

Key Points
Somatic CUX1MT in MNs are heterozygous. CUX1 haploinsufficiency contributes to MDS pathogenesis and worse survival.
Both CUX1MT and CUX1DEL lead to impaired base excision repair function and contribute to more accumulation of somatic mutations.

Abstract
Somatic mutations of the CUT-like homeobox 1 (CUX1) gene (CUX1MT) can be found in myeloid neoplasms (MNs), in particular, in myelodysplastic syndromes (MDSs). The CUX1 locus is also deleted in 3 of 4 MN cases with −7/del(7q). A cohort of 1480 MN patients was used to characterize clinical features and clonal hierarchy associated with CUX1MT and CUX1 deletions (CUX1DEL) and to analyze their functional consequences in vitro. CUX1MT were present in 4% of chronic MNs. CUX1DEL were preferentially found in advanced cases (6%). Most MDS and acute myeloid leukemia (AML) patients with −7/del(7q) and up to 15% of MDS patients and 5% of AML patients diploid for the CUX1 locus exhibited downmodulated CUX1 expression. In 75% of mutant cases, CUX1MT were heterozygous, whereas microdeletions and homozygous and compound-heterozygous mutations were less common. CUXMT/DEL were associated with worse survival compared with CUX1WT. Within the clonal hierarchy, 1 of 3 CUX1MT served as founder events often followed by secondary BCOR and ASXL1 subclonal hits, whereas TET2 was the most common ancestral lesion, followed by subclonal CUX1MT. Comet assay of patients’ bone marrow progenitor cells and leukemic cell lines performed in various experimental conditions revealed that frameshift mutations, hemizygous deletions, or experimental CUX1 knockdown decrease the repair of oxidized bases. These functional findings may explain why samples with either CUX1MT or low CUX1 expression coincided with significantly higher numbers of somatic hits by whole-exome sequencing. Our findings implicate the DNA repair dysfunction resulting from CUX1 lesions in the pathogenesis of MNs, in which they lead to a mutator phenotype.
Introduction
The CUT-like homeobox 1 (CUX1; on 7q22.1) gene has been characterized genetically as a haploinsufficient tumor-suppressor gene1-4 (reviewed in Ramdzan and Nepveu5 ). Paradoxically, CUX1 gene copy number and expression are increased in many cancers and are associated with poorer prognosis.6-8 Strikingly, 1 of 3 tumor cell lines with CUX1 loss of heterozygosity (LOH) also exhibit amplification of the remaining allele during tumor progression.5 CUX1 codes for 2 main isoforms.9 The p110 CUX1 isoform is generated by proteolysis of the full-length protein and functions as a transcriptional repressor or activator depending on promoter context.10-12 The full-length p200 CUX1 protein is abundant, containing 4 DNA-binding domains, 3 Cut repeats (CR1, CR2, CR3), and a Cut homeodomain, with rapid “on” and “off” binding kinetics.13,14 Although p200 was originally identified as a CCAAT-displacement protein that represses transcription by competition for binding-site occupancy,15,16 recent studies established that it also functions as an auxiliary factor in base excision repair (BER).17,18 Specifically, the Cut repeat domains stimulate the enzymatic activities of the 8-oxoguanine DNA glycosylase-1, OGG1.17 CUX1 knockdown or genetic inactivation cause a delay in oxidative DNA damage repair and enhance radiation sensitization, whereas CUX1 overexpression accelerates DNA repair and confers radio resistance.17-19
No familial syndrome had been attributed to germline CUX1 variants/mutations.20-23 Somatic mutations of the CUX1 gene (CUX1MT) have been reported in 0.7% to 5% of tumors depending on the tissue of origin, including truncations and deleterious missense mutations.24-27 LOH of the CUX1 gene also has been reported in uterine leiomyoma (14%) and breast cancer (18%), whereas CUX1 overexpression has been ubiquitous in many carcinomas.5,28 In a mouse model, overexpression of CUX1 causes myeloproliferative syndromes (MDSs) that are myeloproliferative neoplasm (MPN)–like myeloid neoplasms (MNs) with increased neutrophils.29
CUX1 deletions may lead to haploinsufficient expression.30 To date, somatic missense CUX1MT are reported in a small fraction of −7/del(7q) secondary acute myeloid leukemia (AML; sAML),23 especially sAML from antecedent MPN.3 CUX1MT were also described in 2% of MDSs20 and 3% of chronic myelomonocytic leukemia (CMML).31 Previously, CUXMT have been reported in 15% to 25% of primary AML (pAML) and MPNs as well as in therapy-related leukemias.5,24,32 Of note, using deep sequencing, small CUX1MT clones are more frequent than predicted by exome sequencing.24,26 Our group described the molecular spectrum of primary MN (pMN; no prior cancer), secondary MN (sMN; MN patients with primary malignancy not treated with radiotherapy or chemotherapy), and therapy-related MN (tMN; having a prior cancer treated with chemotherapy, radiation, or both). For sMN vs pMN, CUX1MT were 3.4 times more likely to occur in sMN (P = .0417). For sMN vs tMN, CUX1MT were 2.3 times more likely to occur in sMN (P = .2905). For tMN vs pMN, there was almost no difference in CUX1MT frequency. The odds ratio of CUX1MT in tMN vs pMN was 1.3 (P = .78).33
Given the potential implications of CUX1 lesions in MNs, we explored the genotype/phenotype associations of CUX1 lesions, their genomic context, and functional impact on DNA repair in what is to date the largest, most well-annotated cohort of patients with MNs.
Methods
Patients
Samples and clinical data were obtained from patients in accordance with the protocols and consent approved by the institutional review board of the Cleveland Clinic. Clinical parameters (age, sex, bone marrow [BM] and peripheral blood [PB] counts, diagnosis assigned by 2008 World Health Organization [WHO], and overall survival [OS]) were obtained from medical records.34 MDS subtypes of refractory anemia (RA), RA with ring sideroblasts (RARS), and refractory cytopenia with multilineage dysplasia (RCMD) were classified as lower-risk MDSs. RA with excess blasts (RAEB1 and RAEB2) cases were considered higher-risk MDSs. The cases with CUX1MT have been reclassified according to the 2016 WHO criteria: lower-risk MDSs (n = 12; RCMD [6], refractory cytopenia with unilineage dysplasia [2], −5q [2], RA with ring sideroblasts [1], RCMD with ringed sideroblasts [1]) and higher-risk MDSs (n = 14; RAEB-1 [8], RAEB-2 [5], MDS unclassifiable [1]). There were 6 cases with pAML, 2 cases with AML with myelodysplastic changes, 8 cases with CMML and 5 cases with MDS/MPN unclassifiable in addition to 2 cases with idiopathic myelofibrosis and 1 case with chronic myeloid leukemia. The cases that harbor CUX1 deletions (CUX1DEL) classified as lower-risk MDSs (n = 13) were 1 RARS case and 12 RCMD cases whereas higher-risk MDSs (n = 27) were 20 RAEB-1 and 7 RAEB-2. There were (n = 12) AML cases and (n = 24) AML with myelodysplastic changes. The MDS/MPN cases (n = 8) were 6 CMML and 2 MDS/MPN unclassifiable in addition to 3 cases with idiopathic myelofibrosis. Cases with CUX1MT: high risk (n = 8) were International Prognostic Scoring System (IPSS) intermediate 2 (INT-2; n = 7) and high (n = 1) whereas low risk (n = 18) were IPSS low (n = 12) and INT-2 (n = 6). Cases with CUX1DEL: high risk (n = 25) were IPSS INT-2 (n = 17) and high (n = 8) whereas low risk (n = 15) all were INT-1. CUX1 wild type (CUX1WT) have been reclassified according to the 2016 WHO as shown in supplemental Figure 10. The median follow-up time was 60 months (range, 0.5-311 months). The mutational status of CUX1 was analyzed in BM and PB specimens from 1480 patients, of which 131 samples were analyzed by whole-exome sequencing (WES) and 1349 samples were tested by targeted deep sequencing (TS) for mutations in 60 commonly mutated genes in MNs (supplemental Table 1). The study cohort included lower-risk MDSs (n = 359), higher-risk MDSs (n = 249), pAML (n = 322), sAML (n = 205), MDS/MPN (n = 217), and MPN (n = 128) patients (supplemental Table 2).
Metaphase cytogenetics
Metaphase cytogenetics was performed on BM aspirates. The median number of metaphases analyzed was 20. Chromosomal preparation was performed on G-banded metaphase cells using standard techniques, and karyotypes were described in 50 patients with CUX1MT and in 81 of 87 patients with CUX1DEL and 1088 of 1343 CUX1WT samples according to the International System for Human Cytogenetic Nomenclature.35
SNP-A karyotyping
Genome-wide human SNP 6.0 and gene chip human mapping 250K arrays (Affymetrix) were used for single-nucleotide polymorphism array (SNP-A) of BM DNA as described previously.36 Germline-encoded copy-number variants and nonclonal areas of uniparental disomy (UPD) were excluded from further analyses by a bioanalytical algorithm based on lesions identified by SNP-A karyotyping in an internal control series and reported in the Database of Genomic Variants (http://projects.tcag.ca/variation). In 6 patients, microdeletions of CDR1 (7q22.1; 101458959-10192724) were detected.
WES and TS
For WES, the 50 Mb of protein-coding sequences were enriched from total genomic DNA by liquid-phase hybridization using Sure Select (version 4; Agilent Technology), followed by massively parallel sequencing with an HiSeq 2000 sequencer (Illumina). Data were analyzed using our in-house pipeline for somatic mutation calling (Genomon) as previously described (http://genomon.hgc.jp/exome/en/index.html).
To minimize false-positives and focus on the most prevalent or relevant somatic events, we implemented a rational bioanalytic filtering approach and applied heuristic bioanalytic pipelines. Variants were annotated using Annovar.37 This enabled us to obtain normal population frequencies from public databases (ExAC,1000g).38,39 This also enabled us to find functional consequences of that variant (synonymous, nonsynonymous, stopgain, frameshift, splice site, intronic). Finally, we were able to find the presence of a variant in various somatic and germline databases (Cosmic70 and ClinVar)28,40 (supplemental Tables 3 and 4; supplemental Figure 1). Prioritized variants were validated by TS using MiSeq. Our targeted panel for deep sequencing was based on the TruSeq Custom Amplicon panel (Illumina). To detect germline variants of CUX1MT genes, we selected nonsynonymous variants present in both somatic and germline samples that were possibly deleterious. Furthermore, the putative candidates were prioritized based on population genotypic frequency. For the purpose of this study, nonsynonymous, stopgain, frameshift, and splice site variants known to be pathogenic, rare, or with unreported genotypic frequency in the general population were included.
Clonal architecture analysis
Mean coverage for CUX1MT was 265 times for the samples analyzed by deep sequencing; and, to avoid false-positives, only variants with variant allele frequencies (VAFs) ≥5% were considered for further analysis. For distinction between ancestral and subclonal mutations, VAFs (adjusted for copy number) were compared and the largest clone was deemed as a founder hit in such a case. Clonal burden (accounting for ploidy) rather than VAF was used for comparisons. For the purpose of this study, given resolution limitations due to sequencing deeps we used a cutoff of at least 5% difference between VAFs to identify founder mutations. If the difference in VAFs between 2 mutations was <5%, we referred to them as codominant. When phenotypes of cases with CUX1 deemed dominant or codominant mutations were compared, no phenotypic or clinical differences were identified, suggesting that the lack of resolution is inconsequential with regard to the mutations occurring early in the ontogeny.
Expression array and RNA sequencing
Expression Affymetrix U133 plus 2 CEL files were used for 183 MDS patients and 17 healthy controls in Gene Expression Omnibus GSE19429 in which mean minus standard deviation (SD) of healthy controls was used as a cutoff point of lower expression in MDSs; in the Microarray Innovations in Leukemia (MILE) Study program GSE13159, mean minus SD of healthy controls (n = 82) was used as a cutoff point of lower expression in MDSs (n = 206) and AML (n = 467).
Data sets of RNA sequencing from TCGA cases were downloaded from the TCGA repository (http://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp),6 in which mean minus SD of AML with normal karyotyping (n = 118) was used as a cutoff point of lower expression.
Single-cell gel electrophoresis (comet assay)
Patients’ BM mononuclear cells were maintained at 37°C, 5% CO2, and 3% O2, in Iscove modified Eagle medium supplemented with 10% fetal bovine serum and a combination of cytokines: stem cell factor, fms-related tyrosine kinase 3 ligand, interleukin 3, and thrombopoietin at 10 ng/mL each. The cells were kept in culture for 7 days before H2O2 treatment. Trypan blue staining was used to estimate cell viability before comet assay. Cell viability range was 73% to 92%. To ensure reproducibility of single-cell gel electrophoresis (comet) assays performed on different days, cells from healthy donor (HD) 12393 was aliquoted into 3 frozen vials and a fresh vial was used. To measure DNA strand breaks, comet assays were performed using cells treated with 10 µM or 50 µM H2O2 on ice for 20 minutes. After treatment, cells were allowed to recover at 37°C in fresh medium for the indicated periods of time before harvesting. Comet assays were carried out using precoated slides according to the manufacturer’s protocol (Trevigen). The slides were stained with propidium iodide and visualized with an Axiovert 200M microscope with an LSM 510 laser module (Zeiss). Comet tail moments were measured on a minimum of 50 cells using the Comet Score software (TriTeck Corp).
Cell culture and virus production
Hs578T were maintained in Dulbecco's modified Eagle medium (Wisent) and HCC1419 maintained in RPMI 1640 medium (Wisent), both supplemented with 10% fetal bovine serum (tetracycline-free; Invitrogen) and penicillin-streptomycin (Invitrogen). All cells were grown at 37°C, 5% CO2, and atmospheric oxygen O2. Lentiviruses were produced by cotransfecting 293-FT cells with the following plasmids: pTRIPZ-DoxOnshCUX1 plasmid (Open Biosystems), packaging plasmid psPAX2, and envelop plasmid pMD2G. The medium of the transfected cells containing the lentivirus were collected for 3 days, starting 48 hours posttransfection.
Protein extraction and immunoblotting
In vitro 8-oxoG cleavage assay
Double-stranded 32-mer oligonucleotides containing an 8-oxoguanine 8-oxoG at the X position, 5′*CCGGTGCATGACACTGTXACCTATCCTCAGCG-3′, were labeled with 32P-γ adenosine triphosphate at the 5′ end of the top strand (*) using polynucleotide kinase and used in cleavage and DNA-binding assays. Cleavage reaction total cell extracts were performed at 37°C as described by Paz-Elizur et al.41 To visualize the 32-mer substrate and 17-mer product, DNA was loaded on a prewarmed 20% polyacrylamide-urea gel (19:1) and separated by electrophoresis in Tris-borate and EDTA (pH 8.0) at constant 20 mAmp. The radiolabeled DNA fragments were visualized by storage phosphor screen (GE Healthcare).
Radiation survival
Radiation survival of 533 cancer cell lines comprising 26 cancer types was measured using a high-throughput profiling platform as previously described.42,43 The area under the curve was estimated by trapezoidal approximation and the survival values for each trapezoid were multiplied by the dose interval, [f(X1) + f(X2)/2] × ΔX, summed and rescaled by multiplying by (7/log210) so that integral survival is defined from 0 (completely sensitive) to 7 (completely resistant). The association between CUX1MT and the radiation response profile was determined using the information coefficient (IC) values.44,45
Statistics
Kaplan-Meier statistics were applied to assess the effects of CUX1MT on OS. Groups were compared using the log-rank test. For comparison of the occurrence of mutations and/or correlation with other clinical features between disease groups, categorical variables were analyzed with the Fisher's exact test. All P values were 2-sided and values <.05 were considered statistically significant. Analyses were performed using GraphPad Prism 7. For gene-expression analysis, GraphPad Prism 7 was used to create histograms of log2 expression and the unpaired Student t test was used for comparison of changes in expression profile. P < .05 was set as the threshold of clinical significance. The Student t test was performed for pairwise continuous variable comparisons.
Study approval
Informed consent was obtained per protocol approved by the institutional review board (5024 and 16-020) of the Cleveland Clinic and in accordance with the Declaration of Helsinki. Clinical parameters, including blood counts, demographics and survival times, were obtained from medical records.
Results
CUX1 mutations and deletions in MNs
Our study consists of 1480 patients with MNs including lower-risk MDSs (24%; n = 359), higher-risk MDSs (17%; n = 249), pAML (22%; n = 322), sAML (14%; n = 205), MDS/MPN (15%; n = 217), and MPN (9%; n = 128) (supplemental Table 2). Following next-generation sequencing (NGS), only alterations determined to be somatic were included in further analyses, whereas CUX1 germline variants (6 of 1480; 0.4%) of unknown significance were excluded (supplemental Table 5). Somatic alterations predicted to be functionally consequential were selected for further analysis (supplemental Table 3). Following these analytic steps, somatic CUX1MT were asserted in 4% (n = 55 mutations; 50 cases) of the total cohort and CUX1DEL were found in an additional 6% of cases (53 were in monosomy 7 and 28 del7q and 6 microdeletions; Figure 1A). Notably, 12 of 14 truncating mutations occurred upstream of the Cut homeobox. In knockout mouse models, these proteins would not be imported into the nucleus and would not be processed into the p110 CUX1 isoform. Thus, these truncating mutations would affect both isoforms (Figure 1A; supplemental Table 4). Of CUX1MT, 75% were missense, 18% frameshifts, and 7% were nonsense alterations. The ratio of loss-of-function/missense mutations as well as the absence of mutational hotspots are consistent with the previous notion that CUX1 functions as a tumor-suppressor gene and not an oncogene.46 All CUX1MT were heterozygous except for 6% (n = 3) homozygous (UPD) and 7% (n = 4) compounded heterozygous cases (Figure 1A; supplemental Table 4).
![Figure 1. Description of CUX1 mutations in MNs. (A) Mapping of SNP-A karyotyping showing (n = 83) cases of deleted CUX1 gene (red: 49 monosomy 7, 28 deleted 7q, 6 microdeletions; blue: 4 cases compound heterozygous [monosomy 7 and CUX1 mutation]; green: 3 UPD 7q). CUX1 region indicated with horizontal rectangle. Pie chart shows the proportion of missense and truncating CUX1 mutations. CUX1 map showing mutation positions and absence of mutational hotspots. (B) Expression of CUX1 in MDS (n = 183) subtypes. A discrete subset of MDS cases with del7/7q (n = 7) exhibits low CUX1 expression defined as mean minus SD of healthy controls (n = 17; P = .004) and compared with MDSs with normal karyotyping (n = 94; P = .0001). A bar graph shows percentage of cases with CUX1 low expression in healthy controls, MDSs with normal karyotyping, and MDSs with del7/7q. CUX1 was most commonly underexpressed in MDSs with del7/7q. The paired Student t test was used to compare means across samples. P < .05 was considered statistically significant. NK, normal karyotyping.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/14/10.1182_bloodadvances.2018028423/2/m_advances028423f1.png?Expires=1771076938&Signature=XbxbnLN~yi2~ZNaaJpL0X~~u7Amo1rM0KH5Uw51-Ij0JgA~AmB8MrPZAG8tjCVsi15TqkriKxv9qYBSBC~Wox8UcrIsfMxmz7sIJDPNBKcwpUc4oDRnkiS5swrmiCSObiyLhNnC0zVVAy-fP07tvSrg6eKJmMlJO8y7bg7BH33giSZZGatP-W0pfZj1OzIh9dNjsVWqWb0D5s~6FnvgT8Rq1sI2jcI80E~oA1FGRn8bwj3LveiivbdEqO4QWVx8Pc885kweMaAK4nrZZYOR88C58eG0ZSHZtb1yjGZ6l7foImvsM6txbnsoWpb3serKEUE3rqXBY4u5Xn6BC~3BKQw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Description of CUX1 mutations in MNs. (A) Mapping of SNP-A karyotyping showing (n = 83) cases of deleted CUX1 gene (red: 49 monosomy 7, 28 deleted 7q, 6 microdeletions; blue: 4 cases compound heterozygous [monosomy 7 and CUX1 mutation]; green: 3 UPD 7q). CUX1 region indicated with horizontal rectangle. Pie chart shows the proportion of missense and truncating CUX1 mutations. CUX1 map showing mutation positions and absence of mutational hotspots. (B) Expression of CUX1 in MDS (n = 183) subtypes. A discrete subset of MDS cases with del7/7q (n = 7) exhibits low CUX1 expression defined as mean minus SD of healthy controls (n = 17; P = .004) and compared with MDSs with normal karyotyping (n = 94; P = .0001). A bar graph shows percentage of cases with CUX1 low expression in healthy controls, MDSs with normal karyotyping, and MDSs with del7/7q. CUX1 was most commonly underexpressed in MDSs with del7/7q. The paired Student t test was used to compare means across samples. P < .05 was considered statistically significant. NK, normal karyotyping.
Description of CUX1 mutations in MNs. (A) Mapping of SNP-A karyotyping showing (n = 83) cases of deleted CUX1 gene (red: 49 monosomy 7, 28 deleted 7q, 6 microdeletions; blue: 4 cases compound heterozygous [monosomy 7 and CUX1 mutation]; green: 3 UPD 7q). CUX1 region indicated with horizontal rectangle. Pie chart shows the proportion of missense and truncating CUX1 mutations. CUX1 map showing mutation positions and absence of mutational hotspots. (B) Expression of CUX1 in MDS (n = 183) subtypes. A discrete subset of MDS cases with del7/7q (n = 7) exhibits low CUX1 expression defined as mean minus SD of healthy controls (n = 17; P = .004) and compared with MDSs with normal karyotyping (n = 94; P = .0001). A bar graph shows percentage of cases with CUX1 low expression in healthy controls, MDSs with normal karyotyping, and MDSs with del7/7q. CUX1 was most commonly underexpressed in MDSs with del7/7q. The paired Student t test was used to compare means across samples. P < .05 was considered statistically significant. NK, normal karyotyping.
Clinical characterization of CUX1 lesions
CUX1MT were most common in higher-risk MDSs (28%; 14 of 50), followed by MDS/MPN (26%; 13 of 50), lower-risk MDSs (24%;12 of 50), pAML (12%; 6 of 50), MPN (6%; 3 of 50), and sAML (4%; 2 of 50), with MDSs (52%) being the most common disorder reported in the context of CUX1MT compared with AML (16%) and MDS/MPN (32%; P = .02; supplemental Figure 2A,C); CUX1DEL were also most commonly detected in higher-risk MDSs (31%; 27 of 87) and sAML (28%; 24 of 87) compared with lower-risk MDSs (15%;13 of 87), pAML (14%;12/87), MDS/MPN (9%; 8 of 87) and MPN (3%; 3 of 87, P < .0001) (Table 1; supplemental Figure 2C). On further analyses comparing CUX1MT vs CUX1DEL, MDS/MPN were more commonly associated with CUX1MT (26% vs 9%; P = .02), whereas sAML was more commonly associated with CUX1DEL (4% vs 28%; P = .002). Complex karyotype (21%) and del5q (21%) more commonly coincided with CUX1DEL vs CUX1MT and CUX1WT (P = .01, P = .0001, respectively). CUX1DEL patients tended to have higher blast percentages and lower platelet counts than the CUX1MT or CUX1DEL cases (Table 1). Monoallelic (heterozygous, hemizygous, and microdeletions) vs biallelic inactivation of CUX1 (UPDs and compound heterozygous) did not display any clinical phenotypic differences (supplemental Figure 2D). VAF of CUX1 alterations was higher MDS/MPN (mean; 39.3%) vs MDSs and AML (26.5% and 27.3%; P = .05; supplemental Figure 2B).
Characteristics of CUX1MT, CUX1DEL, and CUX1WTcases
| CUX1MT | CUX1DEL | CUX1WT | |
|---|---|---|---|
| No. | 50 | 87 | 1344 |
| Sex, male | 50 | 55 | 59 |
| Diagnosis, n (%) | |||
| MDSs | |||
| Low-risk | 12/50 (24) | 13/87 (15) | 335 (25) |
| High-risk | 14/50 (28) | 27/87 (31)† | 208 (15) |
| AML | |||
| Primary | 6/50 (12) | 12/87 (14) | 304 (23) |
| Secondary | 2/50 (4) | 24/87 (28)**† | 179 (13) |
| MDS/MPN | 13/50 (26)* | 8/87 (9) | 196 (15) |
| MPN | 3/50 (6) | 3/87 (3) | 122 (9) |
| Cytogenetics, n (%) | |||
| Normal | 25 (50) | 0 | 509 (38) |
| Complex | 4 (8) | 19 (21)‡ | 147 (11) |
| Del (5q) | 4 (8) | 18 (21)ठ| 96 (7) |
| Del (7q/−7) | 5 (10) | 81 (100) | 0 |
| Del (20q) | 0 | 7 (8) | 54 (4) |
| (+8) | 3 (6) | 3 (3) | 81 (6) |
| −Y | 1 (2) | 2 (2) | 27 (2) |
| Median blood counts | |||
| ANC, ×109/L | 10 | 4 | 6 |
| Hb, g/dL | 10 | 9 | 11 |
| Platelets, ×109/L | 125 | 76 | 133 |
| PB blast, % | 9 | 12 | 8 |
| BM blast, % | 15 | 20 | 14 |
| CUX1MT | CUX1DEL | CUX1WT | |
|---|---|---|---|
| No. | 50 | 87 | 1344 |
| Sex, male | 50 | 55 | 59 |
| Diagnosis, n (%) | |||
| MDSs | |||
| Low-risk | 12/50 (24) | 13/87 (15) | 335 (25) |
| High-risk | 14/50 (28) | 27/87 (31)† | 208 (15) |
| AML | |||
| Primary | 6/50 (12) | 12/87 (14) | 304 (23) |
| Secondary | 2/50 (4) | 24/87 (28)**† | 179 (13) |
| MDS/MPN | 13/50 (26)* | 8/87 (9) | 196 (15) |
| MPN | 3/50 (6) | 3/87 (3) | 122 (9) |
| Cytogenetics, n (%) | |||
| Normal | 25 (50) | 0 | 509 (38) |
| Complex | 4 (8) | 19 (21)‡ | 147 (11) |
| Del (5q) | 4 (8) | 18 (21)ठ| 96 (7) |
| Del (7q/−7) | 5 (10) | 81 (100) | 0 |
| Del (20q) | 0 | 7 (8) | 54 (4) |
| (+8) | 3 (6) | 3 (3) | 81 (6) |
| −Y | 1 (2) | 2 (2) | 27 (2) |
| Median blood counts | |||
| ANC, ×109/L | 10 | 4 | 6 |
| Hb, g/dL | 10 | 9 | 11 |
| Platelets, ×109/L | 125 | 76 | 133 |
| PB blast, % | 9 | 12 | 8 |
| BM blast, % | 15 | 20 | 14 |
ANC, absolute neutrophil count; Hb, hemoglobin.
P < .05, **P < .01 compare CUX1MT vs CUX1DEL, †P < .01 compares CUX1DEL vs CUX1WT, ‡P < .05, §P < .001 compare CUX1MT vs CUX1DEL vs CUX1WT.
Clinical characterization of CUX1 lesions
CUX1MT were most common in higher-risk MDSs (28%; 14 of 50), followed by MDS/MPN (26%; 13 of 50), lower-risk MDSs (24%;12 of 50), pAML (12%; 6 of 50), MPN (6%; 3 of 50), and sAML (4%; 2 of 50), with MDSs (52%) being the most common disorder reported in the context of CUX1MT compared with AML (16%) and MDS/MPN (32%; P = .02; supplemental Figure 2A,C); CUX1DEL were also most commonly detected in higher-risk MDSs (31%; 27 of 87) and sAML (28%; 24 of 87) compared with lower-risk MDSs (15%; 13 of 87), pAML (14%;12 of 87), MDS/MPN (9%; 8 of 87), and MPN (3%; 3 of 87; P < .0001) (Table 1; supplemental Figure 2C). On further analyses comparing CUX1MT vs CUX1DEL, MDS/MPN were more commonly associated with CUX1MT (26% vs 9%; P = .02), whereas sAML was more commonly associated with CUX1DEL (4% vs 28%; P = .002). Complex karyotype (21%) and del5q (21%) more commonly coincided with CUX1DEL vs CUX1MT and CUX1WT (P = .01, P = .0001, respectively). Patients with CUX1DEL had a median blast count of 12% (range, 0% to 96%) in PB and 20% (range, 0% to 97%) in BM whereas patients with CUX1MT had a median blast count of 9% (range, 0% to 86%) in PB and 15% (range, 0% to 78%) in BM (Table 1). Monoallelic (heterozygous, hemizygous, and microdeletions) vs biallelic inactivation of CUX1 (UPDs and compound heterozygous) did not display any clinical phenotypic differences (supplemental Figure 2D). VAF of CUX1 alterations was higher in MDS/MPN (mean, 39.3%) vs MDSs and AML (26.5% and 27.3%; P = .05; supplemental Figure 2B).
Expression signature of CUX1
Comparison of gene-expression levels between disease subgroups (after defining the low expression as mean − 1 × SD of healthy controls) showed that CUX1 is underexpressed in −7/del(7q) cases compared with controls (P = .004) and MDS cases with normal karyotype (P = .0001). Thus, 71% (n = 5/7) of MDS cases with −7/del(7q) showed haploinsufficient expression of the CUX1 gene (Figure 1B). CUX1 was also found to be underexpressed in AML −7/del(7q) cases compared with AML with normal karyotype (P < .0001) (supplemental Figure 2E). In total, 15% of MDS cases and 5% of AML cases without −7/del(7q) showed low expression of CUX1, whereas none of the core-binding factor AML cases displayed low expression of CUX1 (Figure 1B; supplemental Figure 2E).
Genetic background of CUX1 lesions
While studying the genetic background of CUX1MT, CUX1DEL and CUX1WT cases, we found multiple genes that coincide with CUX1MT and CUX1DEL (Figures 2 and 3). For instance, TET2 (P = .02), ASXL1 (P = .004), and 13 other somatic genes illustrated in Figure 3 are significantly mutated with CUX1MT/DEL. RAS (7%) and OGT (1%) mutations were mutually exclusive in CUX1DEL (Figure 2; supplemental Figure 3). When CUX1 mutations/deletions (CUX1MT/DEL) were combined as a group, we found that BCOR (62.5% vs 0% vs 37.5%; P = .05), TET2 (52% vs 26% vs 22%; P = .2), and ASXL1 (46% vs 14% vs 41%; P = .2) were more represented in MDS cases compared with AML and MDS/MPN (Figure 2). To evaluate the effects of CUX1 mutations in respect to other gene mutations, we performed multivariate analyses of cases with CUX1MT and CUX1WT with the same molecular profile. A panel of genes (ASXL1, BCOR, DNMT3A, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2) known to impact survival outcomes and age was included. CUX1MT had no significant impact on survival, age, and AML progression as an independent factor (supplemental Figure 4).
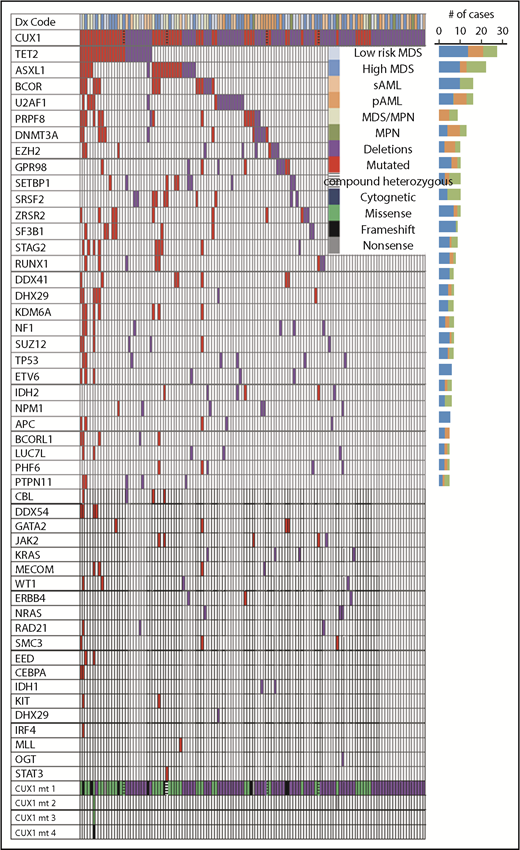
Association of CUX1MTand CUX1DELwith somatic genetic events in MNs. Mutational analysis obtained from WES and TS, karyotyping, and disease type obtained from medical chart review were summarized in a waterfall plot showing the association between diagnoses, karyotyping, number, and type of CUX1 mutations and correlation with 48 commonly mutated genes per each sample. Bar graph (right side) shows the number of cases. CUX1MT and CUX1DEL were 50 and 87, respectively. Red and purple colors indicate presence of mutations and deletions, respectively. Legend summarizes the disease type, cytogenetic status, and types of mutations.
Association of CUX1MTand CUX1DELwith somatic genetic events in MNs. Mutational analysis obtained from WES and TS, karyotyping, and disease type obtained from medical chart review were summarized in a waterfall plot showing the association between diagnoses, karyotyping, number, and type of CUX1 mutations and correlation with 48 commonly mutated genes per each sample. Bar graph (right side) shows the number of cases. CUX1MT and CUX1DEL were 50 and 87, respectively. Red and purple colors indicate presence of mutations and deletions, respectively. Legend summarizes the disease type, cytogenetic status, and types of mutations.
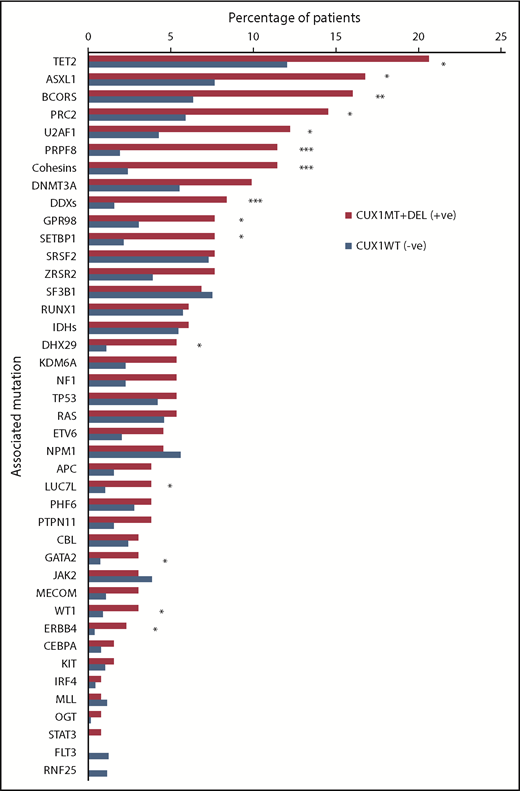
Mutational spectrum of CUX1MT/DELand CUX1WTin MNs. WES and TS were used to detect mutations in CUX1 and other genes commonly mutated in MNs. Stacked bar chart shows the frequency of somatic mutations and gene families in CUX1MT/DEL (n = 137) and CUX1WT (n = 1344). The Fisher's exact test was used to calculate levels of statistical significance. *P < .05, **P < .01, ***P < .001.
Mutational spectrum of CUX1MT/DELand CUX1WTin MNs. WES and TS were used to detect mutations in CUX1 and other genes commonly mutated in MNs. Stacked bar chart shows the frequency of somatic mutations and gene families in CUX1MT/DEL (n = 137) and CUX1WT (n = 1344). The Fisher's exact test was used to calculate levels of statistical significance. *P < .05, **P < .01, ***P < .001.
Clonal architecture and dynamics of CUX1 mutations
Clonal hierarchy analysis was performed to determine whether CUX1MT were dominant or secondary genetic events (Figure 4). The median VAF of CUX1MT was 23% (range, 5% to 100%) being significantly higher in dominant (median, 40%; range, 5% to 100%) vs subclonal CUX1MT (median, 17%; range, 6% to 69%; P = .01; Figure 4C). In 36% (n = 18), CUX1MT were dominant. In cases with dominant CUX1MT (24%; n = 12), the most common secondary hits were BCORs (25%; n = 3) followed by ASXL1 (17%; n = 2), TET2 and DNMT3A (8%; n = 1) each (Figure 4B). Sixty-four percent (n = 32) of CUX1MT were subclonal within this group. TET2MT was the most common first hit (22%; n = 7), followed by BCORs and SF3B1 (each 9%; n = 3), DDX41, IDH2, PRPF8, and U2AF1 each (6%; n = 2) (Figure 4B). There was no characteristic clinical phenotype in dominant vs subclonal CUX1MT (Figure 4C).
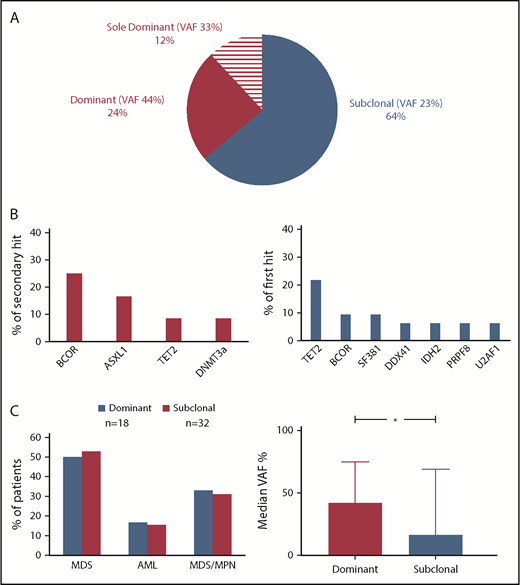
Clonal architecture analysis. (A) For distinction between ancestral and subclonal mutations, VAF (adjusted for copy number and zygosity) were compared and the largest clone was deemed founder. Pie chart shows the frequency of dominant (n = 12), sole dominant (n = 8), and subclonal (n = 32) CUX1 mutations. (B) Bar charts show the frequency of secondary hits in dominant CUX1MT (left panel) and the frequency of first hits in subclonal CUX1MT (right panel). (C) Bar charts show the disease type of the patients harboring dominant and subclonal CUX1MT (left panel) and the median VAF percentage in dominant and subclonal CUX1MT (right panel). The Student t test was used to compare the median VAFs between dominant and subclonal CUX1MT (P = .01). P < .05 was considered statistically significant.
Clonal architecture analysis. (A) For distinction between ancestral and subclonal mutations, VAF (adjusted for copy number and zygosity) were compared and the largest clone was deemed founder. Pie chart shows the frequency of dominant (n = 12), sole dominant (n = 8), and subclonal (n = 32) CUX1 mutations. (B) Bar charts show the frequency of secondary hits in dominant CUX1MT (left panel) and the frequency of first hits in subclonal CUX1MT (right panel). (C) Bar charts show the disease type of the patients harboring dominant and subclonal CUX1MT (left panel) and the median VAF percentage in dominant and subclonal CUX1MT (right panel). The Student t test was used to compare the median VAFs between dominant and subclonal CUX1MT (P = .01). P < .05 was considered statistically significant.
Prognostic impact of CUX1 lesions
CUX1MT were associated with worse survival compared with CUX1WT (median OS; 56 vs 94 months; P = .02) (Figure 5A), as did CUX1DEL (OS; 46 vs 94 months; P = .004; Figure 5A). The combination of both CUX1MT/DEL showed worse survival vs CUX1WT (OS; 49 vs 94 months; P = .0004; Figure 5B). Truncating CUX1MT had a worse prognosis compared with missense CUX1MT, CUX1DEL, and CUX1WT (OS; 39 vs 60 vs 46 vs 94 months; P = .002; Figure 5B). The difference in OS between truncating CUX1MT and CUX1WT was most striking (OS; 39 vs 94; P = .01; Figure 5C). Other survival data analyses were not statically significant (supplemental Figure 5A-E).
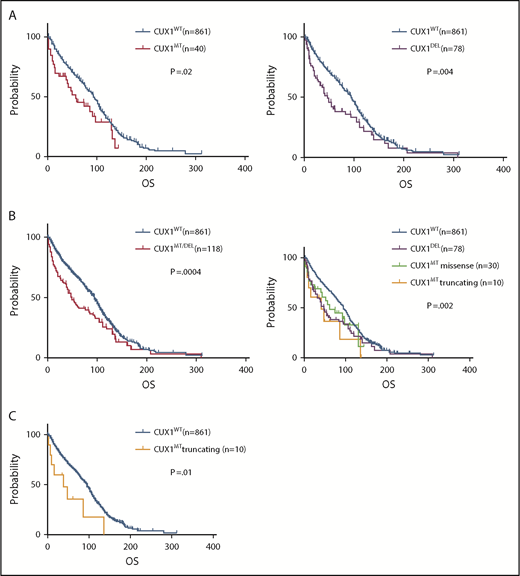
Effect of CUX1 mutational status on OS. Kaplan-Meier survival curves show the OS of (A) patients carrying CUX1MT vs that of patients carrying CUX1WT (P = .02, left), OS of CUX1DEL patients vs CUX1WT patients (P = .004, right). (B) OS of patients with CUX1MT/DEL vs that of patients with CUX1WT (P = .0004, left), OS of patients with CUX1DEL vs OS of patients with CUX1MTmissense vs OS of patients with CUX1MT truncating vs OS of patients with CUX1WT (P = .002, right). (C) OS of patients with CUX1MT truncating vs that of patients with CUX1WT (P=.01). GraphPad Prism 7 was used to estimate OS.
Effect of CUX1 mutational status on OS. Kaplan-Meier survival curves show the OS of (A) patients carrying CUX1MT vs that of patients carrying CUX1WT (P = .02, left), OS of CUX1DEL patients vs CUX1WT patients (P = .004, right). (B) OS of patients with CUX1MT/DEL vs that of patients with CUX1WT (P = .0004, left), OS of patients with CUX1DEL vs OS of patients with CUX1MTmissense vs OS of patients with CUX1MT truncating vs OS of patients with CUX1WT (P = .002, right). (C) OS of patients with CUX1MT truncating vs that of patients with CUX1WT (P=.01). GraphPad Prism 7 was used to estimate OS.
Functional consequences of CUX1 defects
To assess the functional consequences of CUX1MT/DEL, we obtained BM cells from patients and healthy subjects and analyzed them for the presence of defects in DNA repair machinery. To compare their capacity to repair oxidative DNA damage, H2O2-treated cells were analyzed by single-cell gel electrophoresis (comet assay). The comet assay can be performed in various conditions to measure multiple types of DNA damage (Figure 6). Under alkaline conditions (pH 14), double-strand breaks (DSBs), single-strand breaks (SSBs), basic sites, and several types of altered bases can be measured, whereas at pH 10 only DSBs and SSBs will be detected. However, a prior treatment with the formamidopyrimidine DNA-glycosylase (FPG) cleaves 7,8-dihydro-8-oxyguanine, formamidopyrimidines, and oxidized pyrimidines and thus these DNA modifications can also be assessed.
![Figure 6. CUX1 inactivation impairs the DNA repair capacity of BM cells. BM mononuclear cells from patients and healthy subjects were maintained in 3% oxygen for 7 days, exposed to 10 µM (A) or 50 µM (B-D) H2O2 for 20 minutes and allowed to recover for the indicated time. Trypan blue staining was used to estimate cell viability and was: 9654 (87%), 9700 (86%), HD (82%), 6982 (78%), HD (88%), 4487 (92%), 6882 (90%), 8560 (78%), HD (88%), 8342 (75%), 8604 (82%), 11353 (73%). Cells were subjected to single-cell gel electrophoresis (comet assay) at pH 14, pH 10, and pH 10 in the presence of the FPG DNA glycosylase. Comet tail moments were scored for at least 50 cells per condition. The Student t test was used to compare groups. Error bars represent standard error. *P < .05, **P < .01, ***P < .001. HDs: 10972, 11353, 12393; (C-5 [38%], 9700 and C-80, 8342): frameshift mutation and UPD; C-77 (66%), 4487: frameshift mutation and microdeletion; C-10, 8604: deletion 7q, NGS3, 6882: UPD7; (C-29, 8560 and C-73, 6982 and 9654): diploid cases.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/14/10.1182_bloodadvances.2018028423/2/m_advances028423f6.png?Expires=1771076938&Signature=lc7KR~cXPPndggwD93979Bq2s0f5Y4k5BWsIsEbdG1bTJA1nIQfHU3rOiAF5HNQX2gjMkmrm8GUiHOa32HZWiCkEzzd3ttZ7eUAailo9mlN0mdJAavTdMJiBrMxAAGL3jk9J0Yx18Jzgcx6C~kbbhyI4Z7kiSdMReq9WdZ8lNrxrTvPkWuDS31fkt168S1NYOgUeyj-OlD8lEhzwfy~rRMuWgq7YXpyAgei3GJxfDqzFR~BXmch~3sOWIS-VelLiYSqYu1TL7PQKMSQ6p2RKXiQfBK18JiMnHkRQrQX-HO9DzJSptY2rBChNG9LRCNccQIHv6awRy-PRfMAEJcYsjg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CUX1 inactivation impairs the DNA repair capacity of BM cells. BM mononuclear cells from patients and healthy subjects were maintained in 3% oxygen for 7 days, exposed to 10 µM (A) or 50 µM (B-D) H2O2 for 20 minutes and allowed to recover for the indicated time. Trypan blue staining was used to estimate cell viability and was: 9654 (87%), 9700 (86%), HD (82%), 6982 (78%), HD (88%), 4487 (92%), 6882 (90%), 8560 (78%), HD (88%), 8342 (75%), 8604 (82%), 11353 (73%). Cells were subjected to single-cell gel electrophoresis (comet assay) at pH 14, pH 10, and pH 10 in the presence of the FPG DNA glycosylase. Comet tail moments were scored for at least 50 cells per condition. The Student t test was used to compare groups. Error bars represent standard error. *P < .05, **P < .01, ***P < .001. HDs: 10972, 11353, 12393; (C-5 [38%], 9700 and C-80, 8342): frameshift mutation and UPD; C-77 (66%), 4487: frameshift mutation and microdeletion; C-10, 8604: deletion 7q, NGS3, 6882: UPD7; (C-29, 8560 and C-73, 6982 and 9654): diploid cases.
CUX1 inactivation impairs the DNA repair capacity of BM cells. BM mononuclear cells from patients and healthy subjects were maintained in 3% oxygen for 7 days, exposed to 10 µM (A) or 50 µM (B-D) H2O2 for 20 minutes and allowed to recover for the indicated time. Trypan blue staining was used to estimate cell viability and was: 9654 (87%), 9700 (86%), HD (82%), 6982 (78%), HD (88%), 4487 (92%), 6882 (90%), 8560 (78%), HD (88%), 8342 (75%), 8604 (82%), 11353 (73%). Cells were subjected to single-cell gel electrophoresis (comet assay) at pH 14, pH 10, and pH 10 in the presence of the FPG DNA glycosylase. Comet tail moments were scored for at least 50 cells per condition. The Student t test was used to compare groups. Error bars represent standard error. *P < .05, **P < .01, ***P < .001. HDs: 10972, 11353, 12393; (C-5 [38%], 9700 and C-80, 8342): frameshift mutation and UPD; C-77 (66%), 4487: frameshift mutation and microdeletion; C-10, 8604: deletion 7q, NGS3, 6882: UPD7; (C-29, 8560 and C-73, 6982 and 9654): diploid cases.
We selected CUX1MT patients with a substantial proportion of clonal cells for CUX1MT: C-5 (38%); C-77 (66%); NGS3 (UPD); C-80 (UPD); C-10 (deletion 7q; 48% nuclei by fluorescence in situ hybridization). CUX1WT cases were also included: C-73, C-29, C-17 (clonal burden for founder SF3B1 [58%], U2AF1 [108%], NOTCH1 [14%] hits). These data indicate the degree of contamination with normal cells and that elimination of normal cells would likely result in detection of more pronounced changes.
Comet assays at pH 10 following treatment with FPG demonstrated that the repair of oxidized bases was delayed in these samples. CUX1 alleles in the 3 patients with a frameshift mutation must produce a truncated CUX1 protein that does not contain the Cut homeodomain and therefore is not imported to the nucleus. Such mutations reproduce the phenotype seen in the Cux1 mouse knockouts47,48 (supplemental Figure 6). Defective DNA repair in the BM cells of these patients is consistent with the reduced expression of functional CUX1 proteins.
To assess whether the DNA repair defects result from CUX1MT (as demonstrated by comet assays) and translate into functional consequences, we chose to study the impact of CUX1MT on radiation sensitivity. We analyzed the radiation survival of a panel of cell lines (n = 533) comprising 26 cancer types using a previously established high-throughput proliferation assay. We found that CUX1MT cell lines (n = 51) had a significantly higher sensitivity to radiation when compared to CUX1WT (IC = −0.166, P = < .001, false discovery rate [FDR] = 0.01) (Figure 7A).
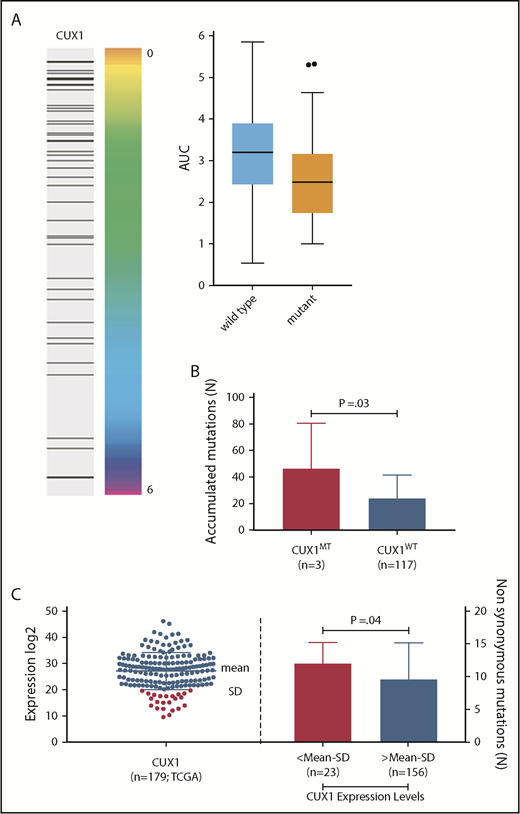
CUX1 mutations and sensitivity to ionizing radiation. (A) Association between integral survival after radiation and CUX1 mutation across 533 cell lines using the information coefficient and as summarized by boxplots (P = .0018; Kolmogorov-Smirnov test). (Right) Black bar represents a mutation in CUX1. CUX1 mutant cell lines were 51. (B) CUX1 inactivation or low expression impacts the accumulation of somatic mutations. Bar graph shows the number of associated mutations in CUX1MT and CUX1WT. All mutations were detected by WES in CUX1MT (n = 3) vs CUX1WT (n = 117). The Student t test was used to compare groups. (C) Dot blot shows the expression levels of CUX1 in the TCGA AML study (n = 179). Bar graph shows the number of associated mutations in cases with CUX1 low expression (n = 23) vs cases with CUX1 high expression (n = 156). The Student t test was used to compare groups; mean minus SD was used as cutoff.
CUX1 mutations and sensitivity to ionizing radiation. (A) Association between integral survival after radiation and CUX1 mutation across 533 cell lines using the information coefficient and as summarized by boxplots (P = .0018; Kolmogorov-Smirnov test). (Right) Black bar represents a mutation in CUX1. CUX1 mutant cell lines were 51. (B) CUX1 inactivation or low expression impacts the accumulation of somatic mutations. Bar graph shows the number of associated mutations in CUX1MT and CUX1WT. All mutations were detected by WES in CUX1MT (n = 3) vs CUX1WT (n = 117). The Student t test was used to compare groups. (C) Dot blot shows the expression levels of CUX1 in the TCGA AML study (n = 179). Bar graph shows the number of associated mutations in cases with CUX1 low expression (n = 23) vs cases with CUX1 high expression (n = 156). The Student t test was used to compare groups; mean minus SD was used as cutoff.
LOH of CUX1 reduces DNA repair capability
Copy-number analysis of 7q22.1 genomic DNA in the HCC1419 cell line (model for CUX1 LOH) reveals loss of 7q22.1. In contrast, 2 CUX1 alleles are present in the Hs578T cell line (supplemental Figure 7A). Immunoblotting analysis showed high CUX1 protein expression in Hs578T cells, but less CUX1 expression in HCC1419 cells and in Hs578T cells expressing CUX1 short hairpin RNA (supplemental Figure 7B). When we carried out an 8-oxoG cleavage assay using cell extracts from these cells, the 17-nt reaction product was not produced as efficiently when the reaction was performed with cell extracts from Hs578T cells in which CUX1 expression was knocked down and was virtually undetectable in the reaction performed with the HCC1419 sample (supplemental Figure 7C; compare lanes 1 and 3 with lane 2).
Impact of CUX1 lesions on accumulation of somatic hits
The results obtained with comet assays prompted us to determine whether CUX1 lesions are associated with accumulation of somatic mutations by studying the mutational burden in samples analyzed by WES. The bioanalytic pipeline adjusted the numbers of hits through comparison with the corresponding germline controls. Samples with CUX1MT harbored significantly higher numbers of somatic hits compared with CUX1WT (median [range], 46 [13-82] vs 23 [2-108]; P = .03, Figure 7B). The 3 CUX1MT cases tended to be older compared with the CUX1WT cohort (80 years vs 69 years; P = .08). Additionally, analyses of numbers of somatic mutations in TCGA samples with low CUX1 expression; defined as mean minus SD of controls, showed a significantly higher number of associated somatic mutations in samples with low CUX1 expression (n = 23) vs normal and high CUX1 expression (n = 156; median [range], 12 [3-17] vs 9 [0-27]; P = .04, Figure 7C).
Discussion
The CUX1 gene appears to be an important tumor-suppressor gene with molecular defects relatively frequently associated with various types of myeloid neoplasia. Our study comprehensively describes the frequency, phenotype/genotype associations, position in clonal hierarchy, and potential functional consequences of relatively large numbers of CUX1 lesions. The high frequency of loss-of-function mutations (frameshifts and stop codons) and deletions suggests that some missense mutations may also be hypomorphic in varying degrees, depending on their configuration and position in the gene. Accumulated evidence supports a model of haploinsufficiency whereby reduced CUX1 expression promotes tumor development. DNA repair pathways have been studied by our group and others and have been ascribed a causal role to the acquisition of genomic instability leading to establishment of dominant clones responsible for MDS progression. Among the 6 major DNA repair pathways (BER, nucleotide excision repair, mismatch repair, homologous recombination, Fanconi anemia/BRCA pathway, and nonhomologous end-joining and translation DNA synthesis) that have been observed to be deregulated in MDSs, BER is the predominant DNA repair pathway involved in handling oxidative DNA damage in MDSs. Because lesions in DNA repair are passed from the founder stem/progenitor cells to the progeny, all clonal cells harbor the defect. Indeed, DNA damage can be found in CD34+ stem/progenitor cells of MDS patients rather than in more differentiated CD34− cells.49 Functional assays demonstrate that CUX1 defects impair specific steps of BER leading to delay in DNA repair. Using extracts from cell lines, we demonstrated that Cux1 knockdown or LOH impair the DNA glycosylase step of the BER pathway. Although CUX1 complementation experiments in hematopoietic cells would be more informative, the impact of CUX1 overexpression on the efficiency of BER has now been demonstrated in cell lines from multiple tissues including mammary, lung, and intestinal epithelial cells, glioblastomas, and fibroblasts.17-19,50 Strikingly, single-cell gel electrophoresis (comet assay) performed on BM mononuclear cells derived from patients with MNs and healthy individuals established that frameshift mutations within CUX1 or deletion of the gene (−7/del7q) cause a delay in the repair of oxidative DNA damage. Reports have shown that CUX1 knockdown or inactivation causes a defect in DNA repair, whereas overexpression of CUX1 or a shorter recombinant protein containing only CUT domains 1 and 2 fused to a nuclear targeting sequence (C1C2-NTS) leads to an acceleration in DNA repair.17-19,50 CUX1 knockdown and rescue by the recombinant C1C2-NTS protein was also shown in a previous study.3,5,26,50 More importantly, the higher sensitivity to radiation exhibited by cell lines carrying CUX1MT suggests that CUX1 loss-associated DNA repair is overwhelmed by radiation leading to increased cell death and thus may also lead to increased repair errors under steady-state condition demonstrated by the higher number of mutational events seen in patients with CUX1MT. The impaired DNA repair function in cells affected by CUX1 lesions may lead to the accumulation of DNA damage and, eventually, point mutations. Indeed, we have shown that ancestral CUX1 lesions may lead to a mutator phenotype and result in the greater accumulation of secondary hits. As such, ancestral CUX1 lesions predispose to subsequent hits, whereas secondary CUX1 hits increase the speed of progression through subclonal accumulation of subsequent defects. This effect may explain the negative prognostic impact of CUX1MT found also in previous studies26,30 as well as CUX1DEL.1,26,30 Similarly, the notion that loss-of-function mutations generate more severe functional defects than missense mutations is also correlated with the differential impact of these types of mutations on outcomes, including OS, as seen in our study and also reported by others.26
The availability of a large cohort of patients allowed for precise assessment of morphologic associations of CUX1 defects. Our results were consistent with previous reports1,5,22,26,30 in that we showed that CUX1MT tended to occur more commonly in MDSs (52%), especially in higher-risk MDSs (28%), whereas CUX1DEL tended to occur more with excess blasts (57%) especially in higher-risk MDSs (33%) and sAML (30%). Previous reports had the same description of CUX1DEL phenotype whereas CUX1MT were reported in MDSs and MPNs generally.26,30 In addition, CUX1DEL were significantly associated with complex karyotyping (23%) and del5q (22%).21,30 However, this larger cohort allowed for the identification of intricate relationships, including the significant role of CUX1 in the pathogenesis of MNs, especially MDSs. LOH of CUX1 displays amplification of the remaining allele, suggesting that decreased expression facilitates tumor development.19 In our analysis, low expression of CUX1 was found in 70% of −7/del(7q) MDSs and 55% of −7/del(7q) AML cases, which is consist with haploinsufficient gene under expression in −7/del(7q).1,30 Overall, we found decreased expression of CUX1 in <20% of patients carrying truncating mutation and deletions. This is consistent with the analysis of results from TCGA of other tumors carrying CUX1 mutations. In many cancers, the few frameshift and nonsense mutations depicted in the lower portion of the dot plot also support the notion that these mutations may cause decreased CUX1 messenger RNA levels (supplemental Figure 8). In addition, in other cancers, both CUX1 LOH and an increase in copy numbers can be observed. One-third of cancer cell lines with CUX1 LOH also display an increase in the copy number of the remaining allele indicating that both genetic events may occur successively during tumor development and progression. Indeed, elevated CUX1 expression has been associated with shorter patient survival in breast, pancreatic, and colorectal cancer.1,6-8,30 In contrast to solid tumors where copy number gain is the prevalent feature, we and others did not observe an increase in CUX1 gene copy number or expression in MDSs and AML samples. These findings suggest that CUX1 functions only as a tumor suppressor in myeloid malignancies.
The subclonal architecture occurring in the setting of co-occurring genetic and molecular abnormalities includes TET2, ASXL1, and BCORs as the most common associated mutations with CUX1MT and the TET2, U2AF1, PRC2 family (EZH2, EED, and SUZ12) and the RAS family (NRAS, KRAS) as the most frequent in CUX1DEL. TET2 was previously described as the most common co-occurring gene with CUX1.30 CUX1 knockdown is synthetic lethal in RAS-transformed cells.18,51 The accumulation of more mutations in CUX1MT cases compared with CUX1WT owing to compromised DNA repair may promote tumor growth and affect survival.5 Indeed, our study shows that CUX1 inactivation causes an impairment in DNA repair proficiency.17-19,50 When we analyzed the sequencing results of our cohort of cases (n = 171), we found that none of our sample had any OGG1 mutations. However, we did find 3 cases carrying CUX1MT in this cohort. When we compared the transversion and transition profiles vs those of CUX1WT patients (n = 168), we found a much higher percentage of G:T in CUX1MT vs WT cases, suggesting a process possibly driven by OGG1 deficiency (supplemental Figure 9).
In conclusion, our study highlights CUX1 gene function impairment, either in a LOH model or with hypomorphic mutations, which leads to specific phenotypes with heterogeneous molecular associations and poor survival outcome. These attributes should be taken into consideration while describing MDS pathogenesis and its molecular background and could be incorporated into new molecular prognostic scoring system models in MDSs.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank The Cancer Genome Atlas portal for providing helpful information for this study.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL118281, R01HL123904, R01HL132071, and R35HL135795, and the Edward P. Evans Foundation. M.E.A. was supported by National Institutes of Health grants KL2TR0002547 (National Center for Advancing Translational Sciences) and R37CA222294 (National Cancer Institute) and VeloSano.
Authorship
Contribution: M.A. and Z.M.R. designed the research, analyzed the data, and wrote the paper; Y.N., S.K.B., N.H., H.M., V.V., T.K., V.A., A. Nazha, M.A.S., and M.E.A. helped with interpretation of the results and edited the manuscript; C.M.K. and B.P.P. performed research procedures; and A. Nepveu and J.P.M. designed the study and wrote the manuscript.
Conflict-of-interest disclosure: M.E.A. discloses research grant support from Siemens Healthcare and research grant, travel support, and an honorarium from Bayer AG. The remaining authors declare no competing financial interests.
Correspondence: Jaroslaw P. Maciejewski, Translational Hematology and Oncology Research, Taussig Cancer Institute, Cleveland Clinic, 9500 Euclid Ave, Desk R40, Cleveland, OH 44195; e-mail: maciejj@ccf.org; or Alain Nepveu, McGill University, Cancer Pavilion, 1160 Pine Ave West, Room 414, Montreal, QC H3A 1A3, Canada; e-mail: alain.nepveu@mcgill.ca.

