Key Points
We generated novel hemophilia A or B mice expressing human protein C.
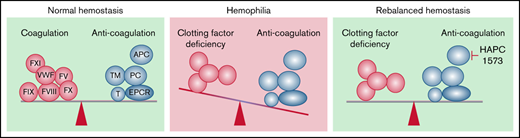
Selectively blocking the anticoagulant activity of human activated protein C improves the clotting defects in hemophilia mice.

Abstract
Hemophilia A and B are hereditary coagulation defects resulting in unstable blood clotting and recurrent bleeding. Current factor replacement therapies have major limitations such as the short half-life of the factors and development of inhibitors. Alternative approaches to rebalance the hemostasis by inhibiting the anticoagulant pathways have recently gained considerable interest. In this study, we tested the therapeutic potential of a monoclonal antibody, HAPC1573, that selectively blocks the anticoagulant activity of human activated protein C (APC). We generated F8−/− or F9−/− hemophilia mice expressing human protein C by genetically replacing the murine Proc gene with the human PROC. The resulting PROC+/+;F8−/− or PROC+/+;F9−/− mice had bleeding characteristics similar to their corresponding F8−/− or F9−/− mice. Pretreating the PROC+/+;F8−/− mice with HAPC1573 shortened the tail bleeding time. HAPC1573 pretreatment significantly reduced mortality and alleviated joint swelling, similar to those treated with either FVIII or FIX, of either PROC+/+;F8−/− or PROC+/+;F9−/− mice in a needle puncture–induced knee-joint bleeding model. Additionally, we found that HAPC1573 significantly improved the thrombin generation of PROC+/+;F8−/− mice but not F8−/− mice, indicating that HAPC1573 enhanced the coagulant activity of hemophilia mice by modulating human APC in vivo. We further documented that HAPC1573 inhibited the APC anticoagulant activity to improve the clotting time of human plasma deficient of FVIII, FIX, FXI, FVII, VWF, FV, or FX. These results demonstrate that selectively blocking the anticoagulant activity of human APC may be an effective therapeutic and/or prophylactic approach for bleeding disorders lacking FVIII, FIX, or other clotting factors.
Introduction
The balance between coagulation and anticoagulation is essential for physiological hemostasis.1-5 Defects in either of these pathways cause bleeding or thrombosis. Coagulation is a cascade of zymogen activation reactions.6 At each stage, a protease precursor is converted to an active protease by cleavage of the precursor molecule. The final protease generated is thrombin, which converts soluble fibrinogen to fibrin, which in turn, together with platelet aggregates, forms a stable hemostatic clot. The coagulation should be tightly balanced by anticoagulants. Protein C is a major physiological anticoagulation factor.7 It is activated from its zymogen by thrombin complexed with thrombomodulin on the surface of endothelial cells.2 Along with protein S and phospholipids as cofactors, activated protein C (APC) downregulates coagulation by degrading activated coagulation factor V (FVa) and FVIII (FVIIIa), thereby suppressing thrombin generation.2
Hereditary genetic defects in genes encoding coagulation factors cause hemophilia.4,8 Among them, hemophilia A and B are X-linked recessive genetic deficiencies of clotting factor VIII (FVIII) or IX (FIX).9 Hemophilia A is the most common type of hemophilia, which affects ∼85% of all patients with hemophilia and is found in 1 in 5000 male births. Many patients have poor quality of life due to frequent spontaneous bleeding inside joints,10 which often leads to permanent joint damage and disability. Even nonsevere hemophilia A patients have an increased risk of developing lethal intracranial bleeding in comparison with the general population.9
The current standard treatment for hemophilia is IV replacement therapy of the deficient coagulation factors either preventively, which could be lifelong, or at the time of bleeding.9 Challenges such as the short half-life of FVIII and FIX products, which require frequent IV infusions, and development of inhibitors have limited the use of the replacement therapies, especially for prophylaxis.9 In recent years, alternative approaches, such as the bispecific-antibody emicizumab that mimics FVIII, have gained considerable success in the management of hemophilia A.4,8,11
Given its important role as an anticoagulant in the regulation of clotting, APC has also been studied as a therapeutic target for hemophilia.4,12 Inspired by the observation that hemophilia patients carrying factor V Leiden mutation exhibit less severe bleeding,13 an engineered human protein C inhibitor was found to promote thrombin generation in vitro and normalized tail bleeding time in hemophilia B mice.12 APC has both anticoagulant and cytoprotective roles.1,14,15 A recent study has tested the therapeutic potential of blocking the human APC anticoagulant activity in a monkey model of acute acquired hemophilia A.16 Whether this approach is effective in chronic hemophilia models, which is more relevant to the hemophilia patient conditions, remains unclear due to lack of appropriate preclinical animal models with genetic deficiency of FVIII or FIX.
To address this question, we generated novel hemophilia A or B mice expressing human protein C by targeted replacement of the mouse Proc gene with the human PROC. We demonstrated that mouse monoclonal antibody (mAb) HAPC1573,17 which selectively blocks the anticoagulant activity of human APC, promoted the thrombin generation in vitro and significantly corrected bleeding symptoms and reduced mortalities of hemophilia mice.
Materials and methods
Descriptions of HAPC1573, quantitative reverse transcription polymerase chain reaction, coagulation assays, platelet count, and enzyme-linked immunosorbent assay are available in the Materials and Methods of the Supplemental Data.
Generation of hemophilia A or B mouse models expressing human protein C (PROC+/+;F8−/− and PROC+/+;F9−/−)
First, to generate mice expressing human protein C (PROC+/+), embryonic stem (ES) cells of C57BL/6 mice were transfected with a targeting vector with standard electroporation procedure (Cyagen Biosciences, Inc). The targeting vector was designed to replace the mouse protein C gene, from start codon ATG in exon 2 to the stop codon TGA in exon 9, with the complementary DNA sequence of human protein C. The exchange vector also contains a neo-resistant gene cassette for positive selection flanked by loxP sites, and a diphtheria toxin A fragment outside the replacement region for negative selection. After electroporation, ES cells were selected by growing in G418-containing media. Cells that survived the drug treatment were propagated and transfected with Cre recombinase–containing plasmid to remove the selection cassette. The engineered ES cells were then microinjected into the blastocysts of an albino C57BL/6 mouse, which were then planted into a foster mouse. PROC+/− and PROC+/+ mice were obtained by breeding the founders and genotyping the offspring.
PROC+/+mice were then crossed with F8−/− mice (Jackson Laboratory, Maine) to obtain PROC+/−;F8+/− mice, which were then crossed with PROC+/−;F8+/− mice to obtain PROC+/+;F8−/− mice. All genotype selections were done by polymerase chain reaction (PCR). PROC+/+;F9−/− mice were generated with the same strategy by crossing PROC+/+ mice and F9−/− mice (Jackson Laboratory).
For pretreatments, human plasma-derived FVIII, FIX, or HAPC1573 was IV injected into different mice 30 minutes prior to the experimental procedures. For the control, mice were injected with phosphate-buffered saline (PBS). Male mice (8-10 weeks in age) were used for all experiments. Mice were bred and maintained in the specific pathogen-free condition in the Laboratory Animal Experimental Center at Soochow University. Mouse studies were approved by the Animal Use Committee of the First Affiliated Hospital of Soochow University.
Protac-induced protein C activation in aPTT assay
0.05, 0.1, 0.05, 0.01, 0.05, 0.06, 0.01, and 0.2 U/mL of Protac (Pentapharm; New Jersey; 113-01) was added, respectively, to the following human plasma samples: FV-deficient plasma (Stago; REF 00744) mixed with 5% normal plasma, FVII-deficient plasma (Stago; REF 00743), FVIII-deficient plasma (Stago; REF 00725), FIX-deficient plasma (Stago; REF 00724), FX-deficient plasma (Stago; REF 00738) mixed with 1% normal plasma, FXI-deficient plasma (Stago; REF 00723), VWF-deficient plasma (Boatman Biotech; REF BMC18), and normal plasma. Various concentrations of HAPC1573 were incubated with the mixture in room temperature for 15 minutes, and the incubated samples (50 µL) were incubated with activated partial thromboplastin time (aPTT) reagent (50 µL; Stago; REF 00595) at 37°C for 4 minutes. Then, 25 mM CaCl2 (50 µL; Stago; REF 00367) was added, and the clotting time was recorded by an automated analyzer (STA Compact Max, Stago) and plotted against the various HAPC1573 concentrations. Data were the average of duplicates of samples.
Tail bleeding time
Mice (male, 8-10 weeks) were anesthetized with 1% pentobarbital sodium solution (200 µL) and then placed in the prone position. The 2 mm distal part of the tail was removed with a scalpel, and the remainder of the tail was immediately immersed in a 50 mL centrifuge tube containing isotonic saline that was preheated to 37°C in a water bath. The tail was kept vertical and below the body horizon. Each animal was monitored for 20 minutes, and in case of any on/off bleeding cycle, the total bleeding time within the period was recorded. If the bleeding did not stop within 20 minutes, the experiment was terminated at 20 minutes to avoid mortality during the procedure according to approved animal protocols.
Knee joint injury by needle puncture
Knee joint bleeding was induced by a needle puncture following published methods.10 Mice (male, 8-10 weeks) were anesthetized with 1% pentobarbital sodium solution. The capsules of the right knee joints of anesthetized mice were punctured under the patella with a 30 × 0.5-G needle to induce joint bleeding. Knee diameter, before the needle injury and every other day during the 2-week period after injury, was measured with an electronic caliper, and the change of joint diameter was calculated as percentage of the before-injury value.10 Survival of mice after the injury was recorded.
Statistics
Values were expressed as means plus or minus standard error of the mean or means plus or minus standard deviation (SD). Survival curves were plotted using the Kaplan-Meier method, and the statistical significance was determined by the log-rank (Mantel-Cox) test. Group comparison was made by analysis of variance (ANOVA) followed by the Newman-Keuls multiple-comparison test or unpaired t test. The other data were analyzed by 1-way and 2-way ANOVA with GraphPad Prism 8.0. P values of <.05 were considered statistically significant.
Results
Generation and characterization of human protein C knock-in hemophilia mice
Mouse and human protein C have only 69% amino acid sequence identity.18 This significant difference indicates that it would not be feasible to study the effects of potential therapeutics related to human protein C in mice. Therefore, we generated a murine genetic knock-in model that expresses human protein C by genetically replacing the mouse protein C gene (Proc) with a human PROC complementary DNA cassette (PROC+/+) (Figure 1A). The PROC+/+ mice were viable and able to breed. We then crossed the PROC+/+ mice with FVIII-deficient (F8−/−) or FIX-deficient (F9−/−) mice to generate preclinical murine models of hemophilia A (PROC+/+;F8−/−) or B (PROC+/+;F9−/−) that express human protein C (Figure 1B). Both PROC+/+;F8−/− and PROC+/+;F9−/− mice exhibited no obvious abnormalities compared with their wild-type (WT) littermate controls. As indicated by the quantitative reverse transcription PCR analyses of liver mRNA expression, PROC+/+ mice had normal expression of F8 and F9 relative to their WT controls. As expected, PROC+/+;F8−/− or PROC+/+;F9−/− livers expressed no detectable F8 or F9 mRNA (Figure 1C-D). In addition, all of the mutant mice expressed PROC transcripts but not the murine Proc transcripts (Figure 1E-F). Expression of human protein C was detected in plasma of these mutant mice (Figure 1G), albeit lower than the level in human plasma. Mice with severe deficiency of protein C have a high rate of mortality.19 In contrast, the PROC+/+ mice had no gross abnormalities, indicating that the human protein C in PROC+/+ mice is expressed and functions at sufficient levels to maintain essential hemostatic function.
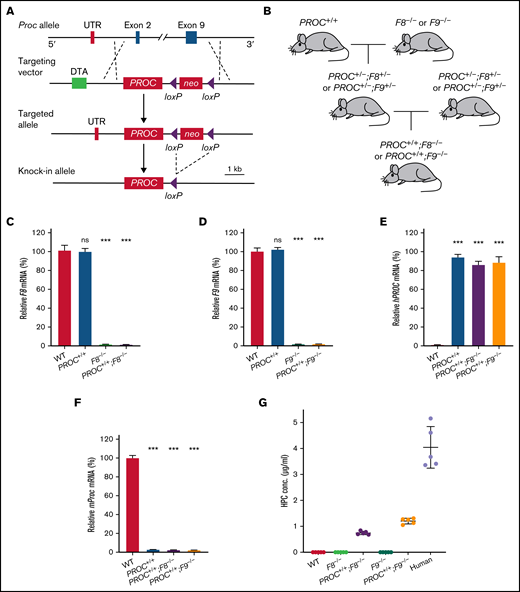
Generation of PROC knock-in hemophilia mice. (A) A schematic of the PROC knock-in strategy. The WT mouse Proc allele with exon 2 to 9 is shown at top, which has the start codon and stop codon, respectively. The targeting vector contains human PROC coding sequence and a downstream neomycin (neo), flanked by LoxP sites, for positive selection. The diphtheria toxin A gene (DTA) was used for negative selection. Once PROC was knocked in, the neo was deleted via Cre-mediated recombination. (B) Breeding strategy to generate PROC+/+;F8−/− and PROC+/+;F9−/− mice. PROC+/+ mice were crossed with mice lacking factor VIII (F8−/−) or factor IX (F9−/−). (C-F) Quantitative reverse transcription PCR analysis of the mRNA expression. Total mRNA was isolated from mice liver tissues. Results were normalized to GAPDH. Bars represent the means and standard errors of 15 biological replicates (5 mice per group and 3 samples per mouse). Liver GAPDH is set as 100%. Statistical significance was determined between WT and the other groups. ***P < .001; ns, P > .05 (1-way ANOVA Test). (G) Human protein C concentrations in the plasma of various mouse models and human were measured using an ELISA kit for human protein C antigen. Data were expressed as mean ± SD. n = 5 individuals. ELISA, enzyme-linked immunosorbent assay; mRNA, messenger RNA; ns, not significant; UTR, sequence corresponding to the untranslated region on mouse protein C mRNA.
Generation of PROC knock-in hemophilia mice. (A) A schematic of the PROC knock-in strategy. The WT mouse Proc allele with exon 2 to 9 is shown at top, which has the start codon and stop codon, respectively. The targeting vector contains human PROC coding sequence and a downstream neomycin (neo), flanked by LoxP sites, for positive selection. The diphtheria toxin A gene (DTA) was used for negative selection. Once PROC was knocked in, the neo was deleted via Cre-mediated recombination. (B) Breeding strategy to generate PROC+/+;F8−/− and PROC+/+;F9−/− mice. PROC+/+ mice were crossed with mice lacking factor VIII (F8−/−) or factor IX (F9−/−). (C-F) Quantitative reverse transcription PCR analysis of the mRNA expression. Total mRNA was isolated from mice liver tissues. Results were normalized to GAPDH. Bars represent the means and standard errors of 15 biological replicates (5 mice per group and 3 samples per mouse). Liver GAPDH is set as 100%. Statistical significance was determined between WT and the other groups. ***P < .001; ns, P > .05 (1-way ANOVA Test). (G) Human protein C concentrations in the plasma of various mouse models and human were measured using an ELISA kit for human protein C antigen. Data were expressed as mean ± SD. n = 5 individuals. ELISA, enzyme-linked immunosorbent assay; mRNA, messenger RNA; ns, not significant; UTR, sequence corresponding to the untranslated region on mouse protein C mRNA.
The aPTTs of plasma from PROC+/+ mice were comparable to that of WT controls. In contrast, the aPTT of PROC+/+;F8−/− or PROC+/+;F9−/− plasma were prolonged, reminiscent of F8−/− or F9−/− mice, respectively (Table 1). Protac-induced aPTT of PROC+/+;F8−/− plasma was also comparable to that of F8−/− plasma (supplemental Figure 1), which further supports that human protein C has sufficient anticoagulant function in the PROC+/+ hemophilia mice. However, the prothrombin times (PTs) of plasma from either PROC+/+;F8−/− or PROC+/+;F9−/− mice were similar to that of WT controls. Additionally, levels of fibrinogen and platelets in these mutant mice were not significantly different than that of their WT littermates. These data demonstrate that the expression of human protein C does not alter the clotting and hemostatic features of these models, which is in agreement with that of hemophilia patients who usually have normal fibrinogen levels and peripheral platelet counts.9
Anti-human APC mAb HAPC1573 significantly improves the hemostatic defects of hemophilia mice
The HAPC1573 is a monoclonal antibody against human APC (murine immunoglobulin G1/κ), which selectively inhibits the anticoagulant activity of human APC but not its cytoprotective activities (supplemental Figures 2-4).17 We used PROC+/+;F8−/− and PROC+/+;F9−/− mice to evaluate its therapeutic potential in correcting bleeding events of hemophilia in vivo. The batch used in this study had an endotoxin level of 0.0733 endotoxin units/100 μg, which is much lower than the allowable value for biologics IV infusion.
We first tested the hemostatic activity of HAPC1573 in PROC+/+;F8−/− mice using tail bleeding time after tail transection. As expected, the bleeding time of PROC+/+;F8−/− mice pretreated with PBS was significantly prolonged relative to that of WT controls (Figure 2A). Treatment with a low dose of human plasma-derived FVIII (0.05 IU per mouse, IV, equivalent to the FVIII level in severe hemophilia A patient) 30 minutes prior to the tail clip partially reduced the bleeding time of PROC+/+;F8−/− mice. Significantly, pretreatment with HAPC1573 (20 µg per mouse, IV) shortened the bleeding time to ∼9 minutes. Pretreatment with both low-dose FVIII (0.05 IU per mouse, IV) and HAPC1573 (20 or 100 µg per mouse, IV) normalized the bleeding time of the PROC+/+;F8−/− mice, similar to that of WT mice (Figure 2A; supplemental Figure 5). In contrast, HAPC1573 did not significantly improve the tail bleeding time of F8−/− mice (Figure 2B), indicating that it functions through interaction with human APC.
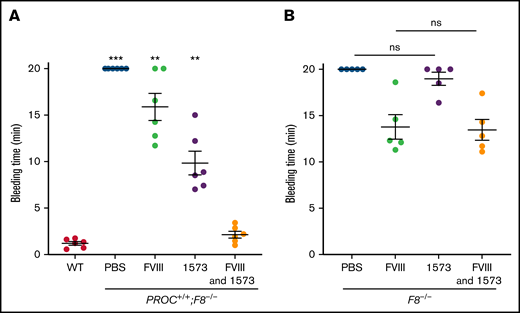
HAPC1573 (1573) reduces tail bleeding time of PROC+/+;F8−/− mice.PROC+/+;F8−/− mice (A) and F8−/− mice (B) were treated with either 0.05 IU per mouse of FVIII, 20 µg per mouse of anti-human APC antibody HAPC1573, or both in combination 30 minutes before the procedure. Tail tip bleeding time was recorded for a maximum of 20 minutes to avoid mortality caused by over bleeding. Pretreatment with PBS was used as a negative control. Data were expressed as mean ± SD. n = 6 mice per group. **P < .01; ***P < .001, ns, P ≥ .05 (1-way ANOVA test). ns, not significant; WT, tail bleeding time of WT mice.
HAPC1573 (1573) reduces tail bleeding time of PROC+/+;F8−/− mice.PROC+/+;F8−/− mice (A) and F8−/− mice (B) were treated with either 0.05 IU per mouse of FVIII, 20 µg per mouse of anti-human APC antibody HAPC1573, or both in combination 30 minutes before the procedure. Tail tip bleeding time was recorded for a maximum of 20 minutes to avoid mortality caused by over bleeding. Pretreatment with PBS was used as a negative control. Data were expressed as mean ± SD. n = 6 mice per group. **P < .01; ***P < .001, ns, P ≥ .05 (1-way ANOVA test). ns, not significant; WT, tail bleeding time of WT mice.
We then performed the knee-joint injury experiment, which is an established model mimicking the intrajoint bleeding commonly associated with hemophilia patients,10 to determine the efficacy of HAPC1573 in controlling intrajoint bleeding of PROC+/+;F8−/− and PROC+/+;F9−/− mice. In each setting, there were 3 experimental groups in addition to WT: PROC+/+;F8−/− treated with or without 2 IU per mouse of FVIII or PROC+/+;F9−/− treated with or without 2 IU per mouse of FIX, which were used as positive or negative controls. The third group was PROC+/+;F8−/− or PROC+/+;F9−/− treated with HAPC1573 30 minutes prior to the knee joint injury, which was induced by needle puncture on the right knee of each mouse. The injured right knees of all groups of mice exhibited bleeding and swelling, whereas the uninjured left knees were normal. In WT group, there was no death within 14 days, whereas in both the PROC+/+;F8−/− group and PROC+/+;F9−/− group without any pretreatment, 7 of the 12 mice in each group died within 5 days due to severe knee bleeding. In contrast, all PROC+/+;F8−/− mice pretreated with FVIII or PROC+/+;F9−/− mice pretreated with FIX survived (Figure 3A,C). In the group of PROC+/+;F8−/− mice pretreated with HAPC1573 (Figure 3A), only 1 out of 15 mice died after the injury, whereas all 12 PROC+/+;F9−/− mice pretreated with HAPC1573 survived (Figure 3C). The PROC+/+;F8−/− mice reached the maximum knee swelling 4 days after injury, which was a ∼120% increase in diameter relative to that prior to the injury. When pretreated with HAPC1573, the maximum knee joint diameter only increased by ∼70% the diameter of the normal knee joint, illustrating a significantly alleviated bleeding compared with the untreated mice (Figure 3B). Pretreatment with FVIII also alleviated the swelling symptom in a degree comparable to pretreatment with HAPC1573 (Figure 3B). In PROC+/+;F9−/− mice, we found that HAPC1573 pretreatment alleviated the knee swelling symptom as effectively as FIX pretreatment (Figure 3D).
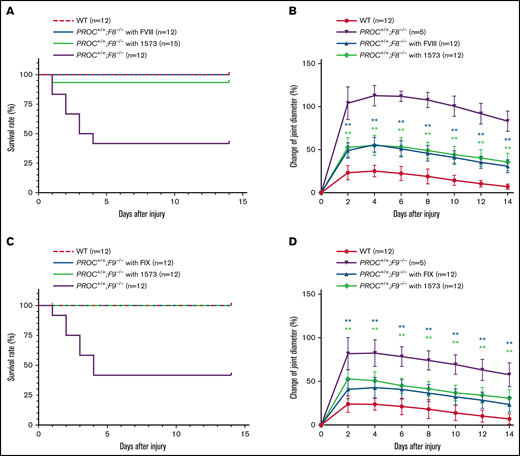
HAPC1573 (1573) antibody treatment significantly improves survival and ameliorates joint bleeding in a knee-joint injury model. The capsules of the right knee joints of anesthetized mice were punctured under the patella with a 30 × 0.5-G needle to induce joint bleeding. Knee diameter, before the needle injury and every other day during the 2 weeks after the injury, was measured with an electronic caliper, and the change of joint diameter was calculated as percentage of the before-injury value. (A,C) The survival rates of mice after the injuries. Survival curves were plotted using the Kaplan-Meier method. (B,D) Changes of knee bleeding represented as percentage of the knee diameter change. Data were expressed as mean ± SD. PROC+/+;F8−/− mice were pretreated for 30 minutes with either 2 IU per mouse of FVIII or 20 µg per mouse of 1573 (A-B). PROC+/+;F9−/− mice were pretreated for 30 minutes with either 2 IU per mouse of FIX or 20 µg per mouse of 1573 (C-D). **P < .01 (log-rank test).
HAPC1573 (1573) antibody treatment significantly improves survival and ameliorates joint bleeding in a knee-joint injury model. The capsules of the right knee joints of anesthetized mice were punctured under the patella with a 30 × 0.5-G needle to induce joint bleeding. Knee diameter, before the needle injury and every other day during the 2 weeks after the injury, was measured with an electronic caliper, and the change of joint diameter was calculated as percentage of the before-injury value. (A,C) The survival rates of mice after the injuries. Survival curves were plotted using the Kaplan-Meier method. (B,D) Changes of knee bleeding represented as percentage of the knee diameter change. Data were expressed as mean ± SD. PROC+/+;F8−/− mice were pretreated for 30 minutes with either 2 IU per mouse of FVIII or 20 µg per mouse of 1573 (A-B). PROC+/+;F9−/− mice were pretreated for 30 minutes with either 2 IU per mouse of FIX or 20 µg per mouse of 1573 (C-D). **P < .01 (log-rank test).
Anti-human APC antibody HAPC1573 corrects defective procoagulant activity of hemophilic plasma
We evaluated if HAPC1573 corrected the procoagulant activity of hemophilic plasma. As expected, pretreating PROC+/+;F8−/− mice with FVIII (2 U per mouse) fully corrected the prolonged aPTT in these mice, and the same correction was also observed in plasma from the PROC+/+;F9−/− mice pretreated with FIX (Table 2). Significantly, pretreatment with 20 µg per mouse of HAPC1573 corrected the prolonged aPTT of plasma from PROC+/+;F8−/− mice. However, the same treatment did not normalize the prolonged aPTT of plasma from PROC+/+;F9−/− mice. The HAPC1573 treatment did not obviously affect fibrinogen levels nor PT of both types of hemophilia mice.
Generation of thrombin is essential for coagulation.6 To further characterize the inhibitory effect of HAPC1573 against anticoagulation, we performed thrombin generation assay with platelet-rich plasma (PRP) from PROC+/+;F8−/− mice pretreated with or without HAPC1573. As expected, WT plasma exhibited normal thrombin generation. Without the pretreatment, there was no obvious thrombin generation in the PRP samples from PROC+/+;F8−/− mice. When pretreated with FVIII or HAPC1573, thrombin generation was increased to the profile similar to that of WT controls (Figure 4). HAPC1573 did not enhance thrombin generation in PRP samples from F8−/− mice, indicating that HAPC1573 improves the plasma coagulant activity of hemophilia mice through modulating human APC in vivo. Interestingly, HAPC1573 did not significantly increase thrombin generation in PRP samples taken from PROC+/+;F9−/− mice either, which is consistent with the aPTT results (Table 2).
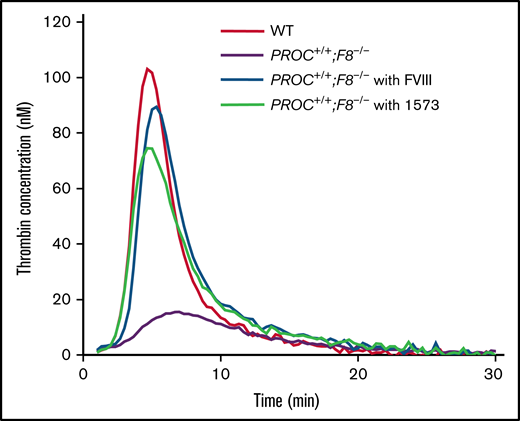
HAPC1573 (1573) antibody treatment restores the thrombin generation of plasma from PROC+/+;F8−/− mice. Platelet-rich plasma was prepared from blood taken from mice (n = 3) in 4 groups as indicated. PROC+/+;F8−/−mice were pretreated with or without FVIII (2 IU per mouse) or 1573 (20 µg per mouse) 30 minutes prior to being euthanized. The profile of thrombin generation was a representative of triplicate samples.
HAPC1573 (1573) antibody treatment restores the thrombin generation of plasma from PROC+/+;F8−/− mice. Platelet-rich plasma was prepared from blood taken from mice (n = 3) in 4 groups as indicated. PROC+/+;F8−/−mice were pretreated with or without FVIII (2 IU per mouse) or 1573 (20 µg per mouse) 30 minutes prior to being euthanized. The profile of thrombin generation was a representative of triplicate samples.
HAPC1573 shortens prolonged clotting time of coagulation factors–deficient plasma through blocking APC anticoagulant activity
The APC anticoagulant pathway serves as a major system to balance coagulation.7 To evaluate if HAPC1573 corrected the clotting deficiency of human plasma with deficiency of FVIII or FIX through its blocking of APC anticoagulant activity, we used the aPTT assay with Protac-induced activation of protein C. Our results show that HAPC1573 exhibited a dose-dependent effect on normalizing the aPTT of FVIII- or FIX-deficient human plasma (Figure 5). In addition, we found that HAPC1573 also shortened the aPTT of the human plasma deficient in FXI, FVII, VWF, FV, or FX as well as normal human plasma in the Protac-induced aPTT assay. These data demonstrate the therapeutic potential of HAPC1573 in the treatment of not only FVIII- or FIX-deficient patients but also patients with deficiency of other types of clotting factors.
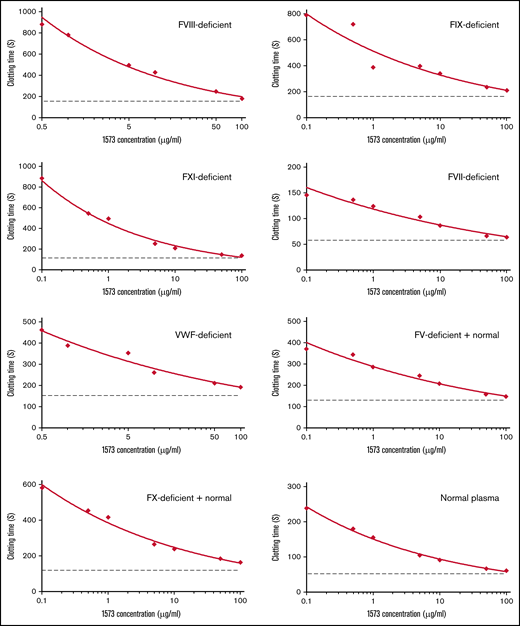
HAPC1573 improves coagulation of human plasma deficient of different types of coagulation factors, respectively, in a dose-dependent manner. Coagulation factor–deficient human plasma and normal human plasma were incubated with 0.1 to 100 μg/mL of HAPC1573 (1573) and Protac, a protein C activator, for 15 minutes, and CaCl2 was added to trigger clotting formation. The clotting time was recorded by an automated analyzer, and data represent the average of duplicates. Dot lines in each assay represent the clotting time without Protac. The coagulation factors deficient in the plasma are as indicated in the top-right corner of each chart. The FV-deficient plasma contained 5% of normal plasma; the FX-deficient plasma contained 1% of normal plasma.
HAPC1573 improves coagulation of human plasma deficient of different types of coagulation factors, respectively, in a dose-dependent manner. Coagulation factor–deficient human plasma and normal human plasma were incubated with 0.1 to 100 μg/mL of HAPC1573 (1573) and Protac, a protein C activator, for 15 minutes, and CaCl2 was added to trigger clotting formation. The clotting time was recorded by an automated analyzer, and data represent the average of duplicates. Dot lines in each assay represent the clotting time without Protac. The coagulation factors deficient in the plasma are as indicated in the top-right corner of each chart. The FV-deficient plasma contained 5% of normal plasma; the FX-deficient plasma contained 1% of normal plasma.
Discussion
In this study, we generated hemophilia A or B mouse models that express human protein C. Using these novel models, we performed preclinical tests of HAPC1573, a mAb specifically blocking the anticoagulant but not other activities of human APC.17 Our data provided in vivo preclinical evidence of therapeutic and prophylactic potentials of HAPC1573 for hemophilia A or B.
Modern hemophilia care has changed from episodic treatment to prophylaxis,4,8 which has significantly improved the prognosis of patients with severe hemophilia. However, conventional prophylaxis requires frequent IV infusions of the replacement factor. This approach requires tremendous resources, causes risks of infection and thrombosis, and is prohibitively expensive.4,8 Furthermore, ∼30% of severe hemophilia A patients develop neutralizing antibodies (inhibitors) against infused factor VIII that renders the replacement therapy ineffective.9 Recently, nonfactor therapeutics, especially antibody-based therapies, have emerged as innovative approaches for the hemophilia prophylactic applications.4,8 Antibody therapeutics such as Hemlibra, which have longer infusion intervals, subcutaneous injection routes, and cause fewer immunogenic reactions, have shown great success in the management of hemophilia A.4,8,20 However, Hemlibra is a bispecific antibody that binds to both activated FIX and factor X, mimicking the function of FVIII.20 Therefore, it is only applicable to hemophilia A but not other types of hemophilia such as hemophilia B. Recently, rebalancing therapies, which block the activities of the natural anticoagulants, such as functional blocking antibodies to protein C and TFPI and an APC-specific serpin, have been developed to enhance coagulation.4,12 Protein C is a major target as it is a primary anticoagulant.7 However, other than its anticoagulant activity, APC also exhibits cytoprotective activities, which include anti-apoptosis, anti-inflammation, and endothelial barrier protection.15 To overcome this issue, we previously generated mAb HAPC1573 that selectively blocks the anticoagulant activity but spares the cytoprotective activities of human APC.17 A recent report shows that humanized HAPC1573 provides in vivo protection against bleeding in an acquired hemophilia A monkey model.16 The monkey model used in this study is an acute hemophilia model, which is quite different from the chronic nature of the human hemophilia A. In addition, APC cleaves activated FVIII and factor V and thus downregulates thrombin generation in the common clotting pathway.1,7 Therefore, HAPC1573 may also be therapeutically applicable for hemophilia B and other types of hemophilia in addition to hemophilia A.
To test the therapeutic potential of therapeutic candidates targeting human APC in a preclinical hemophilia model, we generated a mouse knock-in line (PROC+/+) in which mouse protein C is replaced by the human protein C. By breeding PROC+/+ with F8−/− or F9−/− mice, we also generated hemophilia mice expressing human protein C. These models, to our knowledge, are the first humanized protein C mouse models for in vivo testing of therapeutic candidates targeting human APC. Lack of protein C in mice results in lethal perinatal consumptive coagulopathy.21 In contrast, PROC+/+ mice were viable and able to breed. Hematological measurements, including aPTT, PT, fibrinogen level, and platelet count, were comparable to those of WT mice (Table 1). PROC+/+;F8−/− or PROC+/+;F9−/− mice do not exhibit a more severe bleeding phenotype than F8−/− or F9−/− mice, although they are susceptible to lethal bleeding after tail snipping or knee joint injury similar to F8−/− or F9−/− mice.10,22-24 For its activation, protein C primarily interacts with its endothelial protein C receptor (EPCR). The amino acid sequences of the region interacting with protein C between human EPCR and mouse EPCR are highly conserved (91%).25 Mouse EPCR functionally interacts with human activated protein C in vitro26,27 and in vivo in mice.28 It is of note that some previous in vitro studies show that human plasma-derived or recombinant human APC had a lower anticoagulant activity compared with mouse APC in mouse plasma.29-31 Therefore, it is possible that the human protein C may not function as optimally as mouse protein C in PROC+/+ mice, although our data described above indicate that the human protein C is sufficient to maintain essential anticoagulant function.
Using the PROC+/+ hemophilia models, we found that HAPC1573 effectively corrected the coagulation defects of hemophilia A or B mice challenged with tail transection or knee joint injury and rescued these mice from knee joint injury–induced mortality. HAPC1573 only binds and inhibits the APC but not its proenzyme form.17 As we have shown previously, HAPC1573 blocks the anticoagulant activity of APC without affecting its cytoprotective, anti-inflammatory, and cell-signaling functions.15,17 A previous study shows that humanized HAPC1573 binds to the autolysis loop of APC that comprises a known APC-FVa interface.16 Therefore, it is likely that HAPC1573 inhibits APC’s anticoagulant function through interfering with the interaction between APC and FVa.
The potential therapeutic effects of HAPC1573 are additionally demonstrated in the in vitro coagulation assays. HAPC1573 significantly reduced the prolonged aPTT and enhanced the thrombin generation of PRP samples from PROC+/+;F8−/− mice. In contrast, HAPC1573 did not enhance thrombin generation in PRP samples from F8−/− mice, indicating that HAPC1573 improves the plasma coagulant activity of hemophilia mice through interacting with APC. Protein C is activated by binding of thrombin with thrombomodulin on vascular endothelium. Under physiological conditions, a low level of APC is present in the plasma due to activation by residual thrombin binding to thrombomodulin.32 Therefore, PROC+/+;F8−/− mice should have a low level of residual activated form of the human protein C in the plasma. HAPC1573 could block the interaction between this residual amount of human APC and activated factor V and thus enhance the coagulant activity of the latter, correcting the thrombin generation profile of PROC+/+;F8−/− mice. These results indicate that human protein C in PROC+/+;F8−/− mice can be activated by mouse thrombin-thrombomodulin complex. The fact that PROC+/+ mice are healthy and have normal aPTT also supports this conclusion.
Interestingly, HAPC1573 pretreatment did not correct the prolonged aPTT nor improve the thrombin generation of plasma from PROC+/+;F9−/− mice. As described above, under physiological conditions, there is a very low level of APC in the plasma.32 Therefore, the plasma of PROC+/+;F9−/− mice without challenge should have a low level of APC. In the aPTT assay, FVIIIa in PROC+/+;F9−/− plasma might compete with FVa to interact with the mAb-APC complex. As a result, the FVa cofactor activity might not be affected.
Noticeably, HAPC1573 was able to shorten the clotting time of human plasma deficient of factors V, VII, X, XI, and VWF in the presence of protein C activation, suggesting that the APC-targeted therapeutic has a broad spectrum of applications in correcting the coagulation defects caused by many other types of hereditary deficiency of coagulation factors. In the Protac-induced aPTT assay, we demonstrated the concentration-dependent effects of HAPC1573 on shortening aPTT in both FVIII- and FIX-deficient human plasma. However, HAPC1573 at the concentration of 50 µg/mL did not reach the maximum inhibitory effects on APC anticoagulant activity in this in vitro experiment. This observation is consistent with a previous study,16 which indicates that a high dose at 30 mg/kg of a humanized version of HAPC1573 was found to provide maximal protection in an acute acquired monkey hemophilia model. Therefore, it is important to develop therapeutic antibody mutants with greater affinity performance than the native HAPC1573. To this end, we have performed systemic mutagenesis of every amino acid in the complementarity-determining region of HAPC1573. Based on our promising preliminary data, we anticipate obtaining humanized HAPC1573 mutants with greater affinity that are sufficient for potential human clinical trials in the future.
Factor or nonfactor replacement therapies for the treatment of hemophilia may carry a risk of thrombotic side effects if not appropriately administrated. For example, thrombotic complications were reported when emicizumab was used in combination with activated prothrombin complex concentrate alone or concurrent with activated recombinant factor VII.33 In our study, we found that the maximum inhibitory effect of aPTT by the highest concentration of HAPC1573 in the presence of protein C activation could not shorten the aPTT caused by deficiency of various coagulant factors in the absence of protein C activation; this suggests that inhibiting APC anticoagulant activity by HAPC1573 has little, if any, risk of thrombotic side effects.
In summary, we generated novel hemophilia A or B mouse models expressing human protein C, which is a valuable tool to facilitate future studies of human protein C–targeted therapeutics for hemophilia. Our results show that selectively blocking the APC anticoagulant activity is a promising therapeutic and/or prophylactic strategy for hemophilia A and B, as well as for hemophilia patients with deficiency of other clotting factors.
Acknowledgments
The authors thank Cyagen Biosciences, Inc. for their technical assistance on generating mouse models.
This work was supported by funds from the Jiangsu Provincial Key Medical Center (YXZXA2016002) (C.R. and D.W.), the Priority Academic Program Development of Jiangsu Higher Education Institutions (C.R. and D.W.), and Anhui Provincial Key Research and Development Project, China (grant no. 202104j07020020).
Authorship
Contribution: J.X., M.J., F.Y., L.C., C.T.E., D.W., and L.X. designed research; F.Y., J.H., M.J., J.Y., G.Z., Z.M., L.C., L.Z., L.H., S.N., Z.X., and X.Z. performed research; J.X., M.J., F.Y., Y.J., J.Y., B.Z., X.B., C.R., Y.H., N.L.E., D.W., C.T.E., and L.X. analyzed data and contributed to the manuscript preparation; and J.X., D.W., Y.J., and L.X. wrote the paper.
Conflict-of-interest disclosure: L.C., J.Y., G.Z., Z.X., and J.X. are employees of the Shanghai RAAS Blood Products Co., Ltd. L.C., J.Y., Z.X., J.X., and C.T.E. are inventors of patents and patent applications related to HAPC1573 (US patents No. 8153766 and 9127072; inventors, J.X. and C.T.E.; assignee, Oklahoma Medical Research Foundation) and humanized protein C mouse models (patent application: US2021/0120789 A1).
Correspondence: Jun Xu, Shanghai RAAS Blood Products Co., Ltd., 2009 Wangyuan Rd, Shanghai 201401, China; e-mail: junxu@raas-corp.com; Depei Wu, 188 Shizi St, Suzhou 215006, China; e-mail: wudepei@suda.edu.cn; and Charles T. Esmon, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: charles-esmon@omrf.org.