patients underwent indium-111–based biodistribution and dosimetry on day −28. Twenty-four to 48 hours before infusion of radiolabeled antibody, rituximab 250 mg/m2 was administered IV, historically given to improve biodistribution by depleting normal B cells as potential targets of RIT. Before these infusions, however, serum rituximab levels were measured by using an enzyme-linked immunosorbent assay. For those with rituximab levels >10 µg/mL, these rituximab infusions were omitted. This strategy was based on preclinical work which showed that a rituximab concentration of this level can compete with available CD20-binding sites on tumor cells and affect the efficacy of CD20-targeted RIT.23 Initially, all patients received infusions of trace-labeled indium-111–ibritumomab tiuxetan to establish favorable biodistribution before proceeding to the 90Y-labeled therapy infusion. However, after 16 consecutive patients exhibited favorable biodistribution even with omission of rituximab, we subsequently omitted this step and proceeded directly to therapy infusion after enrollment. Organ dosimetry was calculated in 5 patients using methods previously described.24,25 On day −14 (±2 days), 90Y-ibrutimomab tiuxetan (1.5 mCi/kg actual body weight; maximum, 120 mCi) IV was administered. 90Y-ibrutimomab tiuxetan was provided by Acrotech Biopharma and Cardinal Health. On day −4 to day −2, fludarabine 30 mg/m2 IV (3 doses total) was administered. On day 0, a total of 2 Gy TBI at 6 to 7 cGy/min from a linear accelerator was administered, followed by the infusion of unmodified granulocyte colony–stimulating factor mobilized PBSC from an HLA-identical related or unrelated donor. The treatment schema is illustrated in Figure 1. The entire conditioning regimen and including 90Y-ibrutimomab tiuxetan were administered in the outpatient clinic. Supportive care was provided per standard institutional practice.

Immunosuppression
Cyclosporine 4 mg/kg by mouth twice daily was initiated on day −3. For related donors, this was continued to day 56 and tapered off by day 100; for unrelated donors, this was continued to day 100 and tapered off by day 180. If GVHD was present, immunosuppression followed standard practice guidelines and/or was at the direction of the primary treating physician. Whole-blood trough level was targeted to 400 ng/mL through day 28, then 120 to 360 ng/mL thereafter, but this could be adjusted at the discretion of the attending physician. Equivalent IV doses of cyclosporine were also used at the discretion of the attending physician.
Mycophenolate mofetil (MMF) 15 mg/kg was administered orally, twice daily beginning 5 to 10 hours following PBSC infusion on day 0 for related donors and 3 times daily for unrelated donors. For related donors, MMF was continued at full dose through day 27 and discontinued without a taper. For unrelated donors, MMF was continued at full dose through day 40 and tapered off by day 96. Equivalent IV doses were used at the discretion of the attending physician, as were dose adjustments for various reasons (eg, toxicity).
End points and statistical analysis
The primary end point of this study was PFS at 1 year. Secondary end points included response and mortality rates at 100 days posttransplant and overall survival (OS). One-year PFS of 30% was considered a benchmark for potential efficacy based on historical data. The study was designed to enroll 24 patients, which provided 80% power to observe a 1-year PFS that is statistically higher (at the one-sided significance level of 0.05) than 30% if the assumed-true 1-year PFS is 54%. However, enrollment was halted at 20 patients due to lack of funding. The sample size of N = 24 provided 80% power to observe a statistically significant (at the one-sided significance level of .05) improvement over the fixed benchmark of 1-year PFS of 30%, if the assumed-true 1-year PFS was 54%. With 20 patients, this power was reduced to 74%; however, the observed outcome provided reasonably strong evidence of a potential signal (lower 95% confidence limit, 31%) with the 20 patients treated. Toxicities were captured and scored based on the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.26 Consensus criteria were used to grade acute and chronic GVHD.27,28 Probabilities of PFS and OS were estimated by using the Kaplan-Meier method; SAS was used to generate plots.
Results
Patients
A total of 20 patients were enrolled from September 2012 to June 2019. Baseline characteristics are listed in Table 1. The median age was 52.5 years (range, 36-66 years), and 35% were female. The median number of prior regimens was 4.5 (range, 2-14), with 60% of patients having documented stage III/IV disease at time of enrollment. Of these prior therapies, 70% received a prior ASCT, 20% received prior CAR-T therapy, and 65% had chemosensitive disease at time of transplant. The most common histology was DLBCL not otherwise specified (N = 16 [80%]), with one each of high-grade BCL with MYC, BCL2, and BCL6 rearrangements (“triple-hit lymphoma”), gray-zone lymphoma (or BCL unclassifiable with features intermediate between DLBCL and classical Hodgkin lymphoma), T-cell/histiocyte-rich large BCL, and a CD20+ classical Hodgkin lymphoma.
Dosimetry and therapy delivered
Baseline and pretherapy rituximab levels were available for 16 patients, with a median baseline level of 44.15 μg/mL (range, 0-91 μg/mL) and a median pretherapy level of 53.60 μg/mL (range, 29.90-200.00 μg/mL). Only 4 patients had a baseline rituximab level <10 μg/mL and thus received rituximab before RIT. Patients received a median activity of 113.6 mCi of 90Y-ibritumomab tiuxetan (range, 71.2-129.2 mCi). Available calculated doses of absorbed radiation to critical organs were reported for 5 patients and are listed in Table 2, including the liver (range, 205.80-2410.71 cGy), kidneys (range, 139.65-1473.92 cGy), bone marrow (range, 1094.23-2388.75 cGy), spleen (range, 499.44-8798.40 cGy), and one for lungs (1960 cGy). There were no apparent associations between biodistribution and pretherapy rituximab levels. The PBSC donor type included 10 (50%) matched related and 10 (50%) matched unrelated, with a median cell dose of 7.94 × 106 CD34+ cells/kg (range, 4.3-34.48) infused.
Response, primary, and secondary end points
After transplant, 15 patients (75%) were in a CR, 1 patient each (5%) obtained a partial response and stable disease, and 3 patients (15%) displayed progressive disease as their best response (Table 3). Ten patients (50%) underwent transplant while in CR and maintained CR as best response. Of the 10 patients (50%) with measurable disease before transplant, 5 (50%) achieved CR, 3 (30%) exhibited progressive disease, and 1 each displayed a partial response and stable disease. Eight patients (40%) ultimately relapsed, including 5 who were in CR before transplant. The estimated 1- and 5-year PFS was 55% (95% CI, 31-73) and 50% (95% CI, 27-69), respectively, with a median PFS of 1.57 years. At a median follow up of 5.12 years among the 10 surviving patients (range, 0.86-7.35 years), the estimated 1- and 5-year OS was 80% (95% CI, 54-92) and 63% (95% CI, 38-81), with a median OS of 6.45 years (Figure 2).

Engraftment, early and late toxicities, and nonrelapse mortality
All patients achieved absolute neutrophil count recovery, defined as an absolute neutrophil count >500/μL, and the median time to recovery was 16 days (range, 7-24 days). All patients achieved platelet recovery, defined as platelet >20 000/μL, and the median time to recovery was 9 days (range, 0-21 days). As expected, most patients (80%) experienced at least one grade 3 nonhematologic toxicity within the first 100 days (Table 4), the majority of which were reversible. The estimated 1-year nonrelapse mortality rate was 10% (Figure 3), with 1 patient dying at day 79 from disseminated adenovirus infection resulting in acute respiratory distress syndrome and 1 at day 89 after development of severe gastrointestinal GVHD, complicated by bacteremia. Three additional patients died of causes unrelated to progressive disease, all of which occurred ≥6 years posttransplant: 1 patient died of metastatic squamous cell carcinoma, presumably lung primary, at day 2533; another patient died of bacterial peritonitis secondary to hepatitis C virus cirrhosis and liver GVHD at day 2355; and the last patient died of multiorgan failure, not otherwise specified, at day 2184. No secondary acute myeloid leukemia or myelodysplastic syndrome was reported.

Graft-versus-host disease
Seventeen patients (85%) developed acute GVHD at a median of 37 days (range, 13-91 days), 13 (65%) developing grade 2 GVHD and 4 (20%) developing grade 3 GVHD. No grade 4 GVHD was reported. The skin and gastrointestinal tract were the most commonly involved organs, and 5 patients (25%) developed >1 organ involvement. Nine patients (45%) developed chronic GVHD, declared at a median of 345 days (range, 41-608 days). Six patients (30%) had involvement of ≥3 organs. The most commonly involved organs were the skin, liver, gastrointestinal tract, mouth, eyes, lung, and joints/fascia.27,28
Discussion
We present the findings of our phase 2 study assessing the safety and efficacy of megadose 90Y-ibritumomab tiuxetan combined with fludarabine and 2 Gy TBI before HLA-matched allogeneic hematopoietic stem cell transplantation for patients with R/R aggressive BCL.
Doses of 90Y-ibritumomab tiuxetan similar to our protocol have been previously studied but not before an allograft and not in combination with chemotherapy or TBI. In the ASCT setting, Nademanee et al29 investigated high-dose 90Y-ibritumomab tiuxetan conditioning with etoposide and cyclophosphamide in CD20+ NHL before ASCT. A median of 71.6 mCi (range, 36.6-105 mCi) was administered to deliver a target dose of 1000 cGy to normal organs and was well tolerated, with a reported 2-year estimated OS and relapse-free survival of 92% and 78%, respectively. Ferrucci et al30 conducted a feasibility and toxicity pilot study of escalating doses of 90Y-ibritumomab tiuxetan followed by ASCT in 13 patients, 11 of whom had less than CR before ASCT. Five patients were treated at dose level 3 of 90Y-ibritumomab tiuxetan 56 MBq/kg (1.5 mCi/kg) 13 days before ASCT, and treatment was well tolerated. For patients with poor-risk NHL and deemed ineligible to receive standard BEAM conditioning (BCNU, etoposide, ara-C, melphalan), Devizzi et al31 presented their 5-year update for 60 patients treated with myeloablative doses of 90Y-ibritumomab tiuxetan (0.8 mCi/kg or 1.2 mCi/kg) before ASCT; they reported a 32.5% cumulative incidence of relapse, 1.7% nonrelapse mortality, and a 5-year PFS and OS of 62.7% and 72.9%, respectively.
Bethge et al21 published results of their multicenter, phase 2 dose-escalation study of 90Y-ibritumomab tiuxetan at 2 dose intermediate levels (0.6 mCi/kg and 0.8 mCi/kg) in 20 patients, combined with RIC using fludarabine, melphalan, and alemtuzumab followed by allogeneic transplant. The DLBCL cohort comprised 13 patients, 7 of whom received dose level 1 and 6 received dose level 2; however, the estimated 3-year OS and event-free survival rates were 15%. Our previously investigated study using standard-dose90Y-ibritumomab tiuxetan 0.4 mCi/kg (capped at 32 mCi) along with fludarabine and low-dose TBI before an allogeneic hematopoietic stem cell transplantation series included 40 patients with persistent high-risk BCL, 14 of whom had DLBCL; the findings show the feasibility and safety of this approach, with an estimated cumulative rate of nonrelapse mortality at 30 months of 16%.20 Longer follow-up of this study indicated that this strategy improved outcomes over an eligibility-matched cohort with indolent B-cell NHL receiving fludarabine low-dose TBI conditioning alone and allogeneic hematopoietic stem cell transplantation.32 Unfortunately, for those patients with aggressive B-cell NHL, there were no long-term disease-free survivors regardless of early posttransplant remission status.22 Cabrero et al33 published the results of their GELTAMO trial, also investigating 90Y-ibritumomab tiuxetan dosed at 0.4 mCi/kg as part of an RIC regimen for allogeneic hematopoietic stem cell transplantation. Eighteen high-risk cases of NHL, including 6 patients with DLBCL, were enrolled, and after a median follow-up of 46 months, the estimated 1-year PFS was 50%, and 4-year OS and PFS were both 44%; however, the nonrelapse mortality at 1 year was 28%.
These earlier studies of RIT before both autologous and allogeneic transplant clarified the potential benefits and, despite the need for improved outcomes, supported the hypothesis that even higher doses of 90Y-ibritumomab tiuxetan RIT could further prolong remissions and increase PFS, particularly for aggressive subtypes of BCL, allowing greater time to establish a robust GVL effect. Notably, the dose of 90Y-ibritumomab tiuxetan used in the current study was 3.75× than we previously tested and higher than any study before allogeneic transplant. Despite this approach, it still seemed to be safe, feasible, and effective in treating this high-risk group of patients. With an estimated 1-year PFS in this study of 55%, this exceeded our prespecified benchmark of 30%, thereby yielding what we consider to be a sufficiently strong signal to deem the current treatment worthy of further study. Furthermore, after a median follow-up of >5 years among survivors, no patients developed secondary acute myeloid leukemia/myelodysplastic syndrome, even with this high dose of RIT. Short- and long-term nonhematologic toxicities were also comparable to those of standard RIC regimens.
Despite the promise of RIT-based conditioning regiments for lymphomas, allogeneic stem cell transplantation has fallen out of favor due to the introduction of CD19-directed CAR-T therapy approved after 2 prior lines of therapy. Unfortunately, longer follow-up has shown that only up to 40% to 50% of patients achieve durable and meaningful responses after CAR-T therapy.7-10,34 The remaining patients will die of their underlying lymphoma, posing new challenges in the treatment paradigm for this especially high-risk group.11 Other T cell–mediated approaches for R/R NHL remain an active area of research, particularly bispecific T-cell engaging antibodies. Data from the ongoing phase 1 Study NP30179 investigating the clinical activity of single-agent glofitamab (CD20-CD3 bispecific antibody) with the anti-CD20 antibody obinutuzumab was recently published, reporting on 171 heavily pretreated patients with R/R NHL who achieved an overall response rate of 53.8% (CR, 36.8%) among all doses, and those receiving the recommended phase 2 dosing achieved an overall response rate of 65.7% (CR, 57.1%).35 The safety profile indicated cytokine release syndrome and immune effector cell–associated neurotoxicity syndrome-like symptoms, albeit at generally lower rates and grades than those with CAR-T therapy; therefore, glofitamab may add to the growing armamentarium of therapies in the R/R setting. Long-term data are lacking compared with CAR-T therapy and allogeneic stem cell transplantation; however, its early signs of feasibility and efficacy warrant further investigation, particularly in sequencing or combination approaches. In contrast, high-dose RIT-based conditioning regimens before allogeneic stem cell transplantation remain a viable form of therapy with long-term efficacy data in high-risk patients, even ones entering transplant not in CR.
Although the numbers are small, 4 patients in our study received prior CD19-directed CAR-T therapy, between 118 and 770 days before transplant, and one remains alive and in CR at last follow-up despite entering transplant with active disease. Of the remaining 3 patients, 1 achieved CR and 2 achieved stable disease after transplant, but all developed progressive disease between 28 and 50 days and died between 64 and 403 days after transplant.
In conclusion, our study is the first to show the feasibility, safety, and potential efficacy of megadose (3.75× standard) 90Y-ibritumomab tiuxetan in addition to fludarabine and low-dose TBI followed by an HLA-matched allogeneic hematopoietic stem cell transplant for patients with aggressive BCL. The observed 1-year PFS of 55%, the outpatient delivery, and the short- and long-term nonhematologic toxicities comparable to standard RIC regimens indicate that this approach should be evaluated further. Even as novel cellular therapies, T-cell engagers, and other strategies continue to be developed for this patient population, these data suggest that megadose RIT conditioning regimens have the potential to play a role in the curative-intent algorithm for aggressive BCL.
Acknowledgments
The authors acknowledge April Ingram for editorial support with the manuscript. In addition, they thank Robyn Haaf, Sally Lundberg, Monina Almeda, Lacey Hedin, Jenn Davies, Barbara Pender, Heather Rasmussen, and the patients who participated in this trial.
This research was supported in part by the National Institutes of Health, National Cancer Institute (P30CA015704, P01CA078902, P01CA044991, and R01CA076287), the Leukemia and Lymphoma Society (SCOR grant 7040), the Lymphoma Research Foundation, Frank and Betty Vandermeer, and Sonya and Tom Campion.
Authorship
Contribution: R.D.C., O.W.P., and A.K.G. conceived the study; V.A.C., R.D.C., T.A.G., and A.K.G. wrote and edited the manuscript; T.A.G. performed statistical analysis; and S.D.S., B.M.S., D.J.G., J.J.O., S.A.T., M.M., D.R.F., and D.G.M edited the manuscript.
Conflict-of-interest disclosure: R.D.C. reports spouse employment with Seattle Genetics, stock or other ownership in Seattle Genetics, consultancy or advisory role for Pfizer and Amgen, and research funding from Pfizer, Merck, Amgen, Kite a Gilead Company, and Vanda Pharmaceuticals. T.A.G. is a Data and Safety Monitoring Board member for Kiadis Pharmaceutical and Pharmacyclics; and consults for Regimmune. S.D.S. has received research funding from Acerta Pharma BV, AstraZeneca, Beigene, De Novo Biopharma, Genentech, Incyte Corporation, Merck Sharp and Dohme Corp., Pharmacyclics, and Portola Pharmaceuticals; reports advising fees from AstraZeneca, Millennium/Takeda, Beigene, Karyopharm, KITE pharma, Incyte Corporation, and ADC Therapeutics; and spouse receives research support from Ayala, Bristol Myers Squibb, Merck Sharp Dohme Corp, and Ignyta. B.M.S. has received research funding from Bellicum and consultant fees from Actinium Pharmaceutical. D.J.G. has received research funding, has served as an advisor, and has received royalties from Juno Therapeutics, a Bristol-Myers Squibb company; has served as an advisor and received research funding from Seattle Genetics; has served as an advisor to GlaxoSmithKline, Celgene, Janssen Biotech, and Legend Biotech; and has received research funding from SpringWorks Therapeutics, Sanofi, and Cellectar Biosciences. J.J.O. receives research funding from Actinium Pharmaceuticals. D.G.M. has received grants and honoraria from Juno Therapeutics, Celgene, Kite Pharma; has participated in advisory board meetings and honoraria for Amgen, BioLineRx, Genentech, Gilead Sciences, Incyte, Janssen, Kite Pharma, Legend Biotech, Mustang Bio, MorphoSys, Novartis, Pharmacyclics and Umoja; has rights to royalties from Fred Hutchinson Cancer Research for patents licensed to Juno/BMS; is a member of the A2 Biotherapeutics Scientific Advisory Board with stock options and compensation; and is a member of the Navan Technologies Scientific Advisory Board with stock options and compensation. A.K.G. has received research funding from Merck, I-Mab bio, IgM Bio, Takeda, Gilead, AstraZeneca, Agios, Janssen, BMS, Seattle Genetics; and has provided consultancy/honoraria/advising for Incyte, Kite, MorphoSys, ADC, Acrotech, Merck, Karyopharm, Nurix Inc, Cellectar, Janssen, SeaGen, Epizyme, and I-Mab bio. The remaining authors declare no competing financial interests.
Correspondence: Ajay K. Gopal, Clinical Research Division, Fred Hutchinson Cancer Research Center, Division of Medical Oncology, University of Washington, 825 Eastlake Ave E, LG650 Seattle, WA 98109; e-mail: agopal@uw.edu.