

Key Points
Virions and extracellular vesicles are concomitantly released by SARS-CoV-2–infected cells.
Host-derived tissue factor activity, not viral spike, activates platelets.

Abstract
Platelets are hyperactivated in coronavirus disease 2019 (COVID-19). However, the mechanisms promoting platelet activation by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are not well understood. This may be due to inherent challenges in discriminating the contribution of viral vs host components produced by infected cells. This is particularly true for enveloped viruses and extracellular vesicles (EVs), as they are concomitantly released during infection and share biophysical properties. To study this, we evaluated whether SARS-CoV-2 itself or components derived from SARS-CoV-2-infected human lung epithelial cells could activate isolated platelets from healthy donors. Activation was measured by the surface expression of P-selectin and the activated conformation of integrin αIIbβ3, degranulation, aggregation under flow conditions, and the release of EVs. We find that neither SARS-CoV-2 nor purified spike activates platelets. In contrast, tissue factor (TF) produced by infected cells was highly potent at activating platelets. This required trace amounts of plasma containing the coagulation factors FX, FII, and FVII. Robust platelet activation involved thrombin and the activation of protease-activated receptor (PAR)-1 and -4 expressed by platelets. Virions and EVs were identified by electron microscopy. Through size-exclusion chromatography, TF activity was found to be associated with a virus or EVs, which were indistinguishable. Increased TF messenger RNA (mRNA) expression and activity were also found in lungs in a murine model of COVID-19 and plasma of severe COVID-19 patients, respectively. In summary, TF activity from SARS-CoV-2–infected cells activates thrombin, which signals to PARs on platelets. Blockade of molecules in this pathway may interfere with platelet activation and the coagulation characteristic of COVID-19.
Introduction
Coronavirus disease 2019 (COVID-19) is a respiratory tract infection caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1 Since the onset of this pandemic, millions of people have died.2 Acute respiratory distress syndrome is one of the main characteristics of severe COVID-19, and its manifestation influences the outcome of infection.3 A high degree of generalized inflammation and thrombosis is also reported.1,4-6 Autopsies show that the formation of microthrombi is common in the lung vasculature and other organs distant from the lungs, suggesting a role for dysregulated coagulation.7-10 Furthermore, age, hypertension, diabetes, and coronary heart disease, well-known risk factors for thrombosis and associated with chronic inflammation, are the main determinants of COVID-19 severity and mortality.3,4,11,12
Platelets are anucleated cells at the interface of thrombosis and inflammation.13-15 They are critical in hemostasis, interact with pathogens, and can alert other immune cells.14,15 It is notable that hyperactivated platelets are present in COVID-19.16-22 Hyperactivation is characterized by high expression of P-selectin (CD62P), degranulation (release of platelet factor 4 [PF4] and serotonin [5-HT]), and release of platelet extracellular vesicles (EVs), as well as increased aggregation and adhesion of platelets to other cells, particularly monocytes, and changes in platelet gene expression.16-22
Different laboratories have detected SARS-CoV-2 RNA in association with platelets in patients.16,17,23 While compelling studies confirmed that SARS-CoV-2 could be taken up by platelets, leading to programmed cell death, platelets failed to activate in these conditions.23 Angiotensin-converting enzyme-2 (ACE2), abundantly expressed by epithelial cells in the lungs, is the cognate cell receptor for the SARS-CoV-2 spike protein.24,25 An ACE2-dependent SARS-CoV-2 and platelet interaction has been suggested as a potential mechanism for platelet hyperactivation.26 However, platelet expression of ACE2 could not be conclusively demonstrated,27 and some studies indicated SARS-CoV-2 interacts with platelets through ACE2-dependent and independent mechanisms.23,28 Another putative receptor for a SARS-CoV-2 platelet interaction is CD147.29-31 While evidence of interaction between SARS-CoV-2 and CD147 has been challenged,32 one study suggests that SARS-CoV-2 pseudovirus and recombinant spike proteins activate platelets, characterized by the release of HMGB1+ EVs and soluble P-selectin, in a CD147-dependent manner.33 Another study suggests that spike may engage platelet CD42b, thereby promoting interactions with monocytes and cytokine production by monocytes.34 However, overexpression of spike protein (resulting from vaccination) did not result in platelet activation.35 Thus, there exists conflicting data as to whether and how SARS-CoV-2 activates platelets.
Initiation of the coagulation cascade may also underlie platelet activation. Coagulation abnormalities have been reported in COVID-19,36 which may be the consequence of extensive tissue damage associated with SARS-CoV-2 infection and subsequent engagement of the coagulation cascade. Tissue factor (TF) is the main cellular initiator of coagulation and a highly potent stimulus that is absent in blood under physiological conditions.37-39 Active TF in blood activates the extrinsic (TF) pathway of coagulation, leading to the conversion of prothrombin to thrombin and subsequent fibrin clot formation. Thrombin is a highly potent stimulus of platelet activation and has been previously found to contribute to the H1N1-mediated activation of platelets.40 Several studies detected TF activity in the blood of COVID-19 patients.41-43 Moreover, SARS-CoV-2 spike protein pseudotyped viral infection of human monocyte-derived macrophages induces the expression of active TF in vitro.44 Furthermore, SARS-CoV-2 infection of endothelial cells or endothelial dysfunction have been suggested as a potential source of TF.45
SARS-CoV-2 is an enveloped virus that may acquire host molecules during the budding process.46-51 The propagation of viruses in cell lines is essential, as viral replication requires a cellular host.52 Infected cell supernatants are thus used as a source of the virus, but the potential role of concomitantly released host molecules and EVs must be considered when examining the effects of cell supernatants on target cells. Supernatants containing viruses may be pelleted by centrifugation to reduce the presence of soluble molecules.53,54 Moreover, polyethylene glycol (PEG) precipitation can be used to concentrate viruses from supernatants. However, both centrifugation and PEG precipitation can also enrich EVs.54,55 Given that viruses, protein aggregates, and EVs share biophysical properties,53,54 they cannot be efficiently separated from each other using any of these approaches.53,54 Whether SARS-CoV-2 or host-derived molecules activate platelets has not been studied.
In this study, we examined whether SARS-CoV-2 or components from infected cells could activate platelets.
Methods
A detailed description is provided in the supplemental Material.
Platelet isolation from human donors
Venous blood from healthy donors was collected into citrate anticoagulant solution and centrifuged for 10 minutes at 231g. One mL of platelet-rich plasma (PRP) was transferred to fresh microcentrifuge tubes, and 250 µL of acid-citrate-dextrose (ACD) and ethylene diamine tetraacetic acid (10 mM) were added. PRP was centrifuged (2 minutes at 300g). One mL of the supernatant was transferred to fresh tubes and centrifuged (5 minutes at 1100g). The supernatant was aspirated, and the platelet pellet was resuspended in 100 µL of Tyrode’s buffer (pH, 6.5). Another 900 µL of Tyrode’s buffer (pH, 7.4), 250 µL ACD, and ethylene diamine tetraacetic acid (10 mM) were added. Platelets were then centrifuged for 5 minutes at 1300g. The supernatant was aspirated, and platelets were recovered (200 × 106/mL) in Tyrode’s buffer (pH, 7.4). All steps were performed at room temperature.
Propagation of SARS-CoV-2
SARS-CoV-2 (strain LSPQ, B1 lineage) was obtained from the Laboratoire de Santé Publique du Québec. A549-hACE2 cells were infected at a multiplicity of infection (MOI) of 1 for 1 hour, then washed twice with phosphate-buffered saline (PBS) before the addition of new culture media. The virus was harvested from cell-free media after 4 days and inactivated by the addition of β-propiolactone as described.56
Platelet activation assay
Blood, collected in a 3.2% citrate anticoagulant solution, was centrifuged (2 × 2500g at 15 minutes) to obtain platelet-free plasma (PFP). PFP was incubated in Tyrode’s buffer (pH, 7.4; 10 mM CaCl2) in the presence of stimuli (thrombin, SARS-CoV-2, or controls; 2× concentrated) for 60 minutes. Then, platelets (100 × 106/mL final concentration) were added. All incubations were performed at room temperature. Unless indicated otherwise, platelet activation was assessed after 60 minutes by staining (20 minutes) with antibodies for CD41, activated integrin αIIbβ3*, and CD62P, and platelets were fixed with paraformaldehyde (1%) thereafter.
Results
SARS-CoV-2 activates platelets in a plasma-dependent manner
SARS-CoV-2 was propagated in ACE2-overexpressing human lung epithelial A549 cells. Culture supernatant (C.Med) from noninfected cells used at the same volume as CoV2 was used as a negative control. For safety reasons and convenience, unless specified otherwise, we used β-propiolactone–inactivated SARS-CoV-2 or β-propiolactone–treated C.Med.56 We evaluated the ability of SARS-CoV-2 to induce platelet activation, as assessed by the frequency of platelets expressing the active conformation of αIIbβ3 integrin (αIIbβ3*) and CD62P (αIIbβ3*CD62P), the expression density of αIIbβ3*, and the liberation of EVs (CD41+ EVs). To account for a potential plasma–SARS-CoV-2 interaction independent of a direct virus–platelet interaction, we preincubated virus or C.Med in the presence or absence of recalcified PFP before the addition to washed platelets. The plasma preparations were obtained from nonimmune donors, as determined by the absence of SARS-CoV-2 antibodies (supplemental Figure 1).
Flow cytometric analyses revealed that the presence of SARS-CoV-2 (1:25 vol/vol; 1.9E+05 virus per mL) induced a rapid and significant increase in the percentage of αIIbβ3*CD62P+ platelets (7.5 minutes) and an increase in expression intensity of αIIbβ3* (15 minutes) (Figure 1A). Following a 60-minute stimulation with SARS-CoV-2, the vast majority of platelets expressed αIIbβ3*CD62P, and the levels of CD41+ EVs were increased (Figure 1A). Platelet activation required the presence of plasma (0.5%) (Figure 1A). Platelets failed to activate in response to SARS-CoV-2 (1:25 vol/vol; 1.9E+05 virus per mL) in the absence of plasma or in the presence of C.Med (1:25 vol/vol) in the presence or absence of plasma (Figure 1A). In contrast, platelets efficiently responded to thrombin (1 U/mL) in the presence or absence of plasma (0/0.25/0.5/1%) (supplemental Figure 2).
![SARS-CoV-2 activates platelets in a plasma-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2, red) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med, blue) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 in the presence or absence of PFP (0.5%). Platelets were labeled and fixed after stimulation for 7.5, 15, 30, or 60 minutes, and platelet activation was determined by flow cytometry. N = 3. (B) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and injected into a microfluidics system. The images were taken after 15 minutes and show fluorescently-labeled platelets in the fibrinogen-coated channel. Representative images. N = 4. (C) Platelets were stimulated with C.Med or CoV2 in the presence of PFP (0.5%). N = 4. (D) Platelets were stimulated for 60 minutes with receptor-binding domain (RBD) peptide, S1 subunit of spike, full trimeric spike protein, or thrombin in the presence of platelet-free plasma (0.5%). N = 4. Data are represented as mean ± SD. Statistical analysis: (A, C, D) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. **P < .01, ***P < .001, and ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007444/2/m_advancesadv2022007444f1.png?Expires=1766606662&Signature=idvoiR5W585XwN9Fpugj4N0mynQZYSazt-EOC7jo~EWtcTuzN1niuTA--p5RUoCT6L0xVWAa9nTGGbg6qHqQql6u4zHEQN7CE26Q90IKBY-Y67EJCDeDz2TaCLB7~zqDQsrx~22eMLE1~GfyBbf-AsZ7luu4ulJUUdaG04sQ9tRTCalai1SFFw~y3m3PVLKU9qgmYDT4~~~7P6Lc1~JFb16WBDMU84-TK1NdzeuAltRKSQvVwKLLWjzmR9bYMM2RGBb1Kv9u5qDe-S3AgOX4P8QmQpCRDOlXGhavCtZruwitOXYuIq7Xsmmy-6x2L~QM0pkQYVasxW42OmB~EVX98A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SARS-CoV-2 activates platelets in a plasma-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2, red) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med, blue) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 in the presence or absence of PFP (0.5%). Platelets were labeled and fixed after stimulation for 7.5, 15, 30, or 60 minutes, and platelet activation was determined by flow cytometry. N = 3. (B) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and injected into a microfluidics system. The images were taken after 15 minutes and show fluorescently-labeled platelets in the fibrinogen-coated channel. Representative images. N = 4. (C) Platelets were stimulated with C.Med or CoV2 in the presence of PFP (0.5%). N = 4. (D) Platelets were stimulated for 60 minutes with receptor-binding domain (RBD) peptide, S1 subunit of spike, full trimeric spike protein, or thrombin in the presence of platelet-free plasma (0.5%). N = 4. Data are represented as mean ± SD. Statistical analysis: (A, C, D) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. **P < .01, ***P < .001, and ****P < .0001. ns, not significant.
SARS-CoV-2 activates platelets in a plasma-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2, red) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med, blue) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 in the presence or absence of PFP (0.5%). Platelets were labeled and fixed after stimulation for 7.5, 15, 30, or 60 minutes, and platelet activation was determined by flow cytometry. N = 3. (B) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and injected into a microfluidics system. The images were taken after 15 minutes and show fluorescently-labeled platelets in the fibrinogen-coated channel. Representative images. N = 4. (C) Platelets were stimulated with C.Med or CoV2 in the presence of PFP (0.5%). N = 4. (D) Platelets were stimulated for 60 minutes with receptor-binding domain (RBD) peptide, S1 subunit of spike, full trimeric spike protein, or thrombin in the presence of platelet-free plasma (0.5%). N = 4. Data are represented as mean ± SD. Statistical analysis: (A, C, D) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. **P < .01, ***P < .001, and ****P < .0001. ns, not significant.
As αIIbβ3* expressed on platelets is a major receptor for platelet adhesion to fibrinogen, we tested the ability of platelets to adhere under physiological flow in a fibrinogen-coated microfluidics chamber. Visualization of fluorescently-labeled platelets revealed the formation of large clots induced by SARS-CoV-2 (1:25 vol/vol; 1.9E+05 virus per mL), but only when plasma was present. Clots did not form when platelets were in the presence of C.Med (1:25 vol/vol) and plasma (Figure 1B; supplemental Figure 3).
The response to increasing concentrations of SARS-CoV-2 (1:12.5 to 1:3200 vol/vol; 3.8E+05 to 1.5E+03 virus per mL) was determined in the presence of plasma to assess SARS-CoV-2 platelet activation. Strong platelet activation, based on all measured parameters, was observable with SARS-CoV-2 at 1:12.5 to 1:50 vol/vol (1.5E+03 to 9.5E+04 virus per mL) (Figure 1C). With the exception of CD41+ EVs, which were not significantly increased by SARS-CoV-2 at 1:200 vol/vol dilution (2.4E+04 virus per mL), this lower concentration sufficed to induce the surface expression of αIIbβ3* and CD62P (Figure 1C). In contrast, C.Med did not induce platelet activation within the tested concentration range (1:12.5 to 1:3200 vol/vol) (Figure 1C).
The possibility that the prominently expressed spike protein of SARS-CoV-2 may be able to induce plasma-dependent activation of platelets was examined next. Recombinant full-trimeric SARS-CoV-2 spike protein, the S1 subunit containing the receptor-binding domain (RBD), or a recombinant SARS-CoV-2 RBD peptide, were preincubated in plasma and then added to platelets to assess activation. No platelet activation was observed in response to any of the tested proteins (Figure 1D), which contrasted with the profound platelet activation induced by thrombin (1 U/mL) under the same conditions (Figure 1D). Thus, the combination of SARS-CoV-2 and plasma components activated platelets. This is likely independent of the spike as recombinant SARS-CoV-2 spike proteins failed to activate platelets.
SARS-CoV-2 activates platelets in a TF- and thrombin-dependent manner
We hypothesized that an active component associated with SARS-CoV-2 might be triggering the coagulation cascade. Thus, we incubated plasma deficient in individual coagulation factors (FXIII, FXII, FXI, FX, FIX, FVIII, FVII, FV, FII, FI, Protein C, Protein S, and von Willebrand factor [VWF])57 with SARS-CoV-2 or C.Med, and thereafter added platelets to assess activation (Figure 2A; supplemental Figure 4). Strong activation occurred despite the absence of FXIII, FXII, FXI, FIX, FVIII, FV, FI, Protein C, Protein S, or VWF in plasma (Figure 2A; supplemental Figure 4). However, in the absence of FX, FII, and FVII, SARS-CoV-2 failed to activate platelets, as activation remained at the background level and was not statistically different from that of the negative control (C.Med) (Figure 2A; supplemental Figure 4). These observations point to an involvement of the TF pathway of coagulation.
![SARS-CoV-2 activates platelets in a TF pathway and thrombin-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of platelet-free plasma (0.5%) deficient in individual coagulation factors. The heatmap indicates the mean platelet activation by CoV2 stimulation. The statistical significance symbols indicate comparisons of CoV2 with C.Med-stimulated platelets. N = 5. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and in the presence of d-phenylalanyl-prolyl-arginyl chloromethyl ketone (PPACK) (0/10 µM). CoV2 was preincubated for 15 minutes with PPACK before incubation with PFP. N = 4. (C) Platelets were preincubated for 15 minutes with vorapaxar (20 µM) and BMS-9861200 (20 µM), a PAR-1 and PAR-4 inhibitor, respectively, or DMSO (0.45%). Thereafter, platelets were stimulated with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%). N = 5. Data are represented as mean ± SD. Statistical analysis: (A) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. (B) Paired t-test. (C) Ordinary one-way ANOVA followed by Dunnett’s multiple comparison test. *P < .05, **P < .01, ***P < .001, and ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007444/2/m_advancesadv2022007444f2.png?Expires=1766606662&Signature=XUOisBX1ap40fhpQvuR3FWWkbBnR0Tv~GGpwVTlHLR7EYxZLWDN4oF1Xs1luJ1jd8IjE7PyB1o6wRiz3u83pKqnZiGuxNe0xGV7xAfRUwLiOwaGhBLF4yCsaQgalYKgz6aQ-kZNp-KfapysnQ4HfMZBBPQ3tpo0DCUaEwTJWk3fIDPG6aGe752XofjdU-t~b~v6XQPNmoxBY5lsIjqUzL1J~wRIIPuXEGB4x5Uvdppi6ecX9lSCgay44Z3O8MQC7YppXBZ4138xS9RkS9lX6lTvxAMkyNxHK1JgWmX~oTkSa6O7T3BXaFykLpaLxnz56ZLk-u5JO48ppZlh3YnNnuQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SARS-CoV-2 activates platelets in a TF pathway and thrombin-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of platelet-free plasma (0.5%) deficient in individual coagulation factors. The heatmap indicates the mean platelet activation by CoV2 stimulation. The statistical significance symbols indicate comparisons of CoV2 with C.Med-stimulated platelets. N = 5. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and in the presence of d-phenylalanyl-prolyl-arginyl chloromethyl ketone (PPACK) (0/10 µM). CoV2 was preincubated for 15 minutes with PPACK before incubation with PFP. N = 4. (C) Platelets were preincubated for 15 minutes with vorapaxar (20 µM) and BMS-9861200 (20 µM), a PAR-1 and PAR-4 inhibitor, respectively, or DMSO (0.45%). Thereafter, platelets were stimulated with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%). N = 5. Data are represented as mean ± SD. Statistical analysis: (A) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. (B) Paired t-test. (C) Ordinary one-way ANOVA followed by Dunnett’s multiple comparison test. *P < .05, **P < .01, ***P < .001, and ****P < .0001. ns, not significant.
SARS-CoV-2 activates platelets in a TF pathway and thrombin-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of platelet-free plasma (0.5%) deficient in individual coagulation factors. The heatmap indicates the mean platelet activation by CoV2 stimulation. The statistical significance symbols indicate comparisons of CoV2 with C.Med-stimulated platelets. N = 5. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and in the presence of d-phenylalanyl-prolyl-arginyl chloromethyl ketone (PPACK) (0/10 µM). CoV2 was preincubated for 15 minutes with PPACK before incubation with PFP. N = 4. (C) Platelets were preincubated for 15 minutes with vorapaxar (20 µM) and BMS-9861200 (20 µM), a PAR-1 and PAR-4 inhibitor, respectively, or DMSO (0.45%). Thereafter, platelets were stimulated with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%). N = 5. Data are represented as mean ± SD. Statistical analysis: (A) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. (B) Paired t-test. (C) Ordinary one-way ANOVA followed by Dunnett’s multiple comparison test. *P < .05, **P < .01, ***P < .001, and ****P < .0001. ns, not significant.
Coagulation factors in plasma are present as inactive zymogens, which may become catalytic enzymes when the coagulation cascade is initiated.38,57 The active form of FX is FXa, which is responsible for the conversion of prothrombin (FII) to thrombin. Thrombin mediates the formation of a fibrin clot and is a potent physiological stimulus of platelet activation.57 We hypothesized that thrombin is responsible for the plasma-dependent SARS-CoV-2–mediated platelet activation. To test this, we incubated plasma with SARS-CoV-2 in the presence or absence of the selective and irreversible thrombin inhibitor d-phenylalanyl-prolyl-arginyl chloromethyl ketone before the addition of platelets. The activation was completely blocked by thrombin inhibition (Figure 2B). We further confirmed that SARS-CoV-2 activation of naïve wild-type mouse platelets critically required plasma and thrombin activation (supplemental Figure 5). Murine ACE2 is not recognized by SARS-CoV-2 spike protein,58 and wild-type mouse platelets do not express FcyRIIA, thereby ruling out that platelet activation was ACE2- or Fc-receptor dependent.
Similar to the replication-incompetent fixed virus, infectious SARS-CoV-2 (1:4 or 1:25 vol/vol; 1.2E+06 or 1.9E+05 virus per mL) promptly activated platelets, but only in the presence of plasma (supplemental Figure 6). The activation was blocked by the thrombin inhibitor, demonstrating that the initiation of coagulation and generation of thrombin were not due to the virus fixation (supplemental Figure 6). Moreover, concentrations as high as 1:4/1:25 vol/vol of C.Med, β-propiolactone–treated or untreated, failed to activate platelets even after a 60-minute incubation (supplemental Figure 6). Thus, infectious and fixed SARS-CoV-2 activate platelets in a plasma- and thrombin-dependent manner.
The main cellular receptors for thrombin are PAR-1 and PAR-4, which are expressed by human platelets.59 Hence, we tested if the PAR blockers vorapaxar (PAR-1 blocker) and BMS-9861200 (PAR-4 blocker) could interfere with the activation. Individual receptor blockade by vorapaxar or BMS-9861200 failed to reduce platelet activation (Figure 2C). Treating platelets with both vorapaxar and BMS-9861200 before their addition to plasma-containing SARS-CoV-2 significantly reduced, but did not fully block, platelet activation, as seen by a lower frequency of αIIbβ3*CD62P-expressing platelets (46% [standard deviation (SD), 11.42] compared with 72.7% [SD, 15.73]), 2.3× lower expression of αIIbβ3* and 6.5× lower CD41+ EV release (Figure 2C). The data suggest both PAR receptors are involved. The incomplete blockade indicates that the used inhibitors failed at completely blocking PAR-1 and PAR-4 signaling or that other receptors on platelets, such as GP1bα (CD42b), may respond to thrombin in addition to PAR-1 and PAR-4.60,61
TF is implicated in SARS-CoV-2–mediated platelet activation
The role of factors FX, FII, and FVII pointed to the involvement of the upstream cofactor TF, the primary cellular initiator of coagulation.57,38 While CoV-2 might bear an intrinsic TF-like activity, the most probable explanation was the presence of actual TF associated with virus production. To assess the contribution of TF, the specific levels of TF activity in SARS-CoV-2 and C.Med were determined. To account for potential nonspecific detection of TF activity, a known confounder in these activity assays,62-64 a specific TF-blocking antibody (clone HTF-1) was included in the assay (supplemental Figure 7). While significant TF activity (8.62 pM [SD, 2.8]) was found in SARS-CoV-2 preparations, TF activity was undetectable in C.Med (Figure 3A; supplemental Figure 7), suggesting that (1) CoV-2 infection of cells induced active TF, and (2) SARS-CoV-2 is associated with active TF.
![SARS-CoV-2 activates platelets in a TF-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. TF activity was measured in an undiluted sample. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. Samples were preincubated for 15 minutes with control Ig or HTF-1. The numbers above bars indicate the estimated TF activity in pM. N = 4 (replicates measured in 4 separate assays). n.d. = not detected. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2 was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 6. (C) PF4 measurement by ELISA in platelet supernatant. N = 6. (D) 5-HT (serotonin) measurement by ELISA in platelet supernatant. N = 6. (E) Platelet stimulation with CoV2-Δ in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2-Δ was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 3. Data are represented as mean ± SD. Statistical analysis: (B-D) Wilcoxon test. (E) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. *P < .05 and **P < .01.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007444/2/m_advancesadv2022007444f3.png?Expires=1766606662&Signature=Tp5Mq6BKII-vFdtKhW3iAQQE6kw9syfX3gHzX8EABB8hJfvsvmVzYLrU2bM8QvvHyzeooeh6xvDF1cnTAoUa8giBcOoPbMEwlZ1YFjtxualiJbaPVtbzS5UgREL7AWJCNNnsGBYfaH9o-x3MuE8JGJfZvBJztdRdMW289FKaIcmO7KzbWYje~8KjFEfCIGhXzexpxYUWjiZppZFhULQc-E4xTjoYM9kmCE8MeOmQn~wroI9qGr3ABJzVINupJwpgXCmBpHB-fkZqe7216lxCzMxMcHSlDqP6COSsbOrCN6CoeFxWO0UwXly0Kjhzebv33l0GeF~C7KTipZkdCsh~HA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SARS-CoV-2 activates platelets in a TF-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. TF activity was measured in an undiluted sample. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. Samples were preincubated for 15 minutes with control Ig or HTF-1. The numbers above bars indicate the estimated TF activity in pM. N = 4 (replicates measured in 4 separate assays). n.d. = not detected. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2 was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 6. (C) PF4 measurement by ELISA in platelet supernatant. N = 6. (D) 5-HT (serotonin) measurement by ELISA in platelet supernatant. N = 6. (E) Platelet stimulation with CoV2-Δ in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2-Δ was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 3. Data are represented as mean ± SD. Statistical analysis: (B-D) Wilcoxon test. (E) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. *P < .05 and **P < .01.
SARS-CoV-2 activates platelets in a TF-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. TF activity was measured in an undiluted sample. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. Samples were preincubated for 15 minutes with control Ig or HTF-1. The numbers above bars indicate the estimated TF activity in pM. N = 4 (replicates measured in 4 separate assays). n.d. = not detected. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2 was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 6. (C) PF4 measurement by ELISA in platelet supernatant. N = 6. (D) 5-HT (serotonin) measurement by ELISA in platelet supernatant. N = 6. (E) Platelet stimulation with CoV2-Δ in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2-Δ was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 3. Data are represented as mean ± SD. Statistical analysis: (B-D) Wilcoxon test. (E) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. *P < .05 and **P < .01.
Blockade of TF using an anti-TF antibody completely prevented platelet activation (Figure 3B) and platelet degranulation measured by PF4 and serotonin release (Figure 3C-D). These findings were replicated with a major SARS-CoV-2 variant of concern, as SARS-CoV-2-Δ (1:8/1:25 vol/vol; 1.8E+04/0.57E+04 virus per mL) also induced platelet activation in the presence of plasma (0.5%) and was prevented by TF blockade (Figure 3E). These data indicate that the platelet activation by SARS-CoV-2 was mediated by TF-induced coagulation.
TF is usually indirectly detected by the assessment of its cofactor activity and ability to stimulate coagulation. Femtomolar amounts of TF activity suffice to accelerate blood coagulation37 and, therefore, promote platelet activation. Indeed, we found that stimulating platelets with plasma incubated with TF (lipoprotein) at concentrations as low as 18.8 fM led to increased expression of αIIbβ3* and CD62P (93.03% [SD, 1.7]), while 75 fM sufficed to induce the release of CD41+ EVs (90.7E+06 mL [SD, 29]) (supplemental Figure 8). This suggests that even extremely low concentrations of active TF in plasma are sufficient to trigger full activation of platelets.
SARS-CoV-2 and EVs, but not soluble proteins, are coisolated with TF activity
While electron microscopy (EM) has been successfully used to characterize SARS-CoV-2 viral architecture,65,66 the presence of EVs in virus preparations has been overlooked so far. Using cryo-EM, we confirm the presence of heterogeneous populations of EVs in releasates of SARS-CoV2–infected cells (Figure 4). Most EVs ranged in size from 80 nm to 500 nm and were either isolated or associated in small clusters. The presence of a population of EVs exposing the procoagulant lipid phosphatidylserine was demonstrated by specific binding of annexin-V conjugated with gold particles67 (Figure 4B). Phosphatidylserine-positive EVs constitute more than half of isolated EVs. In addition, a homogenous population of spherical particles was observed, characterized by their narrow size range (80-130 nm), their typical granular density, likely corresponding to the protein and genome package of SARS-CoV-2, and the presence of extended proteins protruding from the particle surface, most likely representing spike projections (Figure 4C). Thus, virus preparations contained both SARS-CoV-2 and EVs.

Cryo-electron microscopy of releasate from SARS-CoV-2–infected cells. (A) A set of EVs, ranging in size from 100 to 500 nm. Some EVs contain smaller EVs, as frequently observed in EV samples from various origins. (B) Two EVs side-by-side. One EV is specifically labeled with Anx5 gold particles, thus exposing the procoagulant lipid phosphatidylserine; the other one is unlabeled, thus does not expose phosphatidylserine. (C) A group of virus particles, characterized by their homogenous granularity and the presence of long protein protrusions (white arrows) corresponding most likely to spike proteins. (A-C) The white stars indicate thin threads of the carbon support film.
Cryo-electron microscopy of releasate from SARS-CoV-2–infected cells. (A) A set of EVs, ranging in size from 100 to 500 nm. Some EVs contain smaller EVs, as frequently observed in EV samples from various origins. (B) Two EVs side-by-side. One EV is specifically labeled with Anx5 gold particles, thus exposing the procoagulant lipid phosphatidylserine; the other one is unlabeled, thus does not expose phosphatidylserine. (C) A group of virus particles, characterized by their homogenous granularity and the presence of long protein protrusions (white arrows) corresponding most likely to spike proteins. (A-C) The white stars indicate thin threads of the carbon support film.
EVs and viruses share many biophysical features, and enveloped viruses may acquire host proteins during the budding process.46 ,, -51,53,54 TF is primarily expressed as a membrane-bound protein that may be associated with EVs, although a soluble isoform exists.38,39 As direct detection of TF at these concentrations is not technically achievable,62,68 whether TF activity specifically associated with EVs, viruses, or soluble proteins was investigated through fractionation by size-exclusion chromatography (SEC).
Concentrated SARS-CoV-2 in A549-hACE2 cell culture supernatant was subjected to SEC, and elution fractions were collected (Figure 5A). The majority of EVs (71.23%) were found in fractions 3 to 5, with fraction 4 (41.70%) containing most EVs, whereas soluble proteins (91.25%) were found in fractions 9 to 20 (Figure 5B). Consistent with quantifications of EVs, the highest levels of 18S rRNA (as a marker for host-derived EV content69 ) were found in fractions 3 to 5 (supplemental Figure 9). The highest concentration of particles expressing SARS-CoV-2 spike protein and SARS-CoV-2 RNA was found in fraction 4 (Figure 5C; supplemental Figure 10). SARS-CoV-2 RNA was particularly enriched compared with 18S rRNA in fraction 4 (threefold, ±SD 0.12), indicating a higher quantity of particles carrying viral content compared with host cell EVs. Thus, EVs and SARS-CoV-2 are indistinguishable using SEC.

TF activity is exclusively found in SARS-CoV-2 and EV-containing chromatography fractions. (A) β-propiolactone–inactivated SARS-CoV-2 obtained from human A549 (ACE2) cell infection was concentrated by Ultrafiltration (10 kDa filter columns) as size-exclusion chromatography dilutes samples up to 10 times. Celltracker-DR–positive (Cell+) particles (EVs/virus) and protein content before and after sample ultrafiltration were quantified by flow cytometry (left y-axis, 3 technical replicates) and BCA assay (right y-axis, 2 technical replicates), respectively. The concentrated sample was then loaded on an SEC column, and fractions of 0.5 mL after the void volume were collected (see cartoon created with Biorender). (B) Celltracker-DR–positive particles (EVs/virus) and protein content of each SEC fraction were quantified by FACS (left y-axis, 3 technical replicates) and BCA assay (right y-axis, 2 technical replicates), respectively. (C) Celltracker-DR–positive and spike-positive (samples labeled by mouse IgG1 anti-spike SARS-CoV-2, fluorescein-conjugated) particles (EV/virus) and SARS-CoV-2 RNA content (left or right y-axis, respectively) were quantified in each SEC fraction. (D) Left y-axis. TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. Samples were preincubated for 15 minutes with control Ig or HTF-1. The numbers above the bars indicate the estimated TF activity in pM. Measured in 3 technical replicates. (D) Right y-axis, wt C57BL/6J mouse platelet stimulation (n = 6) with SEC fractions (1:25 vol/vol) in the presence of pooled, recalcified (10 mM CaCl2) mouse PFP (0.5%). The amount of CaCl2 was twofold higher than in previous platelet stimulations as phosphate contained in SEC buffer (PBS) may chelate free calcium. Data are represented as mean ± SD. Statistical analysis: (D) one-way ANOVA followed by Dunnett’s multiple comparison test. ****P < .0001.
TF activity is exclusively found in SARS-CoV-2 and EV-containing chromatography fractions. (A) β-propiolactone–inactivated SARS-CoV-2 obtained from human A549 (ACE2) cell infection was concentrated by Ultrafiltration (10 kDa filter columns) as size-exclusion chromatography dilutes samples up to 10 times. Celltracker-DR–positive (Cell+) particles (EVs/virus) and protein content before and after sample ultrafiltration were quantified by flow cytometry (left y-axis, 3 technical replicates) and BCA assay (right y-axis, 2 technical replicates), respectively. The concentrated sample was then loaded on an SEC column, and fractions of 0.5 mL after the void volume were collected (see cartoon created with Biorender). (B) Celltracker-DR–positive particles (EVs/virus) and protein content of each SEC fraction were quantified by FACS (left y-axis, 3 technical replicates) and BCA assay (right y-axis, 2 technical replicates), respectively. (C) Celltracker-DR–positive and spike-positive (samples labeled by mouse IgG1 anti-spike SARS-CoV-2, fluorescein-conjugated) particles (EV/virus) and SARS-CoV-2 RNA content (left or right y-axis, respectively) were quantified in each SEC fraction. (D) Left y-axis. TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. Samples were preincubated for 15 minutes with control Ig or HTF-1. The numbers above the bars indicate the estimated TF activity in pM. Measured in 3 technical replicates. (D) Right y-axis, wt C57BL/6J mouse platelet stimulation (n = 6) with SEC fractions (1:25 vol/vol) in the presence of pooled, recalcified (10 mM CaCl2) mouse PFP (0.5%). The amount of CaCl2 was twofold higher than in previous platelet stimulations as phosphate contained in SEC buffer (PBS) may chelate free calcium. Data are represented as mean ± SD. Statistical analysis: (D) one-way ANOVA followed by Dunnett’s multiple comparison test. ****P < .0001.
Next, specific TF activity was uniquely detected in fractions 3 through 5 (Figure 5D; supplemental Figure 11), demonstrating that TF activity is coisolated with EVs and SARS-CoV-2. Accordingly, strong and significant platelet activation (αIIbβ3*CD62P expression) was induced by fraction 4 (1:25 vol/vol) (Figure 5D), thereby coinciding with the fraction containing the highest level of TF activity and both EVs and virus. In sum, EV fractions are enriched in SARS-CoV-2, and these fractions (and not soluble proteins) contain the TF activity responsible for platelet activation.
TF expression is induced during SARS-CoV-2 infection
As TF is the trigger for coagulation and platelet activation, we determined whether TF is associated with SARS-CoV-2 infection. The presence of RNA coding for TF (F3) was investigated in naïve and infected A549-hACE2 cells. Quantitative transcriptomic analyses revealed a 4.57-fold (SD, 2.5) increase in TF mRNA 36 hours postinfection, at a time of robust infection in the absence of apparent cell death and destruction, in comparison with uninfected cells (Figure 6A).
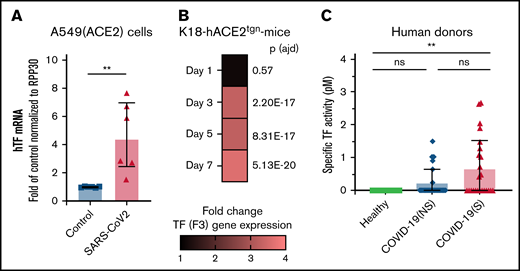
TF expression is associated with SARS-CoV-2 infection in vitro, in a murine infection model, and COVID-19 patients. TF expression (mRNA) or TF activity was assessed in vitro, in mice, and in plasma from human donors. (A) human TF (F3) mRNA expression quantified in SARS-CoV-2–infected A549 (ACE2) cells compared with uninfected (control) A549 (ACE2) cells. mRNA expression levels were normalized to RPP30. (B) Mouse TF (F3) gene expression determined by RNAseq in K18-ACE2tgn mouse lungs infected with SARS-CoV-2 on days 1, 3, 5, and 7 postinfection compared with uninfected K18-ACE2tgn mouse. To consider a differential gene expression statistically significant, thresholds of 0.01 and |log2(3)| were applied for the adjusted P value (P adj) and the minimum fold-change, respectively. N = 4 per group. (C) Human TF activity assessed in plasma from healthy donors (n = 15), COVID-19 patients with nonsevere (n = 25) or severe (n = 25) disease. TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. The numbers above the bars indicate the estimated TF activity in pM. Data are represented as mean ± SD. Statistical analysis: (A) Mann-Whitney test. (C) Kruskal-Wallis test followed by Dunn’s multiple comparison test. **P < .01. ns, not significant.
TF expression is associated with SARS-CoV-2 infection in vitro, in a murine infection model, and COVID-19 patients. TF expression (mRNA) or TF activity was assessed in vitro, in mice, and in plasma from human donors. (A) human TF (F3) mRNA expression quantified in SARS-CoV-2–infected A549 (ACE2) cells compared with uninfected (control) A549 (ACE2) cells. mRNA expression levels were normalized to RPP30. (B) Mouse TF (F3) gene expression determined by RNAseq in K18-ACE2tgn mouse lungs infected with SARS-CoV-2 on days 1, 3, 5, and 7 postinfection compared with uninfected K18-ACE2tgn mouse. To consider a differential gene expression statistically significant, thresholds of 0.01 and |log2(3)| were applied for the adjusted P value (P adj) and the minimum fold-change, respectively. N = 4 per group. (C) Human TF activity assessed in plasma from healthy donors (n = 15), COVID-19 patients with nonsevere (n = 25) or severe (n = 25) disease. TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. The numbers above the bars indicate the estimated TF activity in pM. Data are represented as mean ± SD. Statistical analysis: (A) Mann-Whitney test. (C) Kruskal-Wallis test followed by Dunn’s multiple comparison test. **P < .01. ns, not significant.
The kinetics of infection of K18-hACE2tgn mice by CoV-2 are well described.70-72 We thus infected these mice with SARS-CoV-2. Transcriptomic analysis of RNA isolated from lung tissues revealed that TF gene expression was upregulated starting on day 3 (3.27-fold; P adj = 2.20E-17) postinfection and remained elevated on day 5 (3.24-fold; P adj = 8.31E-17) and Day 7 (3.59-fold; P adj = 5.13E-20) postinfection relative to uninfected mice (Figure 6B).
The presence of specific TF activity was investigated in the blood of COVID-19 individuals with nonsevere disease (n = 25), severe disease (n = 25), and healthy donors (n = 15) (supplemental Table 2). TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. To specifically attribute activity to TF,62-64 plasma samples were preincubated with a TF-blocking antibody or a control antibody (supplemental Figure 12). Compared with healthy donors, TF activity was increased in both patient groups, although activity was only significantly higher in patients with severe disease (Figure 6C). These data indicate that SARS-CoV-2 infection induces TF expression and activity, which may be a mechanism for platelet activation by SARS-CoV-2.
Discussion
We found that supernatants from SARS-CoV-2−infected cells induced platelet activation, as seen by the expression of classical activation markers, degranulation, the release of EVs, and aggregation under flow conditions. This activation was not mediated by SARS-CoV-2 spike proteins and was critically dependent on coagulation factors provided by plasma. We identified thrombin as the ligand for platelet activation and TF activity associated with SARS-CoV-2 as the trigger. This suggests that platelet hyperactivation observed in COVID-19 may be mediated by TF activity induced by SARS-CoV-2 infection rather than direct SARS-CoV-2 platelet interaction. This finding may explain some of the reported coagulation abnormalities in COVID-19 and provide a rationale to specifically target TF, thrombin, and/or PARs when developing treatments for COVID-19. These findings are recapitulated in Figure 7.

Graphical summary of the described mechanism. (A) SARS-CoV-2–infected cells release virus and EVs with associated TF activity. (B) This initiates the TF pathway of coagulation in plasma, thereby converting prothrombin into thrombin. Thrombin signals to platelets via PAR-1 and PAR-4. (C) Platelet activation is characterized by the expression of activated integrin αIIbβ3 and P-selectin, degranulation, aggregation, and the release of EVs. The graphical summary was created with BioRender.com.
Graphical summary of the described mechanism. (A) SARS-CoV-2–infected cells release virus and EVs with associated TF activity. (B) This initiates the TF pathway of coagulation in plasma, thereby converting prothrombin into thrombin. Thrombin signals to platelets via PAR-1 and PAR-4. (C) Platelet activation is characterized by the expression of activated integrin αIIbβ3 and P-selectin, degranulation, aggregation, and the release of EVs. The graphical summary was created with BioRender.com.
SARS-CoV-2 RNA in association with platelets was reported in patients,16,17,23 and viremia in plasma has been detected in 27% of patients hospitalized with COVID-19.73 Increased viral load may be associated with disease severity.73 However, the SARS-CoV-2 viral RNA copy number in blood was in the range of 63 to 6310 copies per mL of blood.73 In contrast, the concentration of platelets is 1.50E+08 to 4.50E+08 per mL of blood in healthy adults. Therefore, the estimated virus-to-platelets ratio ranges from 1 virus per 23 800 (MOI, 4.2E-5) to 7.14E+06 (MOI, 1.4E-7) platelets per mL of blood in individuals with detectable viremia.73 We found that platelets were activated in the presence of SARS-CoV-2 in the range of up to 1 virus per 4200 platelets (23 805 PFU per 100E+06 platelets; MOI, 2.38E-4) and only in the presence of plasma. In contrast, lower concentrations of SARS-CoV-2 (similar to those reported in vivo73 ) were insufficient. Moreover, while other viruses, such as dengue virus74 and human immunodeficiency virus,75 infect,74,75 and replicate74 in platelets, only uptake and degradation by platelets have been demonstrated by electron microscopy for SARS-CoV-2.23 Even at high viral concentrations (1 virus per 10 platelets; MOI, 0.1), no platelet activation was observed in the absence of plasma.23 We cannot exclude that lung megakaryocytes, rather than platelets, may directly interact with SARS-CoV-2,22,76,77 but overall, these observations suggest that direct platelet–SARS-CoV-2 interactions in the blood may be rare and may not explain platelet hyperactivation in COVID-19.
Although more work is needed to address whether and how the mechanism revealed herein is clinically relevant in patients, we found TF activity associated with SARS-CoV-2 infection and that a TF-blocking antibody completely prevented plasma-dependent activation of platelets by SARS-CoV-2 in vitro. Active TF is absent from blood under normal conditions, and even small increases in TF activity have a marked effect.37,39 Several studies reported increased TF expression in COVID-19.41,43,78 -81 We estimated that the level of TF activity associated with SARS-CoV-2 that was required to induce platelet activation was as low as 43 fM (based on Figure 1C and Figure 3A), which was comparable to the activity of recombinant TF required for platelet stimulation (supplemental Figure 8). Guervilly and colleagues41 report that TF activity levels of 78.3 fM and higher in the circulation correlate with disease severity and mortality in COVID-19,41 indicating that such low levels of TF activity in the circulation may be clinically relevant. We also observed that TF expression can be induced in vitro and in vivo in a K18-ACE2tgn murine model of infection. In accordance with other reports, we observed that TF activity in plasma was increased in COVID-19, particularly in individuals with severe disease. TF-activity associated with SARS-CoV-2 infection may thereby present a mode of platelet activation even though the low concentrations of SARS-CoV-2 in the circulation make direct virus–platelet interactions unlikely.
TF is primarily expressed as a membrane-bound protein, although a soluble isoform exists.38,39 However, the soluble form of TF is less procoagulant,38 and efficient propagation of coagulation requires the presence of lipid membranes,38 particularly those exposing phosphatidylserine.38,39 Thus, high levels of TF activity may be associated with phosphatidylserine-exposing cells or EVs.38,39 Intriguingly, SARS-CoV-2 spike protein pseudotyped viral infection of human monocyte-derived macrophage cells can induce the release of EVs with associated TF activity that critically required the decryption (activation) of TF by sphingomyelinase.44,82 It is currently not known whether this novel mechanism of TF decryption is responsible for increased TF activity observed in the plasma of patients with COVID-19.82 While the mechanism of TF upregulation is unknown, it is interesting to consider that TF is structurally related to interferon receptors83 and increases in TF activity, or expression seems to be common in viral infections.84 Furthermore, through the budding process of enveloped viruses, host-derived membrane proteins may be acquired.46-51 Thus, other than in association with EVs, TF might be acquired during the maturation process of SARS-CoV-2. We characterized supernatants of infected cells by electron microscopy and found that these contained both SARS-CoV-2 virions and EVs, which could not be separated from each other by SEC. SEC, however, enabled us to establish the particulate nature of the TF activity. As direct detection of TF protein and separation of virions from EVs are not possible,53,54,62,68 it remains unknown whether TF protein was inherited by virions and/or EVs. However, as both enveloped viruses and EVs comprise host cell membranes, there is currently no rationale for excluding the possibility that TF may be expressed by both EVs and SARS-CoV-2.
In conclusion, we describe a potential mechanism for SARS-CoV-2–mediated platelet hyperactivation and highlight the importance of distinguishing host-derived from viral components when studying the biological activity of viruses. The study suggests further directions for the development of new therapeutic approaches for the treatment of COVID-19.
Acknowledgments
The authors are grateful to the blood donors who participated in this study. The authors thank the next-generation sequencing core facility of the Centre Hospitalier Universitaire de Quebec Research for its excellent service and expertise, the Laboratoire de Santé Publique du Québec, and the Public Health Agency of Canada’s National Microbiology Laboratory for providing the SARS-CoV-2 isolates used in this study. The authors also thank members of the Mammalian Cell Expression Section (NRC-HHT) for their precious help in producing and purifying the S proteins and the financial support from the NRC’s Pandemic Response Challenge Program, and Yannick Doyon (CHUL) for providing reagents and advice.
F.P. is the recipient of a postdoctoral fellowship from Fonds de Recherche du Québec en Santé (FRQS). E.B. is the recipient of the senior award from the FRQS. This work was supported by grants from the Canadian Institutes of Health Research (L.F. and E.B.). This study was supported in part by a COVID-19 Rapid Response grant by the Canadian Institutes of Health Research (CIHR; #OV1-170355) to M.-A.L. Y.Z. was supported by the Cheikh Zaid Foundation.
Authorship
Contribution: F.P., L.F., and E.B. conceived the concept and designed the study; F.P. designed experiments and assays; F.P., I.A., I.D., and E.L. participated in the study design, performed experiments, and were involved in the data extraction; A.R.B. performed electron microscopy analyses and interpretation of the data and contributed to redaction; I.D., E.L., Y.G., Y.Z., L.K., C.B., Y.D., M.-A.L., and L.F. contributed critical reagents, biospecimens, and instruments; A.S.W. and L.F. gave critical advice on experiments; F.P. and E.B. wrote the article; and all authors read and approved the final article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Eric Boilard, Centre de Recherche du Centre Hospitalier Universitaire de Québec, Faculté de Médecine de l'Université Laval and Centre de Recherche ARThrite, 2705 Laurier Blvd, Room T1-49, Quebec, QC G1V 4G2, Canada; e-mail: eric.boilard@crchudequebec.ulaval.ca; and Louis Flamand, Centre de Recherche du Centre Hospitalier Universitaire de Québec-Université Laval, 2705 Laurier Blvd, Room T1-49, Quebec, QC G1V 4G2, Canada; e-mail: louis.flamand@crchudequebec.ulaval.ca.


![SARS-CoV-2 activates platelets in a plasma-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2, red) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med, blue) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 in the presence or absence of PFP (0.5%). Platelets were labeled and fixed after stimulation for 7.5, 15, 30, or 60 minutes, and platelet activation was determined by flow cytometry. N = 3. (B) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and injected into a microfluidics system. The images were taken after 15 minutes and show fluorescently-labeled platelets in the fibrinogen-coated channel. Representative images. N = 4. (C) Platelets were stimulated with C.Med or CoV2 in the presence of PFP (0.5%). N = 4. (D) Platelets were stimulated for 60 minutes with receptor-binding domain (RBD) peptide, S1 subunit of spike, full trimeric spike protein, or thrombin in the presence of platelet-free plasma (0.5%). N = 4. Data are represented as mean ± SD. Statistical analysis: (A, C, D) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. **P < .01, ***P < .001, and ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007444/2/m_advancesadv2022007444f1.png?Expires=1766674042&Signature=HYC0WL~JoXNW8dR0EoD81OGW-3Cz~K45X05vMByFFHOevayyrPx1xTCbB8WtVpzBAYDrvrl2izqtER~lb9bc9tSs9Y6-4t7q88LnNRePAHX4owCHPVie60J4VB04HC25~ehTs7t-BoUFNWP7wFTvhDVKPGqcq0GIwJ7xX~OmP86j6fC3LAIvnIWbxH9rYGUK0A5H0sqtjSgTqD5uQ~xSEOVYalhvaJI5zs0jwo1z7XwXNzgYVhx-dC35JFqlC7DMatdVYHTyEx1jZAV7XbZP4f0qjjpfjs6ePyrovehWJHnemtsnQpF-~uAnWBvoqGEcQg9UuMhXMANXjBk2oyWoJg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![SARS-CoV-2 activates platelets in a TF pathway and thrombin-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) Platelets were stimulated with C.Med or CoV2 (1:25 vol/vol) in the presence of platelet-free plasma (0.5%) deficient in individual coagulation factors. The heatmap indicates the mean platelet activation by CoV2 stimulation. The statistical significance symbols indicate comparisons of CoV2 with C.Med-stimulated platelets. N = 5. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%) and in the presence of d-phenylalanyl-prolyl-arginyl chloromethyl ketone (PPACK) (0/10 µM). CoV2 was preincubated for 15 minutes with PPACK before incubation with PFP. N = 4. (C) Platelets were preincubated for 15 minutes with vorapaxar (20 µM) and BMS-9861200 (20 µM), a PAR-1 and PAR-4 inhibitor, respectively, or DMSO (0.45%). Thereafter, platelets were stimulated with CoV2 (1:25 vol/vol) in the presence of PFP (0.5%). N = 5. Data are represented as mean ± SD. Statistical analysis: (A) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. (B) Paired t-test. (C) Ordinary one-way ANOVA followed by Dunnett’s multiple comparison test. *P < .05, **P < .01, ***P < .001, and ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007444/2/m_advancesadv2022007444f2.png?Expires=1766674042&Signature=DE8gTNN12aETckiwnwaU4zgGJzXqq0Iw~2dpAS44YYcIODiB7xQVM4N9sfZtITtpA5jA1KYhcC5uxvlGyV7FY3LWfPMKWoJssuLJBN54kkc0aEm24te8gqHAcNnl85einoWZXIxN2Sl3ifGp8cGhN~z3LJgUHpT19iBdWP4OD6N15lc907XF-itWYq9na~KcqeXHdB5q7MoKLycMx96Toq5JX21~qv6WOiimp3oSx0pn6xVocGblmAItzwIsokyr1oZvzcnnJe7ziDVZVeokNYtH64aaiAciVPL-vjV2XQXckuOPUomQYRc~ZFuNulLgoqpELyDEwEQhmjLjCdTvYQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![SARS-CoV-2 activates platelets in a TF-dependent manner. Platelet responses to β-propiolactone–inactivated SARS-CoV-2 (CoV2) obtained from human A549 (ACE2) cell infection or conditioned media (C.Med) solution (A549 [ACE2] cell culture supernatant obtained after 4 days of culture and treatment with β-propiolactone [1:1000 vol/vol]). (A) TF activity was assessed by TF/FVII-mediated conversion of FX to FXa and subsequent conversion of FXa substrate. TF activity was measured in an undiluted sample. The specific TF activity indicated is calculated as total TF activity measured in the presence of control Ig minus the total TF activity measured in the presence of anti-TF antibody HTF-1. Samples were preincubated for 15 minutes with control Ig or HTF-1. The numbers above bars indicate the estimated TF activity in pM. N = 4 (replicates measured in 4 separate assays). n.d. = not detected. (B) Platelet stimulation with CoV2 (1:25 vol/vol) in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2 was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 6. (C) PF4 measurement by ELISA in platelet supernatant. N = 6. (D) 5-HT (serotonin) measurement by ELISA in platelet supernatant. N = 6. (E) Platelet stimulation with CoV2-Δ in the presence of control Ig (control Ig, 10 µg/mL) or anti-TF antibody HTF-1 (10 µg/mL) and in the presence of PFP (0.5%). CoV2-Δ was preincubated for 15 minutes with control Ig or HTF-1 before incubation with PFP. N = 3. Data are represented as mean ± SD. Statistical analysis: (B-D) Wilcoxon test. (E) Ordinary two-way ANOVA followed by Sidak’s multiple comparison test. *P < .05 and **P < .01.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007444/2/m_advancesadv2022007444f3.png?Expires=1766674042&Signature=cQLsTBDFpRC0pQgh33TuRPmaTx~IkuJ74MY2tONGplae0ELAr5ViI8uUvLSBmtFFjvKTp8gVUP9yRDw~VArx2IRiLL~89Xa2IAukcQ2~PROuqU6~U3OuACwn7GSeX2sGH3D7YUzxdc32YQrV0uvjisXIGwGl~y3IlG8w2f-QrmoMzU0HdDLnuAzkyTdg2GLwoF-0g25v7NBZ2WYPsY5dsQQu~zliCFISJCSFCAE6hqJ5tUFQQlfVabU4bT~yKJk0ATxBD4h4jTTu2-cHpr6bZaFeKU3xkFZi9IiqBZ8cgKeoJcUwyJGQ8SArfie1iJnSIa~~X2uzgDQtihIHiguu2Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)





