Key Points
Contact/intrinsic and extrinsic pathways of coagulation are both activated in COVID-19 and likely contribute to FIX activation in vivo.
FIXa:AT complex level may be an adverse prognostic biomarker.

Abstract
Coagulation activation is a prominent feature of severe acute respiratory syndrome coronavirus 2 (COVID-19) infection. Activation of the contact system and intrinsic pathway has increasingly been implicated in the prothrombotic state observed in both sterile and infectious inflammatory conditions. We therefore sought to assess activation of the contact system and intrinsic pathway in individuals with COVID-19 infection. Baseline plasma levels of protease:serpin complexes indicative of activation of the contact and intrinsic pathways were measured in samples from inpatients with COVID-19 and healthy individuals. Cleaved kininogen, a surrogate for bradykinin release, was measured by enzyme-linked immunosorbent assay, and extrinsic pathway activation was assessed by microvesicle tissue factor–mediated factor Xa (FXa; MVTF) generation. Samples were collected within 24 hours of COVID-19 diagnosis. Thirty patients with COVID-19 and 30 age- and sex-matched controls were enrolled. Contact system and intrinsic pathway activation in COVID-19 was demonstrated by increased plasma levels of FXIIa:C1 esterase inhibitor (C1), kallikrein:C1, FXIa:C1, FXIa:α1-antitrypsin, and FIXa:antithrombin (AT). MVTF levels were also increased in patients with COVID-19. Because FIXa:AT levels were associated with both contact/intrinsic pathway complexes and MVTF, activation of FIX likely occurs through both contact/intrinsic and extrinsic pathways. Among the protease:serpin complexes measured, FIXa:AT complexes were uniquely associated with clinical indices of disease severity, specifically total length of hospitalization, length of intensive care unit stay, and extent of lung computed tomography changes. We conclude that the contact/intrinsic pathway may contribute to the pathogenesis of the prothrombotic state in COVID-19. Larger prospective studies are required to confirm whether FIXa:AT complexes are a clinically useful biomarker of adverse clinical outcomes.
Introduction
The critical role of coagulation pathways in the pathogenesis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) disease (COVID-19) is supported by a growing number of studies demonstrating alterations of several biomarkers of coagulation activation, some of which have been associated with disease severity,1,2 elevated risk of venous thromboembolism,3,4 and/or presence of pulmonary microvascular thrombi.5,6 Activation of coagulation in the context of infectious disease has long been recognized as an integral part of the host response to pathogens7,8 and involves a complex interplay of immune cells, coagulation factors, and other bioactive compounds, which has recently been termed immunothrombosis. In the early phase of the SARS-CoV-2 pandemic, D-dimer was identified as a biomarker correlating with disease severity.9,10 However, plasma D-dimer generation is dependent upon both the coagulation and fibrinolytic systems, and as such, it does not address the mechanism of coagulopathy in COVID-19. Tissue factor (TF) is a principal trigger of immunothrombosis in acute respiratory distress syndrome,11 and plasma microvesicle-associated TF (MVTF) activity has been demonstrated to correlate with mortality in respiratory viral infections.12 Recently, the association of MVTF with COVID-19 severity13 was reported, suggesting a role for extrinsic activation in this disease. However, because of the lack of an international TF standard, standardization of this assay has not been possible.14 In addition, the assay operates at the limits of sensitivity, with undetectable levels in many healthy individuals. For these reasons, its use has been limited to research laboratories.
Components of the contact system (ie, factor XII [FXII] and FXI) have increasingly been recognized as key mediators in the prothrombotic states observed in multiple clinical contexts, such as sepsis and cancer, despite their lesser roles in hemostasis.15-17 Both FXIa and kallikrein (PKa) can activate FIX after contact activation,18-20 and the TF:FVIIa complex can also directly activate FIX.21 Similarly, FXI may be activated not only by FXIIa but also by thrombin. FXIa is irreversibly inactivated by several serpins, including C1 inhibitor, α1-antitrypsin, and (in the presence of heparin) antithrombin (AT), whereas PKa is primarily inactivated by C1. In turn, FIXa is primarily inactivated by AT, forming a stable covalent FIXa:AT complex. Meticulous sampling techniques and processing must be considered when evaluating plasma for these complexes, because the principal activator of the contact pathway, FXII, undergoes accelerated autoactivation upon contact with an anionic surface.22 Although growing interest in the contact pathway has raised the question of targeting its component proteins for the prevention of thrombotic complications,23,24 the laboratory characterization of these complexes as indices of coagulation activation continues to be challenging.15
To address the mechanism of coagulation activation in individuals presenting with COVID-19, we used a recently described strategy to assess activation of the contact and intrinsic pathways25 and also measured MVTF levels as a biomarker of extrinsic pathway activation.26 In a preliminary secondary analysis, we then sought to determine the associations between levels of these biomarkers and adverse clinical outcomes in a well-characterized study cohort.
Methods
Study population
The study was performed in accordance with the Declaration of Helsinki and approved by the institutional review boards of the University of Campinas and University of North Carolina at Chapel Hill. All patients (or their legal representatives) and all healthy volunteers provided written informed consent before any study procedure. The study population consisted of patients admitted to a single academic hospital with a diagnosis of COVID-19 between 23 April and 14 June 2020 who were enrolled in a clinical trial focused on bradykinin (BK) inhibition in COVID-19 (that trial was registered in the Brazilian Clinical Trials Registry at https://ensaiosclinicos.gov.br/rg/RBR-5s2mqg). In brief, 30 patients with severe COVID-19 were randomly assigned to 1 of the following 3 treatment arms: standard care, icatibant (BK receptor 2 inhibitor), or C1 esterase inhibitor.27 Samples used in our study were obtained before any trial intervention (hereafter referred to as baseline samples). Inclusion criteria included age ≥ 18 years, symptom duration ≤12 days, positive real-time polymerase chain reaction for SARS-CoV-2, presence of typical COVID-19 pneumonia on lung computed tomography (CT) scan, and oxygen saturation ≤94% in ambient air or arterial oxygen partial pressure/fractional inspired oxygen ratio <300 mm Hg. Exclusion criteria were pregnancy, severe renal or liver disease, HIV infection or other immunodeficiency state, previous diagnosis of cancer, ischemic myocardial disease, history of thromboembolic events, hereditary angioedema, or use of any experimental treatment for SARS-CoV-2 infection. Age- and sex-matched healthy individuals from the same geographic regions were recruited from health care employees of the local blood bank, observing the same exclusion criteria.
Sample collection and processing
After ensuring satisfactory venipuncture, blood samples were collected into 3.2% sodium citrate or EDTA K2 tubes immediately after enrollment in the clinical trial (median time from admission, 1 day; range, 0-3 days). Samples were processed within 2 hours of collection by double centrifugation at 1800g for 15 minutes at 22°C to obtain platelet-free plasma. EDTA K2 samples were subjected to only a single centrifugation step. Aliquots (300 μL) were immediately frozen at −80°C until analysis.
Clinical and laboratory outcomes
Clinical and laboratory data were obtained from electronic medical records and case report forms of the clinical trial. The extent of lung disease on admission was based on CT scans performed upon enrollment and scored according to previously published criteria.27
Laboratory evaluation of classical hemostatic parameters
Coagulation screening assays (prothrombin time and activated partial thromboplastin time), coagulation factor activity (fibrinogen, FVIII:C, FIX:C, FX:C, FXI:C, and FXII:C), von Willebrand factor antigen, von Willebrand ristocetin cofactor activity, and AT level were measured in an automated coagulometer (ACL TOP 550 CTS; Instrumentation Laboratory, Bedford, MA) using commercially available assays from the same manufacturer (HemosIL reagents). D-dimer was measured using an immunoturbidimetric assay (Innovance D-Dimer; Siemens Healthcare). P-selectin, urokinase-type plasminogen activator receptor, and plasminogen activator inhibitor 1 (PAI-1) levels were measured using a customized Luminex immunoassay (Procarta Plex multiplex panel; Thermo Fisher Scientific) in a Bioplex 200 instrument (Bio-Rad). Plasmin-antiplasmin levels were measured using a commercially available enzyme-linked immunosorbent assay (Technozym). All assays were performed in citrate-anticoagulated plasma, except for the Luminex assays, which were performed in EDTA-anticoagulated samples.
Protease:serpin complexes
Activation of the contact system and intrinsic coagulation pathway was determined by immunologic assays to detect complexes of proteases and their respective serpin inhibitors, as previously described.25 Briefly, citrated test plasma was thawed at 37°C for 10 minutes before dilution into buffer containing benzamidine and FPR-chloromethylketone to quench any remaining enzymatic activity. Samples were assayed in triplicate and calculated against a standard curve. Complex standards were diluted into deficient plasma for the respective assay; for example, FXIIa:C1 complexes were serially diluted into FXII-deficient plasma.
Measurement of intact and cleaved HK
The immunoassay for detecting intact and cleaved kininogen (HK) was performed as previously described.28
Measurement of MVTF activity
Assessment of the procoagulant capacity of TF-bearing microvesicles was performed as previously described using an assay based on FXa generation by isolated microvesicles in the presence of added FX and FVIIa.29
Detection and enumeration of PDMVs
Platelet-derived microvesicles (PDMVs) were detected and enumerated by flow cytometry using a CytoFLEX cytometer (Beckman Coulter, Carlsbad, CA) as previously described.30 Briefly, 50 μL of platelet-free plasma was thawed at 37°C for 3 minutes and incubated on ice for 30 minutes with 20 μL of a reagent mix containing 1 μL of calcein Violet AM (Thermo Fisher Scientific), 1.5 μL of bovine lactadherin fluorescein isothiocyanate (Haematologic Tech), 1 μL of anti-CD41 allophycocyanin (eBioscience), and 16.5 μL of filtered sterile phosphate-buffered saline (PBS) buffer. Sterile PBS (1930 μL) was added, and the solution was centrifuged at 20 000g for 30 minutes at 4°C. The resulting MV pellet was resuspended in 1000 μL of PBS buffer, and 100-μL aliquots were subjected to flow cytometric analysis. PDMVs were identified as events positive for lactadherin, calcein, and anti-CD41. Polystyrene spherical particles <2000 nm (Rosetta calibration beads; Exometry, Amsterdam, the Netherlands) were used for the quantification of events. PBS buffer was used as negative control, and PDMVs were expressed per events per milliliter.
Statistical analysis
Data are presented as mean ± standard deviation or median (interquartile range), as indicated. Differences in continuous variables were analyzed using the Student t or Mann-Whitney test according to data distribution, assessed by the D’Agostino and Pearson normality test. Correlation was calculated using the Pearson or Spearman correlation coefficient. A P value ≤.05 was considered significant. All statistical analyses were performed using SPSS (version 26; IBM) or GraphPad Prism (version 7.0; GraphPad, Inc.) software.
Results
Study participant information
Thirty patients with COVID-19 and 30 healthy individuals were included in this study. Basic demographic and hematologic characteristics of the study population are shown in Table 1, and patient comorbidities and medications are shown in supplemental Table 1. Clinical characteristics of patients with COVID-19 are shown in Table 2. As expected, at the time of admission, significantly higher levels of several coagulation and fibrinolysis biomarkers were observed in patients at presentation (Table 3). These included elevated levels of multiple contact and intrinsic pathway coagulation factors (FXII, FXI, FIX, and FVIII) measured by 1-stage clotting assays. In addition, as reported by others, von Willebrand factor and fibrinolytic parameters were elevated.1,9,10 However, neither AT chromogenic activity nor P-selectin antigen level differed between patients and controls. Of note, neither baseline differences nor the clinical outcomes evaluated in the study were affected by treatment allocation in the 3 arms of the clinical trial from which baseline samples were obtained for this study (supplemental Table 2).27 Moreover, the standard patient care protocol did not differ by treatment allocation (supplemental Table 3).
Contact and intrinsic pathway activation
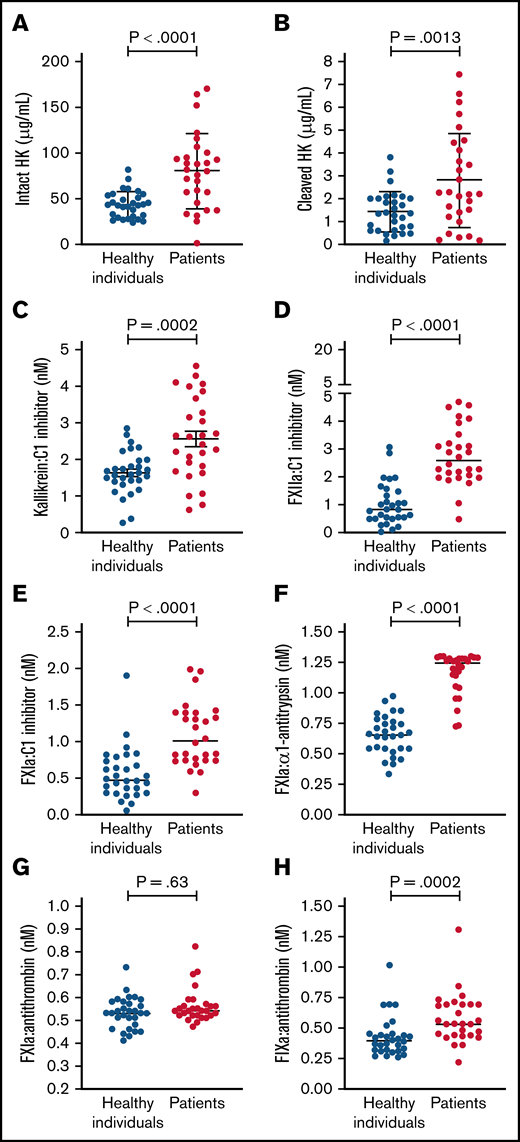
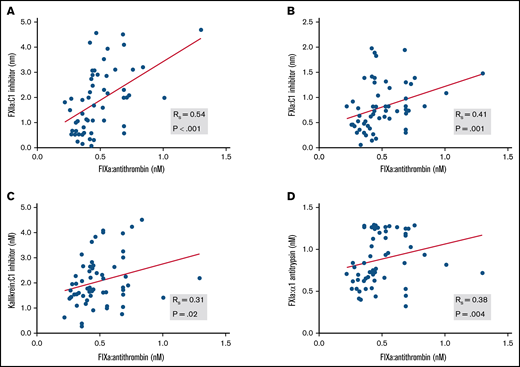
Circulating levels of protease:serpin complexes in patients and controls at baseline are shown in Figure 1. The levels of all complexes in the contact and intrinsic pathways, with the exception of FXIa:AT, were significantly elevated in patients with COVID-19 compared with healthy individuals. These results are indicative of systemic activation of the contact and intrinsic pathways. FIXa:AT level correlated with other protease:serpin complexes in the contact pathway, particularly FXIIa:C1 (Spearman correlation coefficient [rs] = 0.54; P < .001) and FXIa:C1 (rs = 0.41; P = .001), supporting the hypothesis that activation of the contact pathway contributes at least in part to the downstream activation of FIX (Figure 2). Of note, no differences were observed in FIXa:AT level between patients allocated to any of the 3 treatment arms,27 either on admission or at the end of treatment with study drug (day +4; supplemental Figure 1). These data support the fact that the experimental treatment arms had no influence on FIXa:AT level. Lastly, we noted that 14 of the 30 enrolled patients received a single prophylactic 40-mg dose of enoxaparin before baseline laboratory results were obtained. However, neither the serine protease:serpin levels nor any of the other biomarkers demonstrated any trends when comparing those who did vs those did not receive enoxaparin (data not shown).

Contact and intrinsic pathway activation in patients and healthy individuals. Dot plots showing plasma levels of intact high molecular weight HK (A), cleaved HK (B), kallikrein:C1 inhibitor (C), FXIIa:C1 (D), FXIa:C1 inhibitor (E), FXIa:α1-antitrypsin (F), FXIa:antithrombin (G), and FIXa:AT (H). Mean ± standard deviation or median (horizontal bars) depicted for Gaussian and non-Gaussian data, respectively; similarly, P values are from t or Mann-Whitney test according to data distribution (n = 28-30 per group). Notably, because the standard curve typically spans 4 logs for FXIa:α1-antitrypsin complex, many patient samples exceeded the upper limit of detection. Samples from 2 patients were not processed because of critical preanalytic issues (low volume).
Contact and intrinsic pathway activation in patients and healthy individuals. Dot plots showing plasma levels of intact high molecular weight HK (A), cleaved HK (B), kallikrein:C1 inhibitor (C), FXIIa:C1 (D), FXIa:C1 inhibitor (E), FXIa:α1-antitrypsin (F), FXIa:antithrombin (G), and FIXa:AT (H). Mean ± standard deviation or median (horizontal bars) depicted for Gaussian and non-Gaussian data, respectively; similarly, P values are from t or Mann-Whitney test according to data distribution (n = 28-30 per group). Notably, because the standard curve typically spans 4 logs for FXIa:α1-antitrypsin complex, many patient samples exceeded the upper limit of detection. Samples from 2 patients were not processed because of critical preanalytic issues (low volume).

Association between FIXa:AT with other contact and intrinsic pathway protease:serpin complexes. Spearman correlation coefficients are shown for FXIIa:C1 inhibitor (A), FXIa:C1 inhibitor (B), kallikrein:C1 inhibitor (C), and FXIa:α1-antitrypsin (D). Samples from 2 patients were not processed because of critical preanalytic issues (low volume).
Association between FIXa:AT with other contact and intrinsic pathway protease:serpin complexes. Spearman correlation coefficients are shown for FXIIa:C1 inhibitor (A), FXIa:C1 inhibitor (B), kallikrein:C1 inhibitor (C), and FXIa:α1-antitrypsin (D). Samples from 2 patients were not processed because of critical preanalytic issues (low volume).
Correlations between individual biomarkers
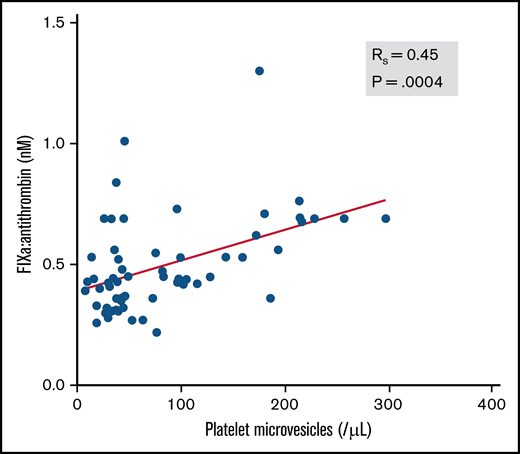
Next, we evaluated whether classical biomarkers of thromboinflammation correlated with circulating FIXa:AT complexes. The correlation coefficient (rs) between FIXa:AT level and D-dimer was 0.481 (P ≤ .001). A commonly measured prognostic biomarker in COVID-19, CRP, was also moderately correlated with D-dimer (rs = 0.51; P = .005) but only at a moderate to low degree with FIXa:AT (rs = 0.37; P = .06). Soluble P-selectin, which has been reported to be elevated in patients with COVID-19,31 was moderately correlated with CRP (rs = 0.64; P < .0001), D-dimer (rs = 0.52; P = .003), and FIXa:AT (rs = 0.41; P = .002). We posited that participation of FIXa in the phospholipid-dependent intrinsic tenase complex on activated platelets might contribute to thrombin-dependent activation of platelets and enhanced release of platelet (CD41+)–derived microvesicles. Indeed, as shown in Figure 3, the FIXa:AT complex was moderately correlated with platelet-derived microvesicle number in plasma (rs = 0.45; P = .0004).

Association between FIXa:AT complex and platelet microvesicles among patients with COVID-19 and healthy individuals. Platelet microvesicles were counted by flow cytometry. Spearman correlation coefficient (n = 58). Samples from 2 patients were not processed because of critical preanalytic issues (low volume).
Association between FIXa:AT complex and platelet microvesicles among patients with COVID-19 and healthy individuals. Platelet microvesicles were counted by flow cytometry. Spearman correlation coefficient (n = 58). Samples from 2 patients were not processed because of critical preanalytic issues (low volume).
Contribution of MVTF activity to FIXa:AT generation
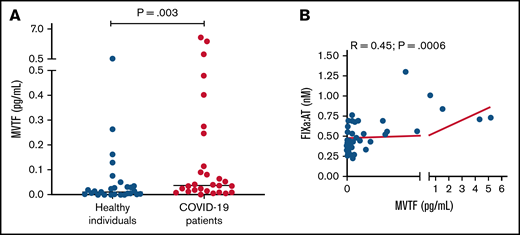
Recent reports have demonstrated that TF-bearing microvesicles are correlated with disease severity and mortality in COVID-19.13,32 To explore whether increased extrinsic pathway activation could also contribute to FIXa generation in COVID-19, we measured MVTF activity in plasma. Figure 4A demonstrates that MVTF activity was significantly elevated in patients with COVID-19 compared with healthy individuals, albeit with significant overlap at lower or undetectable concentrations. However, MVTF activity was moderately but significantly correlated with circulating FIXa:AT complex (r = 0.45; P = .0006; Figure 4B). The latter observation suggests that the TF:FVIIa complex may also contribute to FIX activation in vivo.
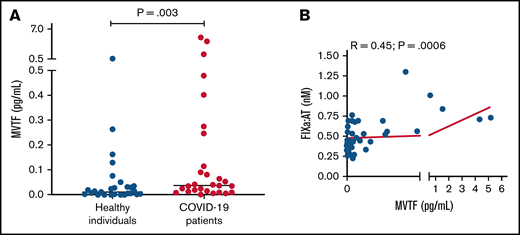
MVTF activity is increased in patients with COVID-19. (A) Levels of TF activity in microvesicles isolated from healthy individuals (blue circles) and patients with coronavirus disease (red circles). (B) Correlation between FIXa:AT complex and MVTF.
MVTF activity is increased in patients with COVID-19. (A) Levels of TF activity in microvesicles isolated from healthy individuals (blue circles) and patients with coronavirus disease (red circles). (B) Correlation between FIXa:AT complex and MVTF.
Plasma analytes as potential markers of disease severity
Finally, we explored whether the circulating level of ≥1 protease:serpin complexes and/or other biomarkers might be associated with clinically relevant adverse outcomes, specifically total length of hospital stay, length of intensive care unit stay, and progression of lung disease based on radiologic CT score. As shown in Table 4, although a number of analytes were significantly associated with ≥1 of these outcomes, only a few were associated with all 3 outcomes. These included several candidate biomarkers that have previously been implicated as prognostic biomarkers, including CRP,33 D-dimer,34 and PAI-1.35 In addition, plasma level of FIXa:AT complex was also associated with all recorded adverse clinical outcomes (Table 4; Figure 5). In sum, these data suggest that FIXa generation, a specific marker of coagulation activation, may be a novel biomarker of clinically significant adverse outcomes in COVID-19.
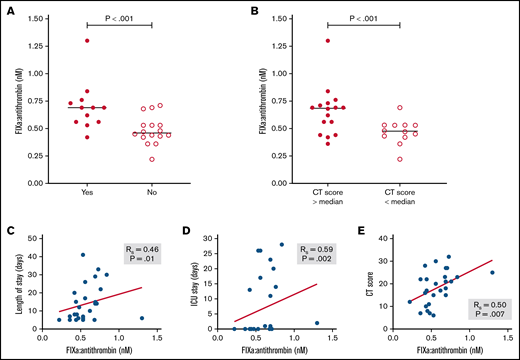
Association of FIXa:AT complex with clinical outcomes among patients with COVID-19. FIXa:AT level in patients divided by need for intensive care unit (ICU) (A) and lung CT score at admission (B). (C-E) P values from Mann-Whitney test. Correlation coefficients between FIXa:AT level with other clinical outcomes. Samples from 2 patients were not processed because of critical preanalytic issues (low volume).
Association of FIXa:AT complex with clinical outcomes among patients with COVID-19. FIXa:AT level in patients divided by need for intensive care unit (ICU) (A) and lung CT score at admission (B). (C-E) P values from Mann-Whitney test. Correlation coefficients between FIXa:AT level with other clinical outcomes. Samples from 2 patients were not processed because of critical preanalytic issues (low volume).
Discussion
The mechanism underlying coagulation activation and its contribution to adverse outcomes in severe SARS-CoV-2 disease (COVID-19) remain elusive. In this exploratory study, circulating levels of protease:serpin complexes indicative of contact and intrinsic pathway activation were observed to be elevated in patients with COVID-19. We demonstrated that levels of the various FXIa:serpin complexes and, to a lesser extent, PKa:C1 complexes were positively correlated with FIXa:AT level, suggesting that both FXIa and PKa might contribute to FIX activation in vivo.18-20 Furthermore, in agreement with previous reports,13,32 we observed that MVTF level was also elevated at baseline and also correlated with level of FIXa:AT, suggesting that the activation of FIX occurs via both the contact/intrinsic and extrinsic pathways. Whatever the mechanism of initial FIX activation, the resultant downstream thrombin generation may then activate FXI, thereby promoting further FIX activation.36
Although activation of the contact pathway by bacterial infection has been described in humans and in animal models,24,37 few studies have investigated whether this pathway is activated in viral infection.38,39 Hyperinflammation associated with COVID-19 results in activation of innate immune cells (neutrophils and monocytes) that interact with platelets and the coagulation cascade, ultimately leading to micro- and macrothrombosis. Generation of neutrophil extracellular traps40,41 and/or release of platelet-derived polyphosphate42 may promote activation of the contact pathway in COVID-19 during immunothrombosis in the microvasculature of the lung and other vascular beds. In addition to its contribution to thrombin generation, contact activation leads to an inflammatory cascade mediated by BK as a product of PKa cleavage of HK. Indeed, the phrase BK storm was coined to describe the dysregulated generation of BK (and its metabolite des-Arg9-BK) in COVID-19 infection, which may enhance pulmonary inflammation and increase vascular permeability.43,44 Although the very short half-life of BK makes it challenging to measure directly in plasma, BK generation can be inferred by quantitation of the more stable cleaved form of HK.28 Our data suggest that in the early stages of infection, levels of cleaved HK are significantly increased in those with COVID-19 compared with healthy individuals, but this is accompanied by an increased concentration of the intact form of HK, likely as a compensatory response. This interpretation is compatible with other data shown here indicating that increased zymogen levels of the other contact factors are also associated with elevated levels of the respective serine protease:serpin complexes (eg, elevated FXII:C and increased FXIIa:C1 complexes were observed in the same samples).
An exploratory aim of this study was to evaluate associations between established and novel biomarkers of coagulation activation and adverse clinical outcomes. Along with D-dimer, CRP, and PAI-1, the concentration of FIXa:AT complex upon admission was associated with total duration of hospitalization, duration of intensive care, and degree of lung injury assessed by CT score. Although involvement of the contact pathway in SARS-CoV2 infection was suggested early in the pandemic,45-48 there are limited data linking activation of this pathway with patient outcomes.49,50 A single study evaluated contact activation biomarkers in conjunction with FIXa:AT complex in patients with COVID-19. Busch et al49 quantified these biomarkers at presentation and during follow-up in patient cohorts with mild, moderate, or severe disease. Levels of PKa:C1, FXIa:α1-antitrypsin, FXIa:AT, and FIXa:AT were all elevated at baseline and on follow-up. Notably, all 3 downstream activation biomarkers (FXIa:AT, FIXa:AT, and TAT) demonstrated a strong association (P < .0001) with COVID-19 disease severity. Even allowing for the different definitions of clinical severity, these findings are in broad agreement with the data presented in our report.
This study has several limitations. First, the sample size was relatively small, although the differences in the levels of measured analytes between patients and healthy individuals were rather profound. Moreover, consecutive enrollment of a well-defined patient population, standardized sample collection with control of preanalytic variables, and structured obtention of clinical and laboratory data in the setting of a randomized controlled trial served to minimize the inherent heterogeneity of observational studies conducted in the early phases of the pandemic. Second, we did not measure longitudinal trends in the selected analytes; however, our primary objective was to assess baseline levels of the contact activation markers and preliminarily evaluate their clinical prognostic potential. Furthermore, we wished to avoid any confounding by treatment allocation that may have been operative at subsequent time points. Finally, although our study did not include a control group of patients with non–COVID-19 pneumonia, it should be noted that the primary objective was not to determine whether any observed changes were specific to COVID-19. Rather, we sought to identify whether ≥1 of the measured analytes might be novel prognostic indicators of adverse clinical outcomes. Indeed, our preliminary analysis suggests that baseline FIXa:AT complex may have such prognostic significance. However, because it is difficult to conclude that a meaningful association exists between these laboratory parameters and clinical outcomes when the latter are <20, a larger appropriately powered prospective study is required to validate this hypothesis.
Acknowledgments
The authors thank the staff members of the hospital and research institutions in which this study was conducted and the patients and healthy volunteers who agreed to participate.
This study was funded by Sao Paulo Research Foundation grants 2016/14172-6 and 2020/05985-9, FAEPEX-Unicamp grant 2404/2020, and Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior–Brasil (finance code 001). The project was also supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant UL1TR002489.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: M.W.H. performed protease:serpin complex assays and microvesicle assays, contributed to data analysis, and drafted the manuscript; F.L. obtained and processed samples, performed coagulation factor assays, and contributed to data analysis; C.R.P.M. obtained and processes samples and performed multiplex assays: A.I. performed protease:serpin complex assays and contributed to data analysis; S.C.H., M.S.B., and I.S. processed samples for microvesicle quantification, designed the strategy, and performed flow cytometric assays; A.C.P., T.A.N., R.G.U., L.C.R., A.F.B., and B.B. recruited and managed patients and obtained patient/outcome data; S.S.J.D. analyzed and scored lung tomographies; S.S. provided resources for measurement of high molecular weight kininogen; J.M.A.-B. provided laboratory support and infrastructure for classical coagulation assays; F.A.O. contributed to study design and data analysis; M.L.M., E.M., and L.A.V. designed and conducted the clinical trial from which patients were recruited; N.S.K. contributed to study design, oversaw and provided resources and infrastructure for protease:serpin complexes assays, contributed to data analysis, and drafted the manuscript; E.V.D.P. designed the study, obtained and processed samples, oversaw and provided resources and infrastructure for coagulation and microvesicle analyses, contributed to data analysis, and drafted the manuscript; and all authors revised and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nigel S. Key, 8008B Mary Ellen Jones Building, CB #7035, Chapel Hill, NC 27599; e-mail: nigel_key@med.unc.edu; and Erich Vinicius De Paula, Rua Carlos Chagas 480, Laboratory of Hemostasis and Inflammation, Campinas, SP 13083-878, Brazil; e-mail: erich@unicamp.br.