

Key Points
Accelerated radiotherapy of B-cell lymphoma enhances immunogenic cell death and antitumor immunity compared with conventional radiotherapy.
CD8+ T cells and CD8+ cross-priming dendritic cells are required for this radiation-induced antitumor immunity.

Abstract
Conventional local tumor irradiation (LTI), delivered in small daily doses over several weeks, is used clinically as a palliative, rather than curative, treatment for chemotherapy-resistant diffuse large B-cell lymphoma (DLBCL) for patients who are ineligible for hematopoietic cell transplantation. Our goal was to test the hypothesis that accelerated, but not conventional, LTI would be more curative by inducing T cell–mediated durable remissions. We irradiated subcutaneous A20 and BL3750 lymphoma tumors in mice with a clinically relevant total radiation dose of 30 Gy LTI, delivered in 10 doses of 3 Gy over 4 days (accelerated irradiation) or as 10 doses of 3 Gy over 12 days (conventional irradiation). Compared with conventional LTI, accelerated LTI resulted in more complete and durable tumor remissions. The majority of these mice were resistant to rechallenge with lymphoma cells, demonstrating the induction of memory antitumor immunity. The increased efficacy of accelerated LTI correlated with higher levels of tumor cell necrosis vs apoptosis and expression of “immunogenic cell death” markers, including calreticulin, heat shock protein 70 (Hsp70), and Hsp90. Accelerated LTI–induced remissions were not seen in immunodeficient Rag-2−/− mice, CD8+ T-cell–depleted mice, or Batf-3−/− mice lacking CD8α+ and CD103+ dendritic cells. Accelerated, but not conventional, LTI in immunocompetent hosts induced marked increases in tumor-infiltrating CD4+ and CD8+ T cells and MHCII+CD103+CD11c+ dendritic cells and corresponding reductions in exhausted PD-1+Eomes+CD8+ T cells and CD4+CD25+FOXP3+ regulatory T cells. These findings raise the possibility that accelerated LTI can provide effective immune control of human DLBCL.
Introduction
Radiotherapy is used for the treatment of chemotherapy-refractory diffuse large B-cell lymphoma (DLBCL) patients who are ineligible for hematopoietic cell transplantation.1 Conventional local tumor irradiation (LTI) is delivered in small daily doses over several weeks to reduce toxicities. Because this treatment achieves palliative responses without durable remissions,1 there is a need for radiotherapy regimens that provide greater efficacy. Radiotherapy protocols that establish antitumor immunity have been shown to induce durable complete remissions in solid tumors.2,3 The goal of the current study was to develop an immunostimulatory LTI regimen that leads to long-lasting remissions in murine models of DLBCL.
Radiation protocols can vary markedly with regard to the number of doses, frequency and duration of dosing, and dose per treatment4 to induce immunosuppressive5,6 and immunostimulatory responses.2,3 Radiotherapy has been shown to induce immunostimulatory responses through immunogenic cell death (ICD) of tumor cells.7 Dying tumor cells upregulate damage-associated molecular patterns (DAMPS), such as calreticulin (CRT)8 and heat shock protein 70 (Hsp70) and Hsp90, which promote phagocytosis and enhance antigen cross-presentation by dendritic cells (DCs), respectively.9,10 Interestingly, a clinical trial showed that vaccines derived from human follicular lymphoma cells treated ex vivo with γ-irradiation and loaded into DCs elicited objective clinical responses when CRT and Hsp90 were expressed on the cell surface.11 Furthermore, CRT has been shown to facilitate T-cell infiltration into tumors through upregulation of vascular adhesion molecules and enhance antitumor responses of immunotherapy.12
In our previous study in the CT26 colon tumor model,3 we observed that single doses of LTI (30 Gy) induced immune-mediated durable remissions. However, conventional LTI with daily low doses of 3 Gy for >1 week induced little or no antitumor immunity, was ineffective, and was associated with an immunosuppressive tumor microenvironment.3 We hypothesized that single doses of irradiation or accelerated LTI, delivered as multiple doses per day with short intervals between doses, would interfere with radiation repair mechanisms and increase the endoplasmic reticular stress of tumors that induce ICD.13 This radiation regimen–induced ICD would be expected to create an immunostimulatory tumor microenvironment that promotes potent antitumor immunity. In addition, accelerated LTI reduces the total duration to 4 days compared with 12 days with conventional LTI, so that radiation was completed prior to immune cell infiltration of tumors to avoid killing of infiltrating T cells.
Here, we have developed an accelerated LTI protocol in which multiple low doses of radiation were given each day. We compared the therapeutic and immunological effects of this regimen with a conventional protocol in which the same individual and total doses of radiation were given daily over a prolonged period to murine A20 lymphoma, a BALB/c mouse homolog of human DLBCL,14 or to C57BL/6-derived BL3750 lymphoma.15 In our mouse models, we found that accelerated, but not conventional, LTI induced T cell–mediated durable complete remissions of the A20 lymphoma and delayed tumor growth of aggressive BL3750 lymphoma, as well as that accelerated LTI induced more potent ICD of lymphoma cells than conventional LTI.
Materials and methods
Animals and tumor cell lines and tumor-staining and monitoring procedures
Five- to 6-week-old BALB/c wild-type, BALB/c RAG2−/−, and BALB/c Batf3−/− male mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Care of all experimental animals was in accordance with institutional and National Institutes of Health guidelines. The A20 lymphoma cell line was purchased from American Type Culture Collection. BL3750 B-cell lymphoma cells were obtained from Thomas Tedder (Duke University).15 Details regarding tumor injection, tumor size measurement, tissue processing, flow cytometric analyses of tumor-infiltrating immune cells, and histopathology can be found in supplemental Materials and methods.
In vivo tumor irradiation
Irradiation was performed with a Phillips X-ray unit operated at 200 kV with a dose rate of 1.00 Gy/min, according to Filatenkov et al.3
Cell depletion and adoptive-cell transfer of T cells
For in vivo depletion of CD8+ cells or CD4+ cells, 300 µg of anti-CD8 monoclonal antibody (mAb; clone 2.43; rat IgG2b) or 500 µg of anti-CD4 mAb (clone GK1.5, rat IgG2b; both from Bio X Cell) was injected 5 times, intraperitoneally, every 3 days, starting 1 day before irradiation. For adoptive transfer of T cells, spleens were harvested from mice that received tumor irradiation and were free of tumor for ≥100 days. Total T cells were obtained from spleen cells using immunomagnetic beads.16 Purified T cells were added to T-cell–depleted (TCD) bone marrow (BM) cells and adoptively transplanted according to the method described by Dutt et al.17
Statistical analysis
Kaplan-Meier survival curves were generated using Prism (GraphPad Software, La Jolla, CA). Statistical differences in animal survival were analyzed by the log-rank (Mantel-Cox) test. Survival was defined as the time point after tumor inoculation when the mice died or were euthanized, according to institutional veterinary guidelines, or when tumors reached a diameter > 2 cm. The statistical significance of differences between mean percentages of cells in tumors and mean cytokine production in tumor lysates was analyzed using an unpaired 2-tailed Student t test of means (Mann-Whitney U test). For all tests, P ≤ .05 was considered significant.
Results
Treatment of A20 lymphoma tumors with accelerated hyperfractionated LTI induces complete remissions
A20 B-cell lymphoma cells (2 × 105) were injected subcutaneously into the hind quarter of BALB/c mice, and tumors were allowed to grow for 21 days. Tumors in untreated mice continued to increase in volume through day 60; mice with tumors >2 cm diameter were euthanized (Figure 1). Because lymphoma cells are sensitive to radiation, we chose a clinically applicable dose of 3 Gy for each treatment. Tumors were given accelerated hyperfractionated LTI with 10 doses of 3 Gy cumulatively delivered over 4 days (3 doses per day with 4 hours between doses for the first 3 days + 1 dose on day 4) or conventional radiation with 10 daily doses of 3 Gy over 12 days (weekend interruption after the first 5 daily doses). By day 60, subcutaneous tumors completely regressed in 16 of 18 mice in the accelerated LTI group (Figure 1B) and in 7 of 11 mice given conventional irradiation (Figure 1C). All untreated mice were euthanized by day 50 as a result of progressive subcutaneous tumor growth (Figure 1D). Some animals in both irradiation treatment groups were killed as a result of progressive subcutaneous tumor growth, and some died with subcutaneous tumors in remission after 60 days with tumor growth in the secondary lymph nodes (inguinal, axillary, or brachial nodes). The survival of tumor hosts at 100 days is shown in Figure 1D. Interestingly, conventional irradiation of the tumor was considerably less effective, based on host survival, than accelerated irradiation (P = .0006) (Figure 1D). There was no obvious hair loss, scarring, or contracture of the skin in the fields of accelerated or conventionally irradiated mice during the 100-day observation period. In contrast to our previous study in a CT26 colon tumor model,3 in A20 tumors, a single dose of LTI (30 Gy) was less effective than accelerated LTI and, by day 60, tumors regressed in 4 of 7 mice (supplemental Figure 1) with hair loss and scarring of the skin in the field of irradiation. Three of 7 mice showed complete remissions at day 100, and 1 had relapse at a distant site. Therefore, this single high dose of irradiation was not used in further studies.
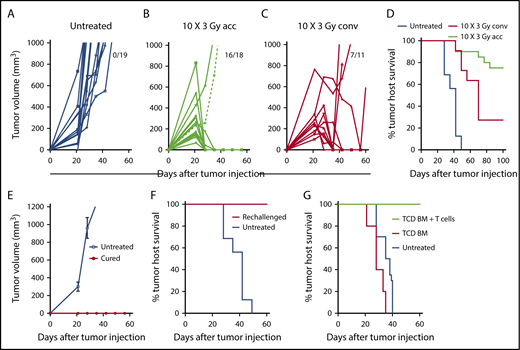
Accelerated LTI, but not conventional LTI, therapy induces potent T cell–mediated durable complete remissions in A20 lymphoma. (A) Changes in individual tumor volumes of A20 lymphomas after subcutaneous (s.c.) flank injection of 2 × 105 lymphoma cells in untreated BALB/c mice. Fraction of mice alive with complete remission of primary tumors at day 60 is shown. (B) Changes in mice treated with accelerated (acc) tumor irradiation (10 × 3 Gy) over 4 days. (C) Changes in mice treated with conventional (conv) daily tumor irradiation over (10 × 3 Gy) 12 days. (D) Tumor host survival of treated and untreated tumors. There were significant differences in survival over 100 days in groups with untreated tumors vs tumors treated with acc irradiation (P < .0001) or conv irradiation (P < .0001), as well as in groups treated with acc irradiation vs conv irradiation (P = .006, Mantel-Cox test). Changes in mean (± standard error) tumor volumes (E) and survival of tumor hosts (F) after tumor cell injection (2 × 105 A20 cells, s.c.) into untreated mice or into mice in complete remission (cured) for ≥100 days after treatment of A20 tumors with accelerated LTI. (G) Survival of untreated mice or adoptive BALB/c hosts given 800 cGy total body irradiation and 5 × 106 TCD BM cells alone or with 6 × 106 splenic T cells from mice in complete tumor remission. Adoptive hosts or untreated mice were injected with lymphoma cells (2 × 105 of A20 cells, s.c.) on the day of BM injection. There was a significant difference in survival between groups with T-cell transplants from cured LTI-treated donors (n = 5) vs transplants of TCD BM alone (P = .0003; n = 5) vs survival of untreated tumor-bearing mice (P < .0001; n = 5).
Accelerated LTI, but not conventional LTI, therapy induces potent T cell–mediated durable complete remissions in A20 lymphoma. (A) Changes in individual tumor volumes of A20 lymphomas after subcutaneous (s.c.) flank injection of 2 × 105 lymphoma cells in untreated BALB/c mice. Fraction of mice alive with complete remission of primary tumors at day 60 is shown. (B) Changes in mice treated with accelerated (acc) tumor irradiation (10 × 3 Gy) over 4 days. (C) Changes in mice treated with conventional (conv) daily tumor irradiation over (10 × 3 Gy) 12 days. (D) Tumor host survival of treated and untreated tumors. There were significant differences in survival over 100 days in groups with untreated tumors vs tumors treated with acc irradiation (P < .0001) or conv irradiation (P < .0001), as well as in groups treated with acc irradiation vs conv irradiation (P = .006, Mantel-Cox test). Changes in mean (± standard error) tumor volumes (E) and survival of tumor hosts (F) after tumor cell injection (2 × 105 A20 cells, s.c.) into untreated mice or into mice in complete remission (cured) for ≥100 days after treatment of A20 tumors with accelerated LTI. (G) Survival of untreated mice or adoptive BALB/c hosts given 800 cGy total body irradiation and 5 × 106 TCD BM cells alone or with 6 × 106 splenic T cells from mice in complete tumor remission. Adoptive hosts or untreated mice were injected with lymphoma cells (2 × 105 of A20 cells, s.c.) on the day of BM injection. There was a significant difference in survival between groups with T-cell transplants from cured LTI-treated donors (n = 5) vs transplants of TCD BM alone (P = .0003; n = 5) vs survival of untreated tumor-bearing mice (P < .0001; n = 5).
To determine whether accelerated LTI resulted in durable immune responses against tumor antigens, mice in remission after 100 days were challenged with a subcutaneous injection of 2 × 105 A20 lymphoma cells in the opposite flank. All of these mice were resistant to rechallenge (6/6 mice), whereas, in naive mice, all tumors grew progressively (Figure 1E). All of the rechallenged mice survived for ≥100 days, whereas control untreated mice challenged in a simultaneous experiment all died by 50 days (Figure 1F). Accelerated LTI of primary tumors did not generate an abscopal antitumor response in unirradiated tumors when tumor cells were injected simultaneously in both flanks (supplemental Figure 2).
To confirm that tumor-reactive T cells are responsible for the observed efficacy of accelerated LTI, purified splenic T cells (6 × 106 cells) obtained from the cured mice after 100 days were adoptively transferred IV into total body irradiated (8 Gy) BALB/c adoptive hosts in combination with 5 × 106 TCD BM cells from normal donor BALB/c mice. The adoptive hosts were challenged with 2 × 105 A20 lymphoma cells injected subcutaneously on the day of BM transplantation, and survival was observed for 60 days. Untreated mice given tumor cells or total body irradiated mice given TCD BM cells alone served as controls. Figure 1G shows that all adoptive hosts given the T-cell injection survived ≥60 days with no detection of tumors, whereas the control groups did not survive beyond 40 days (P < .001) and had progressive tumor growth.
Accelerated LTI–induced remission of A20 lymphoma tumors is dependent on CD8+ T cells and antigen cross-presenting CD8+ DCs
We tested the antitumor activity of accelerated LTI in 3 types of BALB/c mice: immunodeficient Rag-2−/− mice, which are deficient in T and B cells, mice depleted of CD8+ T cells or CD4+ T cells with anti-CD8 mAb or anti-CD4 mAb treatment, respectively, and Batf-3−/− mice selectively deficient in CD8+ and CD8+CD103+ DCs (Figure 2A-E). Supplemental Figure 3 shows the depletion of CD8+ T cells, CD4+ T cells, and CD4+CD25+FOXP3+ cells after antibody treatments. Whereas 16 of 18 wild-type mice given accelerated LTI had complete remissions of subcutaneous tumors at day 60, none of the CD8+ TCD or Batf-3−/− mice developed complete remissions. Only 2 of 8 Rag-2−/− mice had complete remissions. In contrast, 5 of 6 mice in the CD4+ TCD group had complete remissions after accelerated LTI of primary tumors. This result was reflected in the survival of the groups over 100 days (Figure 2F). About 70% of the wild-type mice survived 100 days, in contrast to <20% of the immunodeficient Rag-2−/− mice. Although 5 of 6 mice in the CD4+ TCD group developed complete remissions of primary tumors, 1 of the 5 mice developed tumors in distant lymph nodes that resulted in death, and the others developed liver metastasis (Figure 2F). Figure 2G shows that, on autopsy, there was no distant lymph node tumor spread or spread to the liver by gross examination in wild-type mice (n = 18) given accelerated LTI, and only 4 of these mice exhibited spread to local lymph nodes. In contrast, all Rag-2−/− mice had local lymph node spread, and 2 of 5 had spread to distant nodes but not liver. Interestingly, 5 of 6 Batf-3−/− mice had spread to the liver without spread to the nodes at the time of autopsy. Tumors found in all gross specimens were confirmed by microscopic examination.
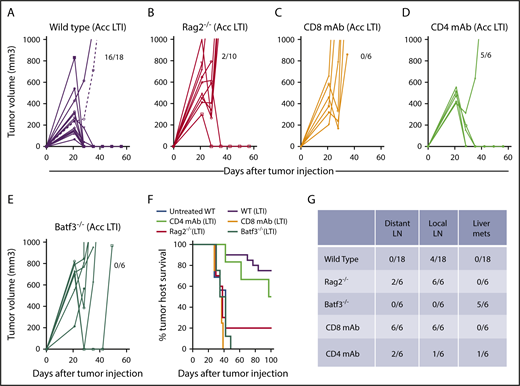
Accelerated LTI is ineffective in immunodeficient Rag2−/−mice, mice depleted of CD8+T cells, and Batf3−/−mice lacking antigen cross-presenting CD8+and CD103+DCs. Mice were injected with 2 × 105 lymphoma cells to induce subcutaneous tumors in the flank. Changes in tumor volume after accelerated LTI in wild-type mice (A), Rag2−/− mice (B), anti-CD8 T-cell mAb–treated mice (C), anti-CD4 T-cell mAb–treated mice (D), and Batf3−/− BALB/c mice (E). Fractions of mice alive and in complete remission at day 60 are shown. (F) Survival of tumor-bearing mice from each group after irradiation or not treatment. There were significant differences in survival in the groups with untreated tumors vs tumors treated with accelerated LTI in wild-type (WT) mice (P < .0001), WT vs Rag2−/− mice (P < .0001), WT vs CD8 TCD mice (P < .0001), and WT vs Baft3−/− mice (P < .0001, Mantel-Cox test). (G) Fraction of mice in (A-E) that had different patterns of tumor spread on autopsy. LN, lymph nodes; mets, metastases.
Accelerated LTI is ineffective in immunodeficient Rag2−/−mice, mice depleted of CD8+T cells, and Batf3−/−mice lacking antigen cross-presenting CD8+and CD103+DCs. Mice were injected with 2 × 105 lymphoma cells to induce subcutaneous tumors in the flank. Changes in tumor volume after accelerated LTI in wild-type mice (A), Rag2−/− mice (B), anti-CD8 T-cell mAb–treated mice (C), anti-CD4 T-cell mAb–treated mice (D), and Batf3−/− BALB/c mice (E). Fractions of mice alive and in complete remission at day 60 are shown. (F) Survival of tumor-bearing mice from each group after irradiation or not treatment. There were significant differences in survival in the groups with untreated tumors vs tumors treated with accelerated LTI in wild-type (WT) mice (P < .0001), WT vs Rag2−/− mice (P < .0001), WT vs CD8 TCD mice (P < .0001), and WT vs Baft3−/− mice (P < .0001, Mantel-Cox test). (G) Fraction of mice in (A-E) that had different patterns of tumor spread on autopsy. LN, lymph nodes; mets, metastases.
Accelerated, but not conventional, LTI induces potent ICD in A20 and BL3750 tumors
The immunogenicity of tumor cells killed in vitro after treatment with chemicals or radiation has been studied in detail.18-20 The molecular changes that are associated with ICD have been attributed to severe reticular endoplasmic stress, resulting in increased surface expression of DAMP molecules by the tumor cells.9,18 Of these, CRT, Hsp90, and Hsp70 are believed to be the most important.9 We studied expression of the DAMPs on tumor cells with or without accelerated or conventional LTI treatment, as described above.
Figure 3A shows representative flow cytometry analysis of CD19+-gated A20 tumor cells after staining for 7-aminoactinomycin D (7AAD) vs annexin V (Ann), 24 hours after completion of accelerated or conventional LTI, and at the same time point without LTI. The 7AAD staining intensity shows the extent of cell membrane permeability and death.21 Ann staining shows the extent of cell membrane expression of phosphatidyl serine, a marker of cells undergoing apoptosis.22 7AADintAnnhi cells (box 2) are late-stage apoptotic cells, 7AADhiAnnhi cells (box 3) are necrotic cells, and 7AAD−Annlo cells (box 1) are nonapoptotic or early apoptotic cells (Figure 3A). Representative staining intensities of CRT, Hsp70, and Hsp90 on the 3 cell populations are shown in Figure 3B. Whereas 7AAD−Annlo cells from box 1 show background staining for DAMPs, cells from boxes 2 and 3 stain positively for DAMPs.
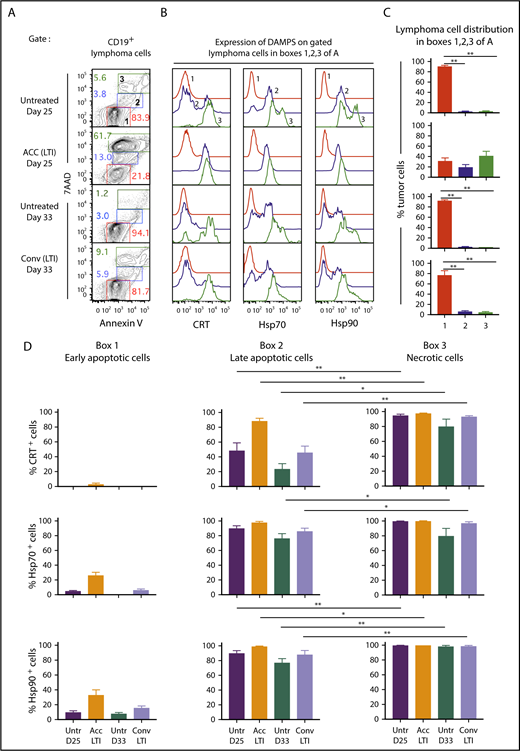
Accelerated LTI is more effective than conventional LTI in the induction of A20 lymphoma cell necrosis with expression of CRT, Hsp70, and Hsp90. (A) Gated CD19+ lymphoma cells from untreated or treated A20 tumors, 24 hours after completion of LTI, were analyzed for 7AAD vs annexin V staining. Gates were drawn based on fluorescence minus 1 controls. (B) Representative 1-color analyses of intensity of staining with CRT, Hsp70, or Hsp90 among gated CD19+ lymphoma cells that are Annlo7AADneg (box 1 in panel A, red), Annint7AADlo (box 2 in panel A, blue), and Annhi7AADhi (box 3 in panel A, green). (C) Percentages (mean ± standard error of the mean) of cells in boxes 1, 2, and 3 in panel A. (D) Mean percentages of CRT+, Hsp70+, or Hsp90+ tumor cells in boxes 1, 2, and 3 in panel A in the 4 groups of mice (n = 5 mice per group). Mean percentages of early apoptotic cells in all groups were significantly different from those in late-apoptotic and necrotic cells. Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
Accelerated LTI is more effective than conventional LTI in the induction of A20 lymphoma cell necrosis with expression of CRT, Hsp70, and Hsp90. (A) Gated CD19+ lymphoma cells from untreated or treated A20 tumors, 24 hours after completion of LTI, were analyzed for 7AAD vs annexin V staining. Gates were drawn based on fluorescence minus 1 controls. (B) Representative 1-color analyses of intensity of staining with CRT, Hsp70, or Hsp90 among gated CD19+ lymphoma cells that are Annlo7AADneg (box 1 in panel A, red), Annint7AADlo (box 2 in panel A, blue), and Annhi7AADhi (box 3 in panel A, green). (C) Percentages (mean ± standard error of the mean) of cells in boxes 1, 2, and 3 in panel A. (D) Mean percentages of CRT+, Hsp70+, or Hsp90+ tumor cells in boxes 1, 2, and 3 in panel A in the 4 groups of mice (n = 5 mice per group). Mean percentages of early apoptotic cells in all groups were significantly different from those in late-apoptotic and necrotic cells. Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
Figure 3C compares the percentages of cells enclosed in boxes 1, 2, and 3 for untreated and irradiated tumors. A mean of 75% to 90% of tumor cells in untreated and conventional LTI–treated mice were in box 1, and <5% to 10% of tumor cells were in boxes 2 and 3. In contrast, a mean of ∼20% and 40% of tumor cells were in boxes 2 and 3, respectively, in mice treated with accelerated LTI, and a mean of ∼30% were in box 1. Thus, there was a marked increase in late-apoptotic and necrotic tumor cells after accelerated LTI compared with other groups. Figure 3D shows the mean percentages of CRT+, Hsp70+, and Hsp90+ cells contained in boxes 1, 2, and 3. There was a significant increase in the percentages of CRT+, Hsp70+, and Hsp90+ cells among 7AADint and 7AADhi populations (boxes 2 and 3) in all groups compared with the 7AAD− population (box 1). In addition, there were significant increases in the mean percentages of CRT+, Hsp70+, and/or Hsp90+ cells in box 3 vs box 2.
Figure 4 compares the percentages of cells that were 7AADint/hiCRThi, 7AADint/hiHsp70hi, or 7AADint/hiHsp90hi (in boxes) in the 4 groups of mice. Whereas unirradiated A20 tumors contained 6% to 12% 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells at day 25, tumors treated with accelerated irradiation contained 75% to 78% of these cells (Figure 4A). At day 33, unirradiated tumor cells contained 1% to 4% of these cells, whereas tumors given conventional irradiation contained 8% to 13% positive cells. As shown in Figure 4B, the mean percentage of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells after accelerated irradiation was significantly increased compared with that in unirradiated or conventionally irradiated tumors. The mean percentages of 7AADint/hiCRThi and 7AADint/hiHsp70hi conventionally irradiated tumors were not significantly different from the percentages in unirradiated tumor cells (Figure 4B).
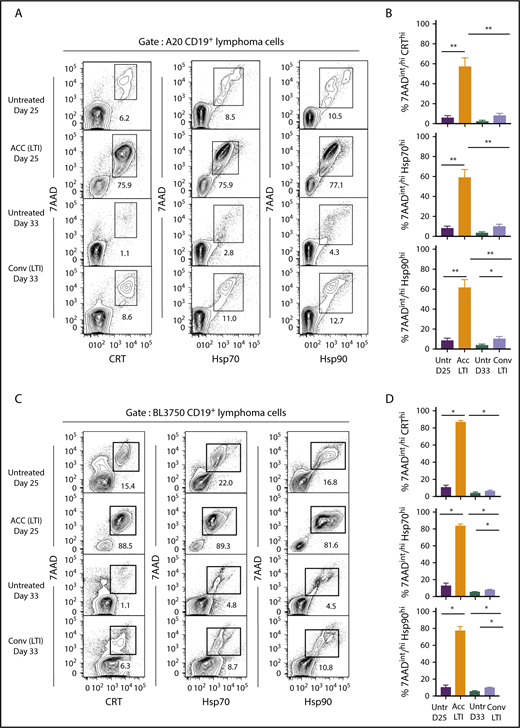
Accelerated, but not conventional, LTI markedly increases the percentages of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hicells in A20 and BL3750 B-cell lymphomas. (A) Representative 2-color FACS patterns of the percentages of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells on CD19+ gated BALB/c A20 lymphoma cells within the boxes from 3 groups of mice. (B) Percentages (mean ± standard error of the mean) of 7AADint/hiCRThi (top panel), 7AADint/hiHsp70hi (middle panel), and 7AADint/hiHsp90hi (bottom panel) cells in panel A. n = 5 mice per group. (C) Representative 2-color FACS patterns of the percentages of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells on CD19+ gated BL3750 lymphoma cells contained in boxes from 4 groups of mice. (D) Percentages (mean ± standard error of the mean) of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells in panel C. n = 4 mice per group. Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
Accelerated, but not conventional, LTI markedly increases the percentages of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hicells in A20 and BL3750 B-cell lymphomas. (A) Representative 2-color FACS patterns of the percentages of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells on CD19+ gated BALB/c A20 lymphoma cells within the boxes from 3 groups of mice. (B) Percentages (mean ± standard error of the mean) of 7AADint/hiCRThi (top panel), 7AADint/hiHsp70hi (middle panel), and 7AADint/hiHsp90hi (bottom panel) cells in panel A. n = 5 mice per group. (C) Representative 2-color FACS patterns of the percentages of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells on CD19+ gated BL3750 lymphoma cells contained in boxes from 4 groups of mice. (D) Percentages (mean ± standard error of the mean) of 7AADint/hiCRThi, 7AADint/hiHsp70hi, and 7AADint/hiHsp90hi cells in panel C. n = 4 mice per group. Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
Similar results were obtained in the BL3750 B-cell lymphoma model in C57BL/6 mice (Figure 4C-D). Accelerated LTI induced significant slowing of tumor growth compared with conventional LTI (supplemental Figure 4), and it induced significantly higher levels of necrosis associated with ICD in A20 and BL3750 tumors. Surprisingly, ICD in A20 tumors was unaffected by immunodeficiency in Rag2−/− and Batf3−/− mice (supplemental Figure 5)
Accelerated LTI results in a significant increase in CD8+ and CD4+ T-cell infiltration into A20 tumors compared with conventional LTI
To determine whether the differences in tumor remissions and ICD between the 2 radiation regimens were associated with differences in the composition of the immune cell infiltrate, we analyzed the tumor-infiltrating mononuclear cells in A20 lymphomas in mice given 10 × 3 Gy accelerated or conventional LTI. Figure 5A compares the representative fluorescence-activated cell sorting (FACS) profiles of CD4+ and CD8+ T cells infiltrating the tumors and expression of PD-1 and Eomes markers that indicate T-cell “exhaustion” among gated CD8+ T cells after accelerated or conventional LTI. The mononuclear cells from the accelerated LTI group contained ∼6% CD8+ T cells and 12% CD4+ T cells, whereas the unirradiated tumors at day 27 contained ∼1% CD8+ T cells and 1.2% CD4+ T cells. In contrast, tumors given conventional LTI contained ∼1% CD8+ T cells and ∼1% CD4+ T cells. At day 35, unirradiated tumors contained <1% CD8+ T cells and ∼1% CD4+ T cells. Figure 5B shows representative FACS profiles of CD4+CD25+FOXP3+ regulatory T (Treg) cells among gated CD4+ T cells in the irradiated and unirradiated tumors. Whereas CD4+CD25+ cells accounted for 74% of CD4+ T cells in tumors after conventional LTI, they accounted for only 28% after accelerated LTI.

Infiltration of CD4+and CD8+T cells and DCs in A20 tumors is markedly increased after accelerated vs conventional LTI. (A) Mice with 21-day tumors received accelerated (ACC) LTI, conventional (Conv) LTI, or no treatment. Tumor-infiltrating mononuclear cells were analyzed 3 days after LTI completion for percentages of CD4+ and CD8+ T cells, as well as expression of PD-1 and Eomes surface markers, among gated CD8+ T cells. Percentages of each subset of cells in boxes on representative 2-color analysis panels are shown, and arrows identify gating strategy. (B) Representative 2-color FACS patterns of CD4 vs CD25 and CD4 vs FOXP3 on gated CD4+ T cells are shown. (C) Mean percentages of tumor-infiltrating CD4+ and CD8+ T cells (upper panels), CD8+ T cells that expressed the “exhausted” phenotype (PD-1+Eomes+) (lower left panel), and CD4+CD25+FOXP3+ Treg cells among CD4+ T cells (lower right panel) (n = 9 or 10). (D) Representative staining for MHCIIhiCD11chi DCs among live mononuclear cells from tumors. These cells were analyzed for expression of CD103. (E) Mean percentages of tumor-infiltrating total MHCII+CD11c+ DCs (left panel) and CD103+ DCs (right panel). Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
Infiltration of CD4+and CD8+T cells and DCs in A20 tumors is markedly increased after accelerated vs conventional LTI. (A) Mice with 21-day tumors received accelerated (ACC) LTI, conventional (Conv) LTI, or no treatment. Tumor-infiltrating mononuclear cells were analyzed 3 days after LTI completion for percentages of CD4+ and CD8+ T cells, as well as expression of PD-1 and Eomes surface markers, among gated CD8+ T cells. Percentages of each subset of cells in boxes on representative 2-color analysis panels are shown, and arrows identify gating strategy. (B) Representative 2-color FACS patterns of CD4 vs CD25 and CD4 vs FOXP3 on gated CD4+ T cells are shown. (C) Mean percentages of tumor-infiltrating CD4+ and CD8+ T cells (upper panels), CD8+ T cells that expressed the “exhausted” phenotype (PD-1+Eomes+) (lower left panel), and CD4+CD25+FOXP3+ Treg cells among CD4+ T cells (lower right panel) (n = 9 or 10). (D) Representative staining for MHCIIhiCD11chi DCs among live mononuclear cells from tumors. These cells were analyzed for expression of CD103. (E) Mean percentages of tumor-infiltrating total MHCII+CD11c+ DCs (left panel) and CD103+ DCs (right panel). Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
The mean percentages of infiltrating total CD4+ and CD8+ T cells were significantly increased in tumors given accelerated LTI compared with those given conventional LTI (Figure 5C). In addition, among the CD4+ T cells, there was a significantly higher percentage of CD4+CD25+FOXP3+ Treg cells in the tumors after conventional LTI compared with accelerated LTI. Whereas 45% to 50% of infiltrating CD8+ T cells in untreated and conventional LTI–treated tumors were PD-1+Eomes+, the percentage was reduced to ∼18% in the tumors treated with accelerated LTI (Figure 5C). Among gated CD8+ T cells in normal spleen, <1% were PD-1+Eomes+ cells (supplemental Figure 6). Interestingly, there was no significant increase in PD-L1 expression in CD19+ lymphoma cells after accelerated LTI, but conventional LTI induced a significant increase compared with untreated controls (supplemental Figure 7A-B). There was a significant reduction in PD-1 expression in CD8+ T cells after accelerated LTI compared with untreated controls (P = .01) and with conventional LTI (P = .005) (supplemental Figure 7C-D).
Representative FACS plots of MHCII+CD11c+ total DCs and MHCII+CD11c+CD103+ DCs in untreated and irradiated tumors are shown in Figure 5D. The percentage of total DCs was highest in the accelerated LTI group. Figure 5E shows that there were significant increases in the percentages of total DCs (MHCII+CD11c+) and CD103+ DCs infiltrating the tumors given accelerated LTI compared with no irradiation or conventional LTI.
Changes in histopathologic appearance, cytokines, and chemokine production of tumors
To further study changes in the composition of CD4+ and CD8+ T cells, we stained histologic sections with anti-CD4, anti-CD8, and anti-CD3 mAbs. We also stained for intracellular expression of HMGB1, which provides maturation signals to DCs23 and chemotactic signals to facilitate DC migration to lymph nodes to cross-present tumor antigens to T cells.24 Figure 6A shows dense infiltration of CD8+ T cells in tumors given accelerated LTI, in contrast to the minimal infiltration seen in untreated or conventionally treated tumors. CD4+ T-cell infiltration was minimal in all 3 groups. The CD3 and merged patterns of CD3, CD4, and CD8 staining followed the pattern of CD8+ T-cell infiltration and were consistent with the flow cytometric analyses shown in Figure 5A and B. Supplemental Figure 7 shows that accelerated LTI induced markedly increased expression of nuclear and cytoplasmic HMGB1 compared with untreated or conventional LTI–treated tumors.

Histopathologic infiltration of CD8+T cells and production of proinflammatory cytokines and chemokines in A20 tumors are markedly increased after accelerated LTI. (A) Immunohistochemistry of A20 tumors that were untreated or treated with conventional or accelerated LTI starting on day 21. Tissue sections obtained from day-27 tumors were stained with anti-CD3, anti-CD4, and anti-CD8 mAbs conjugated with fluorochromes using a 2-stage procedure. CD8+ is red, CD4+ is blue, and CD3+ is green. (B) Lysates from tumor tissues were analyzed for expression of various cytokines and chemokines by Luminex-based assay with or without LTI treatment. Concentrations of cytokines and chemokines per 100 μg of total protein. Data are mean ± SEM (n = 5 mice per group). Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
Histopathologic infiltration of CD8+T cells and production of proinflammatory cytokines and chemokines in A20 tumors are markedly increased after accelerated LTI. (A) Immunohistochemistry of A20 tumors that were untreated or treated with conventional or accelerated LTI starting on day 21. Tissue sections obtained from day-27 tumors were stained with anti-CD3, anti-CD4, and anti-CD8 mAbs conjugated with fluorochromes using a 2-stage procedure. CD8+ is red, CD4+ is blue, and CD3+ is green. (B) Lysates from tumor tissues were analyzed for expression of various cytokines and chemokines by Luminex-based assay with or without LTI treatment. Concentrations of cytokines and chemokines per 100 μg of total protein. Data are mean ± SEM (n = 5 mice per group). Only significant P values are shown. *P < .05, **P < .01, Mann-Whitney U test.
Next, we measured the concentrations of cytokines and chemokines in tumor cell lysates after accelerated LTI, conventional LTI, or no treatment. Interestingly, there were significant increases in the concentrations of IFN-γ, CXCL10, MCP-1, and IFN-β between accelerated LTI and untreated tumors (P < .005). However, there were no differences in the levels of these cytokines and chemokines between conventional LTI and untreated tumors. There was also a significant increase in IFN-γ in accelerated vs conventional LTI (P < .05) (Figure 6B). We did not observe any significant differences in IL-4, IL-6, IL-10, IFN-α, or TNF-α among the 3 groups (data not shown).
Discussion
The objective of this study was to compare the therapeutic and immunological effects of conventional daily tumor radiation of 10 fractions extended over 2 weeks with identical fractions and total dose of radiation given over 4 days in murine B-cell lymphomas. We found that accelerated LTI demonstrated markedly enhanced antitumor immune responses compared with conventional LTI. We observed similar results when CT26 colorectal tumors were treated with a single high dose of (30 Gy) LTI compared with conventional fractionated irradiation.3 Resistance to tumors and antitumor memory T-cell responses could be adoptively transferred in the current study as in our previous study of CT26 tumors.3 Although accelerated LTI–treated mice in remission are resistant to tumor rechallenge, systemic immunity does not develop rapidly enough after accelerated LTI to mediate abscopal antitumor response to simultaneously injected distant tumors. We hypothesize that accelerated irradiation combined with immunotherapy using checkpoint inhibitors may generate a more rapid systemic immune response. We did not determine the role of ICD in the induction of antitumor responses in the previous study. In the present study, we determined whether differences in ICD could explain the differences in the immune responses generated by effective accelerated LTI vs the ineffective conventional LTI.
Accelerated radiation–induced remissions of A20 tumors were not obtained in immunodeficient Rag-2−/− mice, mice depleted of CD8+ T cells after treatment with appropriate mAbs, or Batf-3−/− mice that are deficient in CD8+ DCs and CD8+CD103+ DCs, despite the high sensitivity of lymphomas to radiation-induced cell death. Our results in the lymphoma model are consistent with other studies that show radiation-induced remissions in solid tumors cannot be obtained in immunodeficient mice25 or after depletion of T cells.26 The lack of efficacy of accelerated radiation following CD8+ T-cell depletion and in Baft3-deficient mice lacking CD8α cross-priming DCs indicates that tumor antigen cross-presentation to CD8+ T cells may be critical for radiation-induced remissions in lymphoma and solid tumors. In this study, we observed durable remissions following CD4+ T-cell depletion in accelerated LTI–treated mice. This may be due to depletion of CD4+FOXP3+ Treg cells that suppress antitumor responses. Our study suggests that depletion of Treg cells has a more pronounced effect on antitumor response than the requirement of CD4+ T-cell help.
Tumor cells that have been killed in vitro after treatment with radiation or anticancer drugs can induce potent T cell–mediated immune responses as a result of changes identified as ICD.18,27 The key features of ICD are increased cell surface expression of DAMPs, such as CRT, Hsp70, and Hsp90, and extracellular release of HMGB1 and ATP by dying tumor cells.18,19,28 Our results demonstrate that accelerated LTI induces significantly more ICD in A20 and BL3750 tumors in comparison with conventional LTI, as judged by the percentages of tumor cells expressing high levels of CRT, Hsp70, and Hsp90 in association with intense 7AAD staining. The difference in DAMP expression in tumor cells induced by the 2 irradiation protocols is highly correlated with the percentage of cells undergoing necrosis vs apoptosis, as well as with their respective abilities to induce durable complete remissions. The high expression of DAMPs in tumors treated with accelerated LTI is likely to be related to the inability of the tumor cells to repair endoplasmic reticulum stress within the shorter duration (4 hours) between radiation doses in accelerated irradiation compared with conventional irradiation (24 hours). We hypothesize that reduced ICD in conventional LTI accounts for the increased relapse in mice treated with conventional LTI compared with accelerated LTI. We observed markedly increased necrotic tumor cells (CRTint/hi7AADhi, Hsp70int/hi7AADhi, and Hsp90int/hi7AADhi) following accelerated LTI in Rag2−/− mice compared with untreated controls. This suggests that accelerated LTI is sufficient to induce tumor cell necrosis in the absence of T cells. To our knowledge, this is the first in vivo study that has addressed the role of radiation in inducing ICD; previous studies have been performed on cell lines in vitro.19,20,29
Untreated A20 tumors had increased percentages of CD4+CD25+ Treg cells among infiltrating CD4+ T cells and increased percentages of PD1+Eomes+ exhausted CD8+ T cells compared with normal spleen. The exhaustion of T cells may be due to the action of Treg cells30 and/or chronic tumor antigen stimulation.31 Exhausted CD8+ T cells are dysfunctional, because they have been shown to have reduced cytolytic and cytokine-production capacity.32,33 Accelerated LTI led to a markedly increased infiltration of CD4+ and CD8+ T cells in tumors, with a concomitant reduction in Treg cells. Reduction of exhausted cells within CD8+ T subsets induced by accelerated LTI–treated tumors could be due to a radiation-induced reduction in Treg cells.30 LTI has been shown to increase PD-L1 expression in tumor cells,34 resulting in suppression of radiation-induced antitumor response. This could be overcome by combining LTI with PD-L1 blockade. In our study, conventional LTI, but not accelerated LTI, led to increases in PD-L1 expression in lymphoma cells compared with untreated controls. Significantly higher expression of PD-1 in CD8+ T cells in the conventional LTI–treated tumors compared with accelerated LTI treatment suggests that the PD-1/PD-L1 interaction plays a dominant role in reducing antitumor responses and preventing remissions in conventional LTI but not in accelerated LTI. We observed low levels of infiltrating CD4+ and CD8+ T cells in the conventional LTI–treated group. This is not surprising, because the prolonged daily irradiation over 10 days is likely to have killed tumor-infiltrating T cells that are observed at 7 days after the start of LTI. We observed a similar depletion of tumor-infiltrating T cells in our CT26 tumor irradiation model with conventional LTI that did not effectively treat the tumor. Conventional LTI resulted in markedly reduced T-cell infiltration, even when added to a single dose of 30 Gy.3 Similar results were also reported in a melanoma model.35
Our data suggest that infiltration of tumors with CD103+ DCs after accelerated LTI is likely to be required in the chain of events that leads to the induction of immune-mediated durable remissions, because these DCs are lacking in Batf3−/− mice that failed to respond to accelerated LTI. CD103+ DCs have been shown to traffic to tumors, take up tumor antigens, and migrate to local lymph nodes where they cross-present antigens to CD8+ T cells that mediate antitumor immunity.36,37 We found higher percentages of CD103+ DCs, along with CD8+ T cells, in accelerated LTI–treated tumors compared with conventional LTI.
Accelerated LTI increased intratumoral production of IFN-β, which could enhance the cross-priming capacity of intratumoral DCs and increase the release of IFN-γ and the IFN-γ–inducible chemokine, CXCL10, that promote T-cell infiltration in solid tumors.38 CXCL10 produced by Batf3-dependent CD8+CD103+ DCs is required for effector T-cell trafficking in tumors.39 Our results show that lysates from lymphoma tumors treated with accelerated LTI had significantly higher levels of IFN-γ compared with tumors treated with conventional LTI.
In summary, accelerated LTI, but not conventional LTI, results in a high rate of complete sustained remissions in B-cell lymphomas in mice. We conclude that conventional LTI is ineffective because it does not induce potent ICD and depletes tumor-infiltrating T cells. We hypothesize that conventional LTI is ineffective in inducing durable remissions in DLBCL in humans for the same reasons. Our study raises the possibility that an accelerated radiotherapy regimen can potentially be used with the goal of curing, rather than palliating, relapsed DLBCL. Accelerated irradiation combined with immunotherapy, including checkpoint inhibitors, may further boost antitumor immunity, such that widespread disease may be adequately controlled. Clinical trials are needed to test this hypothesis.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Xiangyue Zhang (Department of Pathology, Stanford University) for providing Batf3-knockout mice and Ometa Herman (Stanford Shared FACS Facility).
This work was supported by grants from the National Cancer Institute, National Institutes of Health (4R01CA16344105 and 5P01CA04960528) (S.S.)
Authorship
Contribution: S.D. designed the protocol, performed experiments, analyzed data, and wrote the manuscript; M.B.A. performed immunofluorescence histopathology; Y.M. performed lymphoma irradiation experiments; A.F. helped to perform initial radiation experiments; K.P.J. designed mAb-staining panels and performed Luminex assays; B.P.I. performed anti-mAb–depletion experiments; R.T. performed HMGB1 staining; J.W. performed Luminex experiments; E.G.E. supervised immunofluorescent histopathology experiments and helped to design protocols; and S.S. supervised all experiments and helped to write the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Suparna Dutt, Stanford University School of Medicine, Division of Immunology and Rheumatology, CCSR Building, Room 2220, 269 Campus Dr, Stanford, CA 94305-5166; e-mail: sdutt@stanford.edu; and Samuel Strober, Stanford University School of Medicine, Division of Immunology and Rheumatology, CCSR Building, Room 2215-C, 269 Campus Dr, Stanford, CA 94305-5166; e-mail: sstrober@stanford.edu.

