Key Points
Elevated IRF8 in granulocyte-monocyte progenitors from Gata2 −77 enhancer mutant embryos cause macrophage-biased differentiation.
Genetically ablating Irf8 reverses fetal liver hematopoiesis defects of Gata2 enhancer mutant embryos, thus establishing a GATA2-IRF8 axis.

Abstract
Cell type-specific transcription factors control stem and progenitor cell transitions by establishing networks containing hundreds of genes and proteins. Network complexity renders it challenging to discover essential versus modulatory or redundant components. This scenario is exemplified by GATA2 regulation of hematopoiesis during embryogenesis. Loss of a far upstream Gata2 enhancer (−77) disrupts the GATA2-dependent transcriptome governing hematopoietic progenitor cell differentiation. The aberrant transcriptome includes the transcription factor interferon regulatory factor 8 (IRF8) and a host of innate immune regulators. Mutant progenitors lose the capacity to balance production of diverse hematopoietic progeny. To elucidate mechanisms, we asked if IRF8 is essential, contributory, or not required. Reducing Irf8, in the context of the −77 mutant allele, reversed granulocytic deficiencies and the excessive accumulation of dendritic cell committed progenitors. Despite many dysregulated components that control vital transcriptional, signaling, and immune processes, the aberrant elevation of a single transcription factor deconstructed the differentiation program.
Introduction
The cloning of sequence-specific DNA binding proteins unveiled master regulators including GATA1, which promotes erythrocyte, megakaryocyte, mast cell, and basophil development, and GATA2, which mediates hematopoietic stem and progenitor cell (HSPC) genesis/function.1-4 During mouse embryogenesis, GATA2 ablation disrupts multilineage hematopoiesis, yielding lethality.3,5 Human GATA2 heterozygous mutations cause GATA2 deficiency syndrome involving cytopenias, immunodeficiency, bone marrow failure, leukemia, and lymphedema.6-10 These mutations alter the GATA2 coding region or “+9.5” intronic enhancer.11-13 Another pathogenic enhancer resides upstream of GATA2 (77 kb in mice; 110 kb in humans).14,15 A 3q21;q26 inversion relocates this enhancer from 1 allele to MECOM1, inducing EVI1 expression and acute myeloid leukemia.16-19 Homozygous deletion of the −77 enhancer reduces Gata2 messenger RNA (mRNA) levels by nearly fivefold in Lin−Sca1−Kit+ progenitors, with reductions occurring in both common myeloid progenitors (CMPs) and granulocyte-monocyte progenitors (GMPs).14 Though hematopoietic stem cell (HSC) production in the aorta-gonad mesonephros of −77−/− embryos is largely unaffected, the mutation depletes megakaryocyte-erythroid progenitors (MEPs), yielding embryonic lethality after embryonic day 14.5 (E14.5).14,20 In addition, −77−/− myelo-erythroid progenitors lose multilineage differentiation and generate excessive macrophages ex vivo.14 Although monocytopenia is common in GATA2 deficiency syndrome that emerges in children or adults, bone marrow macrophages persist.21
Many GATA2-regulated genes have been described in diverse systems.1,20-24 However, the proteins operating downstream of GATA2 that mediate vital GATA2 functions, epistatic relationships governing HSPC functions, and disease phenotypes are incompletely defined. Discovering functionally critical downstream factors and networks has been challenging because of context-dependent permutations of GATA2 mechanisms.1,12,13 In addition, once GATA2-mutant HSCs populate the bone marrow, and bone marrow failure is triggered via ill-defined mechanisms, the pleiotropic and often vast changes complicate mechanistic analyses and interpretations. To circumvent these complexities, we have focused on elucidating hematopoietic mechanisms operational during embryonic development that are sensitive to alterations in GATA2 levels. Furthermore, in the context of bone marrow failure and leukemia predisposition syndromes, it is instructive to consider how variably penetrant genes that are pathogenic in children and adults dysregulate molecular/cellular processes during embryogenesis. Proteomics and single-cell transcriptomics with −77−/− fetal liver progenitors revealed losses of proteins mediating erythroid, megakaryocyte, basophil and granulocyte differentiation, and elevated innate immune proteins, including interferon responsive factor 8 (IRF8).21
IRF8 interacts with other IRFs, PU.1, AP-1, and BatF3 at composite binding sites to control immune cell development, and IRF8 levels dictate target gene selection by regulating transcription factor complex composition.25 Balanced production of monocytes and granulocytes is regulated by IRF8 in lineage-restricted progenitors. GMPs (Lin−Sca-1−cKit+CD34+CD16/32high) are a mix of multipotent and lineage-restricted myeloid progenitors that develop from CMPs (Lin−Sca-1−cKit+CD34+ CD16/32low), which exhibit a broader lineage potential, including erythroid and dendritic cells (DCs). Differentiation of multipotent Ly6C− GMPs into lineage-restricted Ly6C+ progenitors involves upregulation of macrophage colony-stimulating factor receptor (M-CSFR). M-CSFR expression is high in monocyte progenitors (MPs) and low in granulocyte progenitors (GPs).26 GPs have been further characterized as CD81-expressing, committed neutrophil progenitors.27 Bone marrow MPs and GPs are produced in the absence of IRF8, although high IRF8 levels in MPs are required for further monocytic differentiation.26 IRF8 levels also direct differentiation of DC progenitors within the CMP population. In the absence of IRF8, monocyte-DC progenitors (MDPs) fail to differentiate into common DC progenitors (CDPs).28 Production of dendritic cell subtypes from CDPs is also regulated by the level of IRF8. The generation of type 1 conventional DCs (cDC1) requires more IRF8 than cDC2.25,29 IRF8 levels increase during the commitment of monocytic and DC progenitors, and GATA2 and IRF8 are inversely expressed at the single-cell level.21,30
Because IRF8 promotes monocyte development, and its loss favors neutrophil generation,28,31 it is attractive to propose that IRF8 upregulation in −77−/− progenitors unrestrains monocytic differentiation and GATA2 downregulation of IRF8 is critical for multilineage differentiation. Because expression correlations often do not reflect causation, and IRF8 function in GATA2 networks is not understood, IRF8 might be functionally nonredundant, redundant, or insignificant. We asked if Irf8 loss, in the context of the −77−/− allele, rescues the diverse differentiation potential of progenitors. We demonstrate that simultaneous ablation of the −77 enhancer and Irf8 reversed the loss of GPs and promoted granulocytic differentiation of Ly6C− GMPs. Irf8 ablation also reversed the elevated CDP production of −77 mutants and blocked maturation of CMP-derived DCs. Because ectopically high IRF8 resulting from GATA2 deficiency in embryonic progenitor cells disrupted hematopoiesis, this study illustrates how dysregulation of a single component among many dysregulated components of a transcriptome derails a multilineage differentiation process.
Methods
Experimental model
Gata2 −77+/− mice14 and Irf8−/− strain B6(Cg)-Irf8tm1.2Hm/J (Jackson Labs, Bar Harbor, ME) were bred to generate −77+/−;Irf8+/− males and females for timed matings. Animal protocols were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care International regulations.
Flow cytometry
E14.5 fetal liver hematopoietic progenitors were quantified using the LSR Fortessa (BD Biosciences) and Attune (Thermo Fisher Scientific) flow cytometers or sorted using a FACSAria (BD Biosciences). Antibodies were from BioLegend unless indicated. Fluorescein isothiocyanate-conjugated B220 (#103206), TER-119 (#116206), CD3 (#100306), CD5 (#11-0193-85; Thermo Fisher Scientific), CD11b (#101206), CD11c (#117306), CD49b (#108906), Ly6G (#127606), and Sca1 (#108106) antibodies were used for lineage exclusion. Other antibodies were Blue Violet (BV) 605-conjugated CD16/CD32 (#563006; BD Biosciences), BV711-conjugated Ly6C (#128017), phycoerythrin-conjugated CD115 (#135506), eFluor 660–conjugated CD34 (#50-0341; Thermo Fisher Scientific), peridinin chlorophyll-efluor710–conjugated CD135 (#46-1351; Thermo Fisher Scientific), and phycoerythrin-Cy7–conjugated cKit (#105814). Markers for DC analysis were Super Bright 780-conjugated CD11c (#78-0114; Thermo Fisher Scientific) and BV605-conjugated CD86 (105037). Stained cells were washed with phosphate-buffered saline, 2% fetal bovine serum, 10 mM glucose, and 2.5 mM EDTA. Cells were resuspended in the same buffer containing 100 ng/mL 4′,6-diamidino-2-phenylindole for live-dead discrimination.
Quantitative real-time RT-PCR
Total RNA was purified with TRIzol (Invitrogen) and treated with DNAse (Invitrogen). Complementary DNA was prepared with a mixture of random hexamer and oligo (dT) primers incubated with m-MLV Reverse Transcriptase (Invitrogen). Complementary DNA was analyzed by real-time reverse transcriptase polymerase chain reaction (RT-PCR) with a Viia7 real-time RT-PCR cycler (Applied Biosystems). Irf8 primers (GGCAAGCAGGATTACAATCAG and CCACACTCCATCTCAGGAAC) detected exon loss in Irf8−/− embryos. Gata2 primers (GCAGAGAAGCAAGGCTCGC and CAGTTGACACACTCCCGGC) were described previously.14 Values were normalized to the standard curve and 18S control (CGCCGCTAGAGGTGAAATTCT and CGAACCTCCGACTTTCGTTCT).
Cell culture
All primary cell cultures were incubated at 37°C and 5% CO2. Colony assays were performed using M3434 methylcellulose media (StemCell Technologies). Sorted Ly6C− GMPs were plated in duplicate at 1200 cells/plate and cultured for 7 days. Using a blinded strategy, colonies were visually scored as having small, large, or mixed cell content; granulocyte colony-forming unit (CFU-G), macrophase CFU (CFU-M), and granulocyte and macrophage CFU (CFU-GM), respectively. For DC differentiation, sorted fetal liver CMPs (Lin−Sca-1−cKit+FcγRloCD34+) were cultured using the CellXVivo Mouse Dendritic Cell Differentiation Kit (#CDK008; R&D Systems). Cultures of 35 000 to 75 000 CMPs were initiated on the day of sorting in 1 mL of media containing interleukin-4 (IL-4) and granulocyte-macrophage colony stimulating factor (GM-CSF) according to the manufacturer’s protocol. Fresh media was supplied on day 4. On day 6, the media was replaced with fresh media containing IL-4, GM-CSF, and tumor necrosis factor α (TNF-α). Following 24-hour culture in TNF-α-containing media, cells were recovered and analyzed by flow. DC differentiation was assessed using CD11c and CD86 antibodies.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism, version 9.1. Multiple independent cohorts were used in each experiment. Data were evaluated for outliers before analysis with 2-sided Grubbs test. Statistical comparisons were performed using Browne-Forsythe and Welch analysis of variance tests with Welch’s unequal variance t tests. A significance cutoff of P < .05 was used for all analyses. The results are presented as mean ± standard error of the mean.
Results
IRF8 mediates myeloid phenotypes in GATA2-deficient mouse embryos
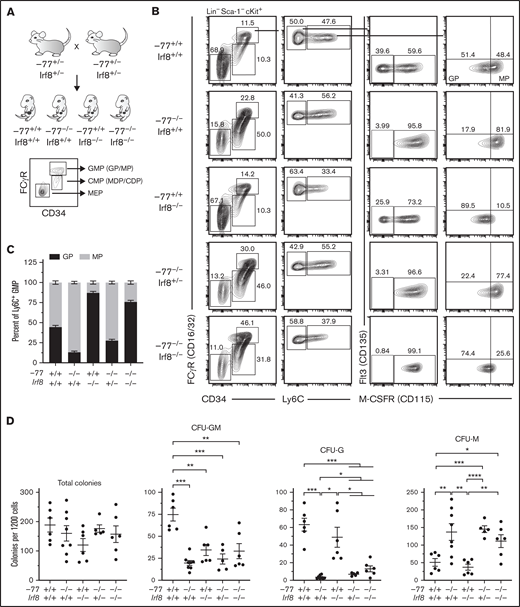
Previously, we demonstrated that the GP:MP ratio of −77−/− fetal liver is skewed toward MPs, and −77−/− progenitors generate excessive macrophages ex vivo.14,21 IRF8, which is upregulated in GATA2-deficient −77−/− progenitors,21 controls survival and differentiation, but not production, of these lineage-committed bone marrow progenitors.32 To determine if elevated IRF8 resulting from −77 enhancer loss causes the GP:MP imbalance, GPs and MPs, and the populations from which they arise (supplemental Figure 1), were analyzed in −77 and Irf8 double-mutant (−77−/−;Irf8−/−) embryos (Figure 1A; supplemental Figure 2A). Bone marrow GPs and MPs comprise the Ly6C+ subset of GMPs and arise from multipotential (monocyte, neutrophil, eosinophil, basophil) Ly6C− GMPs that have little cell surface expression of M-CSFR (CD115) and produce predominately CFU-GM colonies in vitro.26,27 In wild-type fetal liver, only 43% of Ly6C− GMPs were M-CSFR− by flow cytometry; the remaining cells expressed low to moderate levels of M-CSFR (Figure 1B; supplemental Figure 2B). Colony assays performed with wild-type Ly6C− GMPs yielded 42% CFU-GM, comparable to the proportion of Ly6C− GMPs that are M-CSFR–deficient (supplemental Figure 2C, first stacked graph). The remaining colonies comprised solely granulocytes (38%) or macrophage (20%). The progression of GMPs from Ly6C− to Ly6C+ is accompanied by increased surface expression of M-CSFR. M-CSFR− cells are absent, and there are similar proportions of low- and high-receptor-expressing GPs (44%) and MPs (56%), respectively (Figure 1B-C); comparable to what was reported in bone marrow.26

Elevated IRF8 causes aberrant myeloid differentiation potential of GATA2-deficient progenitor cells. (A) Experimental strategy for double mutant in vivo rescue system. Mice heterozygous for −77 enhancer deletion or Irf8tm1.2Hm were bred to generate E14.5 embryos. Fetal liver progenitor populations with select genotypes were analyzed by flow cytometry. (B) Representative flow cytometry analysis of progenitor populations from GMPs (Lin−Sca-1−cKit+CD34+CD16/32high). GPs and MPs are distinguished from multipotential GMPs by expression of Ly6C and from each other by M-CSFR (CD115). (C) Quantitation of the frequency of MPs and GPs within the Ly6C+ GMP populations from 7 litters (−77+/+Irf8+/+, n = 7; −77−/−Irf8+/+, n = 5; −77+/+Irf8−/−, n = 3: −77−/−Irf8+/−, n = 8; −77−/−Irf8−/−, n = 7). Relevant statistical comparisons for panel C are described in "Results." (D) Quantitation of colonies from sorted Ly6C− GMPs plated in M3434 methylcellulose media (STEMCELL Technologies) at 1200 cells/plate. Colonies were scored as mixed-lineage CFU-GM, or single-lineage granulocyte CFU-G, or CFU-M colonies. Ly6C− GMPs were obtained from 4 litters (−77+/+Irf8+/+, n = 6; −77−/−Irf8+/+, n = 8; −77+/+Irf8−/−, n = 6; −77−/−Irf8+/−, n = 5; −77−/−Irf8−/−, n = 6). Error bars for all plots represent mean ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001, ****P < .0001; Welch’s unequal variance t tests.
Elevated IRF8 causes aberrant myeloid differentiation potential of GATA2-deficient progenitor cells. (A) Experimental strategy for double mutant in vivo rescue system. Mice heterozygous for −77 enhancer deletion or Irf8tm1.2Hm were bred to generate E14.5 embryos. Fetal liver progenitor populations with select genotypes were analyzed by flow cytometry. (B) Representative flow cytometry analysis of progenitor populations from GMPs (Lin−Sca-1−cKit+CD34+CD16/32high). GPs and MPs are distinguished from multipotential GMPs by expression of Ly6C and from each other by M-CSFR (CD115). (C) Quantitation of the frequency of MPs and GPs within the Ly6C+ GMP populations from 7 litters (−77+/+Irf8+/+, n = 7; −77−/−Irf8+/+, n = 5; −77+/+Irf8−/−, n = 3: −77−/−Irf8+/−, n = 8; −77−/−Irf8−/−, n = 7). Relevant statistical comparisons for panel C are described in "Results." (D) Quantitation of colonies from sorted Ly6C− GMPs plated in M3434 methylcellulose media (STEMCELL Technologies) at 1200 cells/plate. Colonies were scored as mixed-lineage CFU-GM, or single-lineage granulocyte CFU-G, or CFU-M colonies. Ly6C− GMPs were obtained from 4 litters (−77+/+Irf8+/+, n = 6; −77−/−Irf8+/+, n = 8; −77+/+Irf8−/−, n = 6; −77−/−Irf8+/−, n = 5; −77−/−Irf8−/−, n = 6). Error bars for all plots represent mean ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001, ****P < .0001; Welch’s unequal variance t tests.
Unlike wild-type MPs, −77 enhancer deletion mutant (−77−/−;Irf8+/+) MPs were proportionally more abundant than GPs comprising 87% (P < .0001) of the Ly6C+ population (Figure 1B-C), corroborating our prior findings.21 In addition, M-CSFR-deficient Ly6C−- GMPs were nearly absent from −77−/−;Irf8+/+ fetal liver (7.1-fold lower than wild-type; P < .0001) (Figure 1B; supplemental Figure 2B), and these cells exhibited reduced CFU-GM and CFU-G colony formation relative to wild-type (Figure 1D; supplemental Figure 2C). Unlike −77−/−;Irf8+/+ Ly6C− GMPs, 77+/+;Irf8−/− Ly6C− GMPs retain M-CSFR− and M-CSFR+ populations (Figure 1B). However, M-CSFR levels were reduced in −77+/+;Irf8−/− Ly6C+ GMPs, thereby increasing the proportion of immunophenotypic GPs to 87% (P < .0001) relative to wild-type (Figure 1B-C). Because −77 and Irf8 mutations had opposing effects on the GP:MP ratio, and −77−/− progenitors exhibited elevated levels of Irf8, we tested whether reducing Irf8 expression rescues the −77 mutation-induced defects in GMP populations. Like the −77−/−;Irf8+/+ cells, Ly6C− GMPs from −77−/−;Irf8+/− or −77−/−;Irf8−/− fetal livers predominantly expressed M-CSFR and, in colony assays, CFU-GMs were not rescued (Figure 1B,D; supplemental Figure 2C). However, reducing Irf8 (−77−/−;Irf8+/− or −77−/−;Irf8−/−) increased the proportion of immunophenotypic GPs to levels intermediate between either single mutant (Figure 1B-C).
To assess the differentiation potential of −77 and Irf8 single- and double-mutant progenitors and determine if lowering IRF8 rescues the granulocyte deficiency of −77 mutants, Ly6C− GMPs were scored for colony formation. Though similar numbers of colonies were formed regardless of genotype (Figure 1D), fewer mixed lineage CFU-GMs were produced by −77 (3.8-fold) and Irf8 (2.2-fold) single mutants and CFU-GM were not rescued in −77−/−;Irf8−/− cultures. Consistent with our prior results,14 fewer CFU-G and more CFU-M were produced by −77−/−;Irf8+/+ compared with wild-type. CFU-G and CFU-M from −77+/+;Irf8−/− Ly6C− GMPs were not significantly different than wild-type. Lowering IRF8 levels in −77−/− progenitors increased CFU-G colonies by 2.0-fold (P = .011) and 3.7-fold (P = .036) in cultures of −77−/−;Irf8+/− and −77−/−;Irf8−/− Ly6C− GMPs, respectively, compared with −77−/−;Irf8+/+. This demonstrates that elevated IRF8 levels in −77−/− progenitors causes the loss of granulocytic potential of GMPs, and reducing IRF8 can partially restore granulocytic differentiation of −77−/− GMPs.
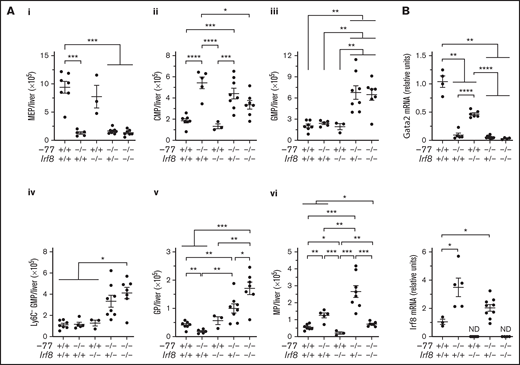
The −77−/− embryos have smaller livers because of loss of Ter119+ erythroid cells and erythroid progenitors, resulting in proportionally higher numbers of CMPs and GMPs.14 To determine if the −77 mutation, with or without loss of IRF8, perturbs absolute cell numbers of these progenitor populations, progenitors were quantified in E14.5 livers. −77−/−Irf8+/+ embryos exhibited a 7.2-fold (P = .0002) reduction in MEPs, and Irf8 loss did not rescue MEPs in −77−/−;Irf8+/− or −77−/−;Irf8−/− fetal livers (Figure 2Ai). Relative to wild-type, −77−/−;Irf8+/+ CMPs increased by 2.9-fold, whereas −77+/+;Irf8−/− CMP levels were unaffected (Figure 2Aii). Elevated CMP levels were retained by −77−/−;Irf8+/− embryos, whereas −77−/−;Irf8−/− CMPs decreased by 38% relative to the −77 single mutant. GMP levels in −77 and Irf8 single-mutant embryos were unchanged relative to wild-type (Figure 2Aiii). However, loss of 1 or both alleles of wild-type Irf8 increased GMPs in −77 mutants by 2.8-fold and 2.7-fold, respectively. Within the GMP compartment, −77−/−;Irf8+/+ Ly6C- GMPs, were unchanged (Figure 2Aiv), despite the loss of M-CSFR− cells (Figure 1B). Of the −77−/−;Irf8+/+ Ly6C+ GMPs, a 2.2-fold (P = .003) decrease in GPs was balanced by a 2.3-fold increase (P = .006) in MPs, thereby retaining normal numbers of total GMPs in −77 mutants. Compared with −77 single-mutant GMPs, −77−/−;Irf8−/− GMPs were increased for Ly6C− cells (3.5-fold) (Figure 2Aiv) and GPs (9.3-fold) (Figure 2Av) and reduced in MPs (59%) (Figure 2Avi). By contrast, −77−/−;Irf8+/− GMPs exhibited elevated GPs (5.4-fold) and MPs (2.1-fold). These results demonstrate that high levels of IRF8 dictate the fate of fetal liver progenitors in −77 mutants.
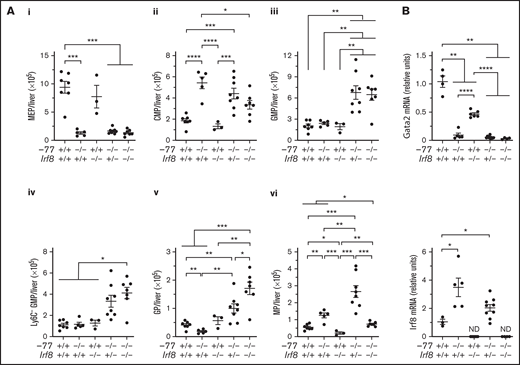
GATA2-IRF8 axis differentially regulates fetal liver progenitor populations. (A) Quantitation of progenitor populations in E14.5 fetal livers obtained from 7 litters (−77+/+Irf8+/+, n = 7; −77−/−Irf8+/+, n = 5; −77+/+Irf8−/−, n = 3; −77−/−Irf8+/−, n = 8; −77−/−Irf8−/−, n = 7). (B) mRNA quantitation from sorted Ly6C− GMPs from 4 to 9 embryos obtained from 10 litters. Real-time PCR primers for quantitation of Irf8 transcripts selectively amplified the wild-type, but not mutant, allele. ND, not detected. Error bars for all plots represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001; Welch’s unequal variance t tests.
GATA2-IRF8 axis differentially regulates fetal liver progenitor populations. (A) Quantitation of progenitor populations in E14.5 fetal livers obtained from 7 litters (−77+/+Irf8+/+, n = 7; −77−/−Irf8+/+, n = 5; −77+/+Irf8−/−, n = 3; −77−/−Irf8+/−, n = 8; −77−/−Irf8−/−, n = 7). (B) mRNA quantitation from sorted Ly6C− GMPs from 4 to 9 embryos obtained from 10 litters. Real-time PCR primers for quantitation of Irf8 transcripts selectively amplified the wild-type, but not mutant, allele. ND, not detected. Error bars for all plots represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001; Welch’s unequal variance t tests.
The −77 enhancer deletion reduces Gata2 expression in CMPs and GMPs and elevates Irf8 levels in a CMP/GMP pool.14,21 We analyzed the relationship between Gata2 and Irf8 expression in Ly6C− GMPs. Gata2 levels were lower in −77−/− single (11-fold) and −77−/−;Irf8−/− double (32-fold) mutant Ly6C− GMPs relative to wild-type (Figure 2B). Gata2 levels were also decreased in Irf8−/− Ly6C− GMPs (2.2-fold). Regardless of genotype, Gata2 expression was reduced in GPs and MPs relative to Ly6C− GMPs; with 33-fold and 46-fold lower expression in wild-type cells, respectively (supplemental Figure 3). As described previously,26,33 Irf8 expression was very low in wild-type Ly6C− GMPs and higher in MPs than GPs (Figure 2B; supplemental Figure 3). However, −77 deletion increased Irf8 expression in Ly6C− GMPs (3.3-fold) and GPs (2.7-fold) but not MPs (Figure 2B; supplemental Figure 3). Because the primers used to measure Irf8 mRNA annealed within the region deleted in the B6(Cg)-Irf8tm1.2Hm/J strain used in our analysis, no Irf8 expression was detected in Irf8−/− embryos. Quantitation of progenitor populations revealed that the proportions of MPs and GPs remain responsive to IRF8 in −77 mutant embryos, and the high-level Irf8 expression, in both multipotential and lineage-restricted −77−/− progenitors, suppresses the expansion of both populations.
IRF8 elevation in GATA2-deficient embryos disrupts mechanisms that restrict production and differentiation of dendritic progenitor cells
In GATA2 deficiency syndrome, DCs are commonly depleted from bone marrow and peripheral blood of children and adults.6,34 As with monocytes, IRF8 promotes DC generation.32,35 IRF8 levels within CDPs determine the DC subtype produced; high and low IRF8 promote cDC1 and cDC2 production, respectively.25 Irf8−/− mice exhibit increased bone marrow MDPs and reduced CDPs.32
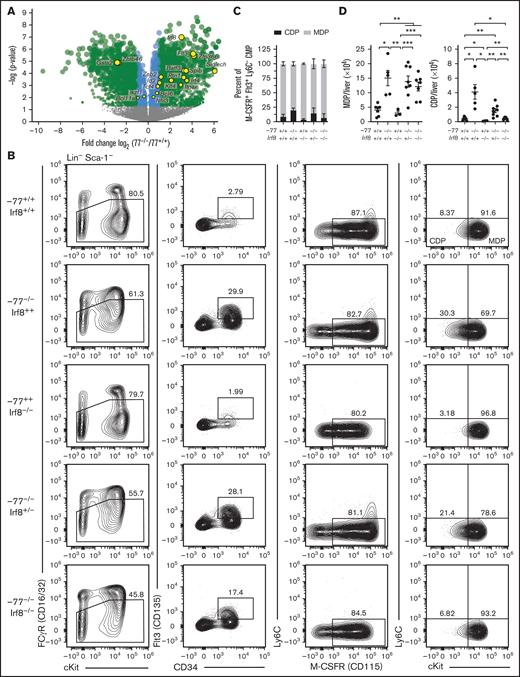
Our prior transcriptomic analyses that identified Irf8 upregulation in −77−/− myeloid progenitors21 also revealed upregulation of additional regulators of DC differentiation (eg, Flt3, Irf4, Batf3, Id2, Zeb2, Zfp366 [DC-Script], and Spib) and/or transcripts enriched in DC progenitor or precursor cells (eg, Siglech, Itgax [CD11c], and Rtn1) (Figure 3A).36-39 Transcripts for other DC-regulatory proteins were either unchanged (eg, Bcl11a, Ikzf1, Nfil3, Bcl6, and Tcf4 [E2-2]) or decreased (Zbtb46), possibly because of broader expression in cell types lost in −77 mutants, such as MEPs. Because IRF8 promotes DC development in adult mice and humans,32,35 GATA2 suppresses IRF8 expression in mouse embryos at the single-cell level21 and high IRF8 corrupts GATA2-deficient (−77−/−) progenitor differentiation (Figure 1D), we asked if the GATA2-IRF8 axis affects DC progenitors. We analyzed DC progenitors using established markers that segregate Flt3+ M-CSFR+ CMPs into MDPs (Ly6C−cKithigh) and CDPs (Ly6C−cKitLow).40 In wild-type E14.5 fetal livers, CDPs were 9.2% of CD16/32lowCD34+Flt3+M-CSFR+Ly6C− progenitors (Figure 3B-C), whereas the proportion of CDPs in Irf8−/− fetal liver was 3.9%. The −77−/− CDPs increased to 20.2%, a 2.2-fold increase over wild-type (P = .007). Biallelic Irf8 and −77 loss restored the proportion of CDPs to resemble wild-type embryos (7.2%, P = .979). In addition to the increased proportion of CDPs to MDPs in −77−/− livers relative to wild-type, the absolute number of both MDPs and CDPs increased in −77 single mutants; 3.7-fold (P = .012) and 10.2-fold (P = .021), respectively (Figure 3D). In −77; Irf8 double-mutants, MDP levels were unchanged relative to −77 single mutants, whereas CDP levels decreased to wild-type levels, consistent with IRF8 promoting CDP production.28
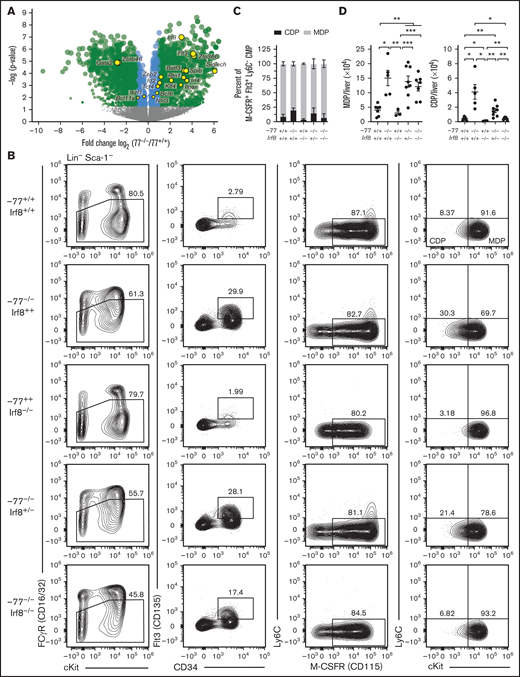
GATA2 suppression of IRF8 levels restricts common dendritic cell progenitor production. (A) Volcano plot of differentially expressed genes mined from transcriptome analysis of lineage-depleted wild-type and −77−/− fetal livers cultured for 3 days as previously described.21 GEO accession: GSE133606. DC genes included factors involved in DC differentiation and/or differentially expressed in DC progenitor populations.38 Gata2 downregulation, resulting from the −77 enhancer deletion, is also depicted. (B) Representative flow cytometry analysis of monocyte-dendritic cell progenitor (MDP) and common dendritic cell progenitor (CDP) populations. Quantitation of the frequency of CDPs and MDPs within the Flt3+M-CSFR+Ly6C− CMP pools (C) and of total cells per liver (D) from 7 litters (−77+/+Irf8+/+, n = 7; −77−/−Irf8+/+, n = 5; −77−/−Irf8+/−, n = 8; −77−/−Irf8−/−, n = 7; −77+/+Irf8−/−, n = 3). Error bars represent mean ± SEM. *P < .05, **P < .01, ***P < .001; Welch’s unequal variance t tests. Relevant statistical comparisons for panel C are described in "Results."
GATA2 suppression of IRF8 levels restricts common dendritic cell progenitor production. (A) Volcano plot of differentially expressed genes mined from transcriptome analysis of lineage-depleted wild-type and −77−/− fetal livers cultured for 3 days as previously described.21 GEO accession: GSE133606. DC genes included factors involved in DC differentiation and/or differentially expressed in DC progenitor populations.38 Gata2 downregulation, resulting from the −77 enhancer deletion, is also depicted. (B) Representative flow cytometry analysis of monocyte-dendritic cell progenitor (MDP) and common dendritic cell progenitor (CDP) populations. Quantitation of the frequency of CDPs and MDPs within the Flt3+M-CSFR+Ly6C− CMP pools (C) and of total cells per liver (D) from 7 litters (−77+/+Irf8+/+, n = 7; −77−/−Irf8+/+, n = 5; −77−/−Irf8+/−, n = 8; −77−/−Irf8−/−, n = 7; −77+/+Irf8−/−, n = 3). Error bars represent mean ± SEM. *P < .05, **P < .01, ***P < .001; Welch’s unequal variance t tests. Relevant statistical comparisons for panel C are described in "Results."
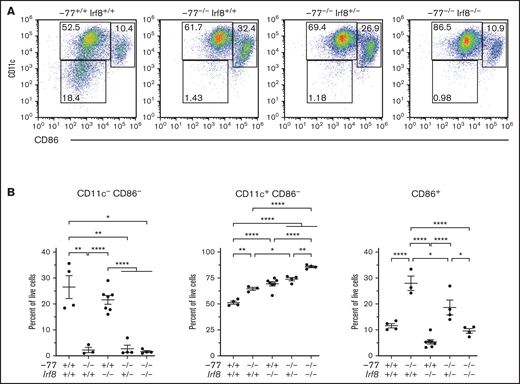
To determine if mutant progenitors are competent to produce DCs, we analyzed DC differentiation using sorted fetal liver CMPs cultured for 7 days in media containing GM-CSF and IL-4 with TNF-α added for the final day of culture (Figure 4A). CD11c, an early DC marker, and CD86, a marker expressed on mature/activated DCs, segregate populations in −77+/+;Irf8+/+ CMP cultures that define progressive DC maturation (Figure 4A-B).41 In −77−/−;Irf8+/+ CMP cultures, CD11c−;CD86− cells are 11.8-fold lower than in wild-type cultures, whereas the more differentiated population of CD86+ cells comprised 28% of the total, a 2.4-fold increase relative to wild-type. Although Irf8 loss in the −77 mutants (−77−/−;Irf8+/−and −77−/−;Irf8−/−) did not rescue the double-negative population, the expanded CD86+ compartment in −77 mutants required Irf8 as the proportion of CD86+ cells in −77−/−;Irf8−/− CMP cultures was decreased to wild-type levels. Consequently, the majority of cells were CD11c+;CD86− (Figure 4A-B). This analysis defines a GATA2-regulated DC population within the CMP pool, and the double-mutant rescue system revealed a GATA2-IRF8 genetic interaction that controls the levels of these cells. GATA2 suppressed MDP levels independent of IRF8, and the elevated IRF8 level of −77 mutants promoted differentiation into CDPs. In addition, progenitors retained the capacity for DC differentiation. Thus, the GATA2-IRF8 axis function extends beyond that of establishing a balance between MPs and GPs within the GMP population.
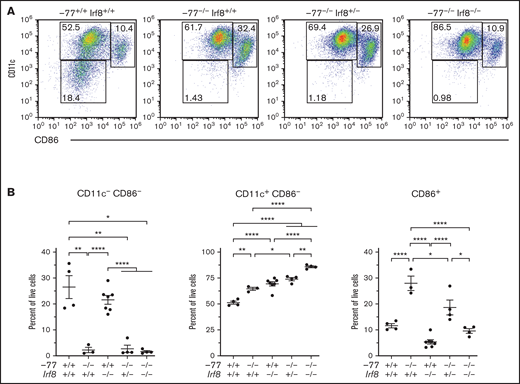
Enhanced dendritic cell differentiation of −77−/− CMPs requires IRF8. (A) Representative flow plots of dendritic cells differentiated from flow-sorted CMPs cultured with GM-CSF, IL-4, and TNFα. (B) Quantitation of dendritic cell populations using early (CD11c) and late (CD86) DC maturation markers. Data were obtained from 3 to 8 embryos (7 litters). The populations were pregated to exclude cell doublets and dead (4′,6-diamidino-2-phenylindole+) cells. Error bars represent mean ± SEM. *P < .05, **P < .01, ****P < .0001; Welch’s unequal variance t tests.
Enhanced dendritic cell differentiation of −77−/− CMPs requires IRF8. (A) Representative flow plots of dendritic cells differentiated from flow-sorted CMPs cultured with GM-CSF, IL-4, and TNFα. (B) Quantitation of dendritic cell populations using early (CD11c) and late (CD86) DC maturation markers. Data were obtained from 3 to 8 embryos (7 litters). The populations were pregated to exclude cell doublets and dead (4′,6-diamidino-2-phenylindole+) cells. Error bars represent mean ± SEM. *P < .05, **P < .01, ****P < .0001; Welch’s unequal variance t tests.
Discussion
Our results unveiled a GATA2-IRF8 axis that orchestrates hematopoietic progenitor generation and function during mouse embryogenesis. Given the conservation of GATA2 and IRF8 mechanisms from mouse to man, and human GATA2- and IRF8-linked hematologic pathologies, these findings inform human disease mechanisms. Because Gata2 and Irf8 mRNAs co-reside in single fetal liver progenitors at reciprocal levels, and reintroducing GATA2 into −77−/− progenitors decreases IRF8,21 GATA2 and IRF8 levels in a single cell are key determinants of this developmental mechanism. GATA2 and IRF8 also control basophil development because GATA2 expression in Irf8−/− bone marrow cells rescues basophil differentiation.42 In this context, however, it is unclear if they function in the same or distinct cells to control differentiation.
Patients with GATA2 mutations can be asymptomatic, and with variable penetrance, the predisposition state can be triggered to progress into bone marrow failure and leukemia.6,34,43 Epigenetic silencing of the wild-type GATA2 allele has been implicated in triggering,44 and additional mutations are likely relevant.45-47 Because clinical hallmarks of GATA2 deficiency syndrome in children and adults include monocyte, lymphocyte, and DC depletion,6,34,48 it is instructive to consider whether GATA2 deficiency in the developing embryo recapitulates these cellular deficits. In a mouse GATA2 deficiency model involving a single-nucleotide variant in an Ets motif of the conserved +9.5 intronic enhancer,49 mutant HSPCs exhibit a nearly normal transcriptome and activity until they are forced to regenerate, which reveals gross transcriptomic and HSC defects.50 Despite the stress-instigated pathogenic activity of this variant, it has little to no impact on hematopoiesis during embryogenesis. Patients with GATA2 deficiency have monocytopenia, yet retain macrophages in bone marrow, and mouse −77−/− GATA2-deficient embryonic progenitor cells generate excessive macrophages.21 DC progenitors accumulate in −77−/− GATA2-deficient fetal liver progenitors, and these cells can generate cells ex vivo with the immunophenotype expected for DCs (Figures 3 and 4). In patients with GATA2 deficiency, DCs are depleted,7,48,51 although depletion has not been described in asymptomatic children or adults. The differential impact of GATA2 mutations in embryos and adults, with or without stressors, has important implications for pathogenesis, analogous to the resistance of embryonic mouse progenitors to the common adult leukemogenic gene Flt3-ITD.52 One could envision GATA2 mutations inducing an indelible epigenomic alteration that serves as a pathogenic predisposition many years thereafter. Consistent with this model, allele-specific chromatin accessibility analysis revealed that a heterozygous +9.5 enhancer deletion in GATA2 deficiency syndrome11 reverted accessible to inaccessible chromatin at the mutant Gata2, but not the wild-type, allele.53
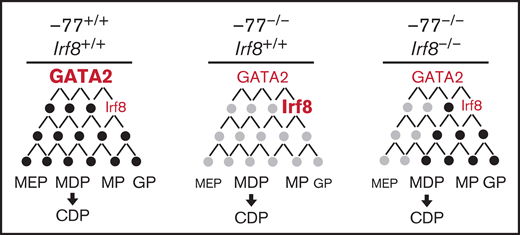
The reduced GATA2 level of −77−/− myelo-erythroid progenitors, which impedes erythroid, megakaryocyte, granulocyte, and basophil developmental trajectories, while maintaining monocyte progenitors, is associated with collapse of the GATA2 network (3161 differentially expressed genes and 434 differentially expressed proteins in −77−/− vs −77+/+ progenitors).21 Because the aberrant network includes many regulatory factors, one would assume that a complex multicomponent mechanism skews differentiation. Intrinsic to this type of mechanism, phenotypic normalization would likely require restoration of physiological expression patterns and protein levels of multiple components. Strikingly, our results with the double-knockout rescue system demonstrated that ablating Irf8 partially normalized the skewed hematopoietic progenitor levels and granulocyte colony activity of −77−/− progenitors (Figure 5), thus establishing a mechanism in which GATA2 suppresses IRF8 levels to ensure normal levels of specific types of hematopoietic progenitors. IRF8 elevation in GATA2-deficient embryos corrupts this mechanism. Applying the strategy innovated to other network components will discriminate vital stem and progenitor cell regulators from those exhibiting intriguing expression patterns that represent inconsequential correlations.

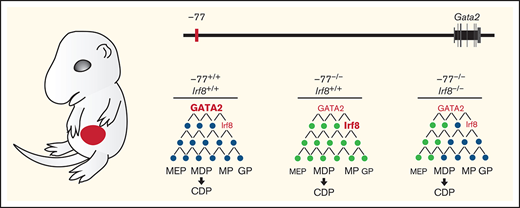
Model illustrating IRF8 function as an essential mediator within GATA2 genetic networks. Physiological levels of GATA2 establish a transcriptome (genes, black circles) with low Irf8 expression, which maintains a balance in myeloid and dendritic cell progenitors. Loss of the −77 enhancer and consequent reduction in GATA2 disrupts the network (gray circles), upregulating Irf8 and increasing proportions of MP, MDP, and CDP populations. Reducing Irf8, in the context of the −77 mutant allele, reversed granulocytic deficiencies and the excessive accumulation of dendritic cell progenitors.
Model illustrating IRF8 function as an essential mediator within GATA2 genetic networks. Physiological levels of GATA2 establish a transcriptome (genes, black circles) with low Irf8 expression, which maintains a balance in myeloid and dendritic cell progenitors. Loss of the −77 enhancer and consequent reduction in GATA2 disrupts the network (gray circles), upregulating Irf8 and increasing proportions of MP, MDP, and CDP populations. Reducing Irf8, in the context of the −77 mutant allele, reversed granulocytic deficiencies and the excessive accumulation of dendritic cell progenitors.
Acknowledgments
The authors thank Mabel M. Jung for graphic design.
This work was funded by NIH grants R01DK68634 and University of Wisconsin Carbone Cancer Center Support Grant P30CA014520), ASH Scholar Award (A.A.S.), Leukemia and Lymphoma Society Career Development Program (A.A.S.), and Edward Evans Foundation.
Authorship
Contribution: K.D.J. and E.H.B. conceived the project, designed the experiments, and wrote the paper; K.D.J. and A.A.S. performed the experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emery H. Bresnick, 4009 WIMR, University of Wisconsin-Madison, Madison, WI 53705; e-mail: ehbresni@wisc.edu.