

Key Points
Contrary to rat platelets, mouse platelets express both RAB32 and RAB38 that play fully redundant roles for dense granule biogenesis.
Combined RAB32 and RAB38 deficiency mimics severe Hermansky-Pudlak syndrome with albinism and profound defects in hemostasis.

Abstract
The biogenesis of lysosome related organelles is defective in Hermansky-Pudlak syndrome (HPS), a disorder characterized by oculocutaneous albinism and platelet dense granule (DG) defects. The first animal model of HPS was the fawn-hooded rat, harboring a spontaneous mutation inactivating the small guanosine triphosphatase Rab38. This leads to coat color dilution associated with the absence of DGs and lung morphological defects. Another RAB38 mutant, the cht mouse, has normal DGs, which has raised controversy about the role of RAB38 in DG biogenesis. We show here that murine and human, but not rat, platelets also express the closely related RAB32. To elucidate the parts played by RAB32 and RAB38 in the biogenesis of DGs in vivo and their effects on platelet functions, we generated mice inactivated for Rab32, Rab38, and both genes. Single Rab38 inactivation mimicked cht mice, whereas single Rab32 inactivation had no effect in DGs, coat color, or lung morphology. By contrast, Rab32/38 double inactivation mimicked severe HPS, with strong coat and eye pigment dilution, some enlarged lung multilamellar bodies associated with a decrease in the number of DGs. These organelles were morphologically abnormal, decreased in number, and devoid of 5-hydroxytryptamine content. In line with the storage pool defect, platelet activation was affected, resulting in severely impaired thrombus growth and prolongation of the bleeding time. Overall, our study demonstrates the absence of impact of RAB38 or RAB32 single deficiency in platelet biogenesis and function resulting from full redundancy, and characterized a new mouse model mimicking HPS devoid of DG content.
Introduction
Platelet granules critically contribute to hemostasis and thrombosis through the release of various soluble factors.1 The α granules mainly contain proteins involved in coagulation and tissue repair. Dense granules (DGs, also called δ-granules) contain high concentrations of small nonprotein molecules such as nucleotides, pyrophosphates, and polyphosphates, as well as calcium and magnesium.2-4 Following strong platelet activation, the contents of α granules and DGs are released to amplify the platelet responses. Defects in DG formation (δ storage pool diseases, δ-SPD) or in platelet secretion are associated with bleeding disorders. Inherited δ-SPD represent a heterogeneous group presenting with either an isolated platelet function defect or more complex symptoms, as in Hermansky-Pudlak syndrome (HPS).5-7 In addition to the bleeding disorder, HPS is associated with oculocutaneous albinism and variable other serious complications such as immune deficiency or lung fibrosis depending whether the mutated gene also affects other organelles and cell types.7,8
DGs belong to the family of lysosome related organelles (LRO), characterized by an acidic pH and the presence of lysosomal-associated membrane proteins (LAMPs), but their biogenesis differs from ubiquitous lysosomes. Because LROs also include, among others, the melanosomes in melanocytes, it has long been suggested that melanosomes and DGs share similar biogenesis routes. This hypothesis was supported by the identification of mutated genes responsible for both albinism and platelet granule defects in HPS syndrome.9-11 DGs are synthesized in stage I megakaryocytes (MKs) and arise from late endosomal/multivesicular bodies (MVBs), the precursors of α granules and DGs and of lysosomes connecting the endocytic and biosynthetic pathways.12,13 In the early/recycling endosomes, cargos are sorted using ubiquitously expressed machinery and adaptor protein (AP) complexes associated with clathrin-coated vesicles, which tether and fuse with the maturing granules. RAB proteins are key mediators of vesicular trafficking. RAB38 and the closely related molecule RAB3214,15 are expressed in a tissue-specific manner and have been proposed to confer cell-type specificity on LROs, notably on melanosomes.10,11,16 This is consistent with the phenotype of the fawn-hooded rat, one of the first rodent models of HPS. The fawn-hooded rat carries a null mutation in Rab38 and exhibits a diluted coat color resulting from impaired melanosome biogenesis, associated with lung defects and a bleeding phenotype resulting from δ-SPD in the absence of DGs.17-20 This has long suggested that RAB38 is the major RAB directing cargos to maturing DGs. However, a spontaneous mutation which arose in the murine RAB38-GTPase domain (chocolate [cht] mouse) was found to cause slight pigment dilution and defective lung lamellar bodies biogenesis but no bleeding phenotype with absolutely normal DGs21-23 suggesting a hypomorphic mutation.
RAB32 and RAB38 have been well investigated in melanosome biogenesis, where they direct the transport of cargos to maturing melanosomes through interaction with a number of intracellular partners involved in trafficking.9,11 Deficiencies in RAB32 and RAB38 proteins or defects in their GTPase activity are responsible for mistrafficking of melanosomal resident proteins, which results in melanosome malformation and reduced melanin pigment synthesis.9,11,24-26 The mechanisms of DG biogenesis have been less extensively studied, mainly because of the difficulty of manipulating MKs. Using a megakaryoblastic cell line, Di Pietro et al proposed a similar role for RAB32 and RAB38 in the transport and tethering of cargo-containing vesicles and/or their fusion with immature DGs.16,27 These studies were conducted in vitro in a cancer cell line, however. Thus, the question remained open as to their actual role in the in vivo setting, especially in view of the different phenotypes of fawn-hooded rats and cht mice.
This was evaluated by generating mice inactivated for Rab32 and Rab38, following our observation that, contrary to rat platelets, mouse and human platelets also express RAB32 in addition to RAB38. In addition, we showed that inactivation of both genes is required to mimic severe HPS, these mice presenting with oculocutaneous albinism, lung morphology abnormalities, and defective DG formation, demonstrating that RAB32 and RAB38 play redundant roles in DGs biogenesis. In line with the DG defects, these mice exhibited a strong impairment in hemostasis and thrombus formation, representing a new mouse model mimicking severe HPS.
Materials and methods
Materials and animals
Materials and animals are listed in the supplemental Data.
Washed platelet preparation, aggregation, and quantification of granule contents
Platelets were washed in Tyrode’s albumin buffer as previously described.28 Aggregation was measured turbidimetrically using a 4-channel APACT 4004 aggregometer (ELITECH, France). To quantify total platelet factor-4 (PF4) and 5-hydroxytryptamine (5-HT), platelets were lysed by 5 successive cycles of freezing and thawing. To quantify secreted granule secretion, platelets were stimulated with the indicated dose of agonist and centrifuged after 3 minutes. PF4 and 5-HT were quantified in the supernatants by enzyme-linked immunosorbent assay (ELISA). Nucleotides were determined by high-performance liquid chromatography (HPLC; Agilent 1260 Infinity) using a Zorbax SAX 1.6 × 250-mm column (Agilent Technologies). β-hexosaminidase was measured as a marker of lysosome content29 in sera from both wild-type (WT) and inactivation of single or both RAB proteins (DKO) mice.
Immunofluorescence on megakaryocytes and confocal observations
RAB32, RAB38, 5-HT, and von Willebrand factor (VWF) immunolabeling were performed on MKs differentiated in liquid culture for 4 days using standard protocols and observed by confocal microscopy (detailed in the supplemental Methods).
Western blotting and immunolabeling
In western blotting experiments, platelets, tissues, or Lin− cells were lysed in the presence of 1% sodium dodecyl sulfate and proteins were separated by electrophoresis and immunoblotted with the indicated antibodies. For immunolabeling, megakaryocytes were fixed in 4% paraformaldehyde (PFA), cytospun, and labeled with appropriate antibodies.
Lung histology
Twenty eight- to 31-week-old mice were euthanized and the lungs and heart were dissected en bloc. The lungs were inflated with 4% PFA fixative administration through the trachea and immediately separated, the right lobe immersed in PFA for histology and the left lobes immersed in glutaraldehyde 2.5% for transmission electron microscopy (TEM) (see supplemental Methods). For histology, paraffin-embedded lung tissue was sectioned (5-µm-thick sections) and stained with either Masson trichrome or hematoxylin and eosin staining. Alveolar space and multilamellar bodies were quantified using ImageJ software.
Flow cytometry for measurement of integrin αIIbβ3 activation, P-selectin, and LAMP1 exposure and mepacrine loading capacity
Washed platelets (5 × 107/mL) were stimulated with thrombin and subsequently blocked with hirudin (100 U/mL), colabeled with anti-GPIbβ antibody and JonA-PE (1/50, recognizing activated αIIbβ3) or P-selectin (5 μg/mL) or LAMP1 (1/100). The mepacrine loading capacity was evaluated by incubating resting or 1 U/mL thrombin prestimulated platelets with mepacrine (10 µM) for 30 minutes. Amounts of incorporated mepacrine were quantified on the basis of flow cytometry measurements (BD Fortessa).
Whole blood perfusion
Platelet thrombus formation was studied by perfusing collagen-coated capillaries with hirudin-anticoagulated blood as described previously,30 and examination of the capillaries was done by scanning electron microscopy or differential interferential contrast.
Bleeding time and thrombosis models
The bleeding time was measured as described prevously.31 A model of thromboembolism was performed in vivo by injecting tissue factor (TF, 165 µL/kg) into the jugular vein of anesthetized animals within an infusion time frame of 3 to 4 seconds. Viability was monitored during the 30 minutes following TF injection. The same dose of TF was used to measure platelet consumption, quantified as the difference between the platelet counts before and 2 minutes after TF injection.32 Two models of mural thrombosis were used, 1 triggered by laser-induced localized deep injury of the mesenteric arterioles, as previously described,33 and detailed in the supplemental Data. The second one was induced by exterior application of FeCl3 7.5% on the carotid artery, as described elsewhere.34 The lesion was performed laterally to measure thrombus height growth in real time. Details are provided in the supplemental Data.
Results
RAB32 and RAB38 are expressed in human and mouse platelets, whereas RAB32 is absent from rat platelets
RAB32 and RAB38 were both found to be expressed in human and mouse platelets as observed by western blot and by reverse transcription-polymerase chain reaction analyses (Figure 1A). In contrast, RAB32 protein or messenger RNA (mRNA) was not detected in rat platelets (Figure 1A). To make sure that this was not unique to a specific rat strain, we compared platelets from Wistar and OFA albino rats, and also from a Brown Norway background, a non-agouti brown rat, and found the same results. RAB32 was nevertheless expressed in other tissues in the rat such as in the kidney, spleen, lungs, and skin (supplemental Figure 1A). Hence our data suggest that even if RAB32 is dispensable for the biogenesis of DGs in the rat, the possibility remains of a role in human and mouse platelets.
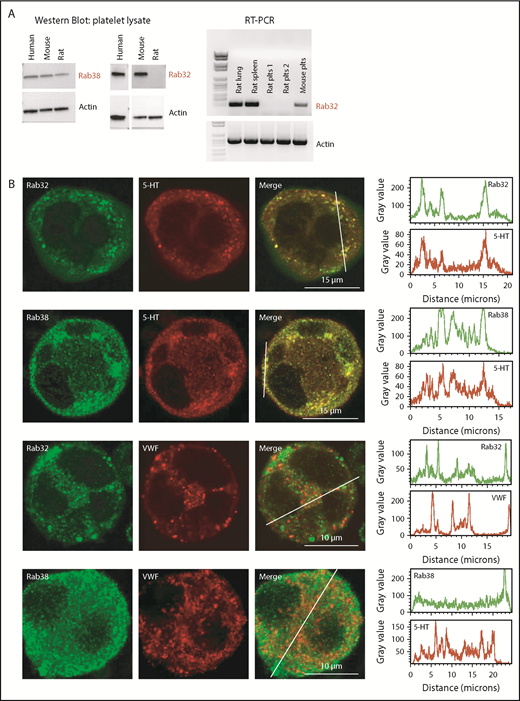
Expression of RAB38 and RAB32 proteins in MKs and platelets. (A, left panel) Western blots of human, mouse, and rat total platelet (Plts) lysates (10 µg) showing expression of RAB38 in all 3 species but not of RAB32 in rat platelets. Actin was present as a loading control. Representative blots from 3 independent experiments. (A, right panel) Reverse transcription polymerase chain reaction amplification of mRNA from rat lung, spleen, and platelets and mouse platelets, indicating absence of RAB32 mRNA in rat platelets. Rat plts 1: OFA rat; rat plts 2: Wistar rat. (B) Confocal images of immunolabeled mouse MKs. Left panels show RAB32 or RAB38 labeling (green), 5-HT or VWF labeling (red), and the merged images. Right panels show for each labeling a line scan of the fluorescence intensity along the drawing line visualized in the merged images. Bar, 5 µm. Images are representative of at least 3 independent labeling experiments.
Expression of RAB38 and RAB32 proteins in MKs and platelets. (A, left panel) Western blots of human, mouse, and rat total platelet (Plts) lysates (10 µg) showing expression of RAB38 in all 3 species but not of RAB32 in rat platelets. Actin was present as a loading control. Representative blots from 3 independent experiments. (A, right panel) Reverse transcription polymerase chain reaction amplification of mRNA from rat lung, spleen, and platelets and mouse platelets, indicating absence of RAB32 mRNA in rat platelets. Rat plts 1: OFA rat; rat plts 2: Wistar rat. (B) Confocal images of immunolabeled mouse MKs. Left panels show RAB32 or RAB38 labeling (green), 5-HT or VWF labeling (red), and the merged images. Right panels show for each labeling a line scan of the fluorescence intensity along the drawing line visualized in the merged images. Bar, 5 µm. Images are representative of at least 3 independent labeling experiments.
Consistent with this hypothesis, we found that RAB32 and RAB38 expression are present throughout murine MK differentiation (supplemental Figure 1B). We observed MK at a stage where maturing granules are present and found that VWF-positive α granules are not or only marginally labeled with either Rab32 or Rab38. By contrast, the majority of 5-HT+ granules were also labeled for RAB38 or RAB32 (Figure 1B), in accordance with a role for RAB32 and RAB38 in DG but not α granule biogenesis. Other vesicles were labeled for RAB38 and RAB32, which could represent early immature DGs or transport vesicles. To further study the role of RAB32 and RAB38 in DG formation, we generated mice in which Rab32 or Rab38 were inactivated.
Effect of Rab32 and Rab38 inactivation on coat pigmentation, lung structure, and granule content
Rab38−/− mice were crossed with Rab32−/− mice to obtain inactivation of single or both RAB proteins [DKO: Rab32−/−;Rab38−/−] (Figure 2A). Heterozygous genotypes expressed intermediate levels of RAB32 or RAB38 (Figure 2A). Inactivation of Rab32 and/or Rab38 did not lead to compensatory overexpression of other RAB proteins in platelets (supplemental Figure 2). Rab32 inactivation had no impact on coat color, whereas Rab38 inactivation led to a light pigment dilution, similar to the cht mice (Figure 2B). However, severe eye and coat color dilution were observed in DKO mice, presenting a light beige pigmentation and red eyes. In contrast, mice heterozygous for Rab32 and deficient in Rab38 [Rab32+/−;Rab38−/−] had an intermediate coat color, whereas [Rab32−/−;Rab38+/−] animals displayed less pigment dilution, very close to that of the cht21 or Rab38−/− mice. Thus, RAB38 and RAB32 were found to be partially redundant for mouse coat color, RAB38 nevertheless appearing to predominate over RAB32.
![Figure 2. Impact of Rab32 and/or Rab38 inactivation on coat pigmentation. (A) Western blots showing the absence of RAB32 and RAB38 in Rab32−/− and Rab38−/− mouse platelets, respectively, and in DKO mouse platelets, and partial RAB32 and RAB38 expression in [Rab38−/−;Rab32+/−] and [Rab38+/−;Rab32−/−] mouse, respectively. Representative of 3 independent western blots. (B) Different coat color dilution according to the mouse genotype.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/15/10.1182_bloodadvances.2019031286/2/m_advances031286f2.png?Expires=1769190854&Signature=36eNO3Pv9xrpXEx6I6DXiyYDZujPiikjwge84xHjzmj5logjg3V0PM9WX221l8ec-fV1a5mEXaZHrwOrGlRvaNlOT0jaiqNEYehSmWALPsrkx4CSnqn59MdTP42TmS9Kp-625qr42ipEojX4O4nZDWz4kXTYpBTYe32ffmv0Z-aAboTsr9HViSLLBSXWv26pqR7nvhvpDDRR6Lp8jwBYiM9UANfO5AsovsKLbr6wYti4Ref-nsuXV8Ulc5Cd4JSvJAqyEUFQKtBTs2hku9ykCWhxuGAgk10Zpzt-dHrtc4bCitDi2tj8mUEbBi352YTQAZXGVdLEusQcsBGEYbj3uw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Impact of Rab32 and/or Rab38 inactivation on coat pigmentation. (A) Western blots showing the absence of RAB32 and RAB38 in Rab32−/− and Rab38−/− mouse platelets, respectively, and in DKO mouse platelets, and partial RAB32 and RAB38 expression in [Rab38−/−;Rab32+/−] and [Rab38+/−;Rab32−/−] mouse, respectively. Representative of 3 independent western blots. (B) Different coat color dilution according to the mouse genotype.
Impact of Rab32 and/or Rab38 inactivation on coat pigmentation. (A) Western blots showing the absence of RAB32 and RAB38 in Rab32−/− and Rab38−/− mouse platelets, respectively, and in DKO mouse platelets, and partial RAB32 and RAB38 expression in [Rab38−/−;Rab32+/−] and [Rab38+/−;Rab32−/−] mouse, respectively. Representative of 3 independent western blots. (B) Different coat color dilution according to the mouse genotype.
Because it was previously reported that RAB38-deficient animals presented with lung defects,18,20,23 we next examined lung tissue in our knockout (KO) strains. Histological observations showed that Rab38−/− and DKO, but not Rab32−/− mice, had impaired lung alveolar structure. Although no clear increase in collagen was detected with Masson trichrome staining, we observed epithelium damage as evidenced by significant thickening of alveolar septa of Rab38−/− and DKO lungs, compared with WT and Rab32−/− mice, suggesting that RAB38 plays a predominant role over RAB32 in lung physiology (supplemental Figure 3A-B). By TEM, we closely observed alveolar type II cells containing the lamellar bodies and found that Rab38−/− and DKO presented with some enlarged lamellar bodies (supplemental Figure 3C-D).
The hallmark of HPS is oculocutaneous albinism coupled with defective platelet DG content, so we next examined platelet DG 5-HT and nucleotide content. 5-HT storage was almost abolished in DKO platelets, whereas [Rab32+/−; Rab38−/−] and [Rab32−/−;Rab38+/−] mice presented a 32% and a 40% decrease in platelet 5-HT content, respectively (Figure 3A). The 5-HT concentration was unaltered in single knockout and double heterozygous platelets (Figure 3A). Platelets contain 2 major compartments for adenosine triphosphate (ATP) and adenosine 5′-diphosphate (ADP), the cytosol, and the DGs. We measured the total nucleotide content of platelets using HPLC. As shown in Figure 3A, DKO mouse platelets displayed a total 75% decrease in ADP and 50% decrease in ATP content. The ATP/ADP ratio was therefore twice that of WT mice (mean ratio ± standard error of the mean [SEM]: 3.05 ± 0.06 for WT [n = 14] vs 6.33 ± 0.24 for DKO [n = 3]), in line with δ-SPD. Once again, [Rab32+/−;Rab38−/−] and [Rab32−/−;Rab38+/−] animals had an intermediate phenotype, with ∼30% and 45% decreases in platelet ADP content, respectively, and 17% and 20% decreases in platelet ATP content, respectively, whereas single KO had normal nucleotide content.
![Figure 3. Impact of Rab32 and/or Rab38 inactivation on platelet granule content. (A) Quantification of DG constituents in mouse whole platelet lysates: 5-HT measured by ELISA (left); ADP (middle) and ATP (right) measured by HPLC separation. Results are the mean ± SEM in at least 3 mice; **P < .01, ***P < .0001 using 1-way analysis of variance (ANOVA). (B) Quantification of α granule constituents in mouse whole platelet lysates: PF4 (left) measured by ELISA, P-selectin (middle) and fibrinogen (right) measured by western blotting (mean ± SEM, n = 3; not significant [ns] using Student t test). (C) Quantification of β-hexosaminidase in mouse sera as a measure of lysosome content (mean ± SEM, n = 5, *P < .05 using Student t test).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/15/10.1182_bloodadvances.2019031286/2/m_advances031286f3.png?Expires=1769190854&Signature=W4MPDZsZBJBmIXD7WlTQXfyOf2bpejU5B7FYbe6TLMEUaMCFbY6-7QoQU9~KaaP0qhKTperUPRTgBlG-MbPelxqPukfga9zbWfZhtDFG3PAh8gUqZoPctyN3BPi1dXzHAcuEtTP3o4-1Vfv5D9X5EVH-5DdTNAQA4xfKuN7rDNVrQuO0naGS4V1sN3oKVwcLwkTjMRBofGt54lCZ1PJF~xXZjibbHVe0g4N1ytitM6tV58zPfuNPUf1fsTIJb0XR4naWLNKU4u26Xcf3kkcF9caFBu9Ne6h-ryYMpN7AjNT3a~bBHj41ZIUOmRbQGL6i8bRPkWKGQczzdyKNNgQLmA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Impact of Rab32 and/or Rab38 inactivation on platelet granule content. (A) Quantification of DG constituents in mouse whole platelet lysates: 5-HT measured by ELISA (left); ADP (middle) and ATP (right) measured by HPLC separation. Results are the mean ± SEM in at least 3 mice; **P < .01, ***P < .0001 using 1-way analysis of variance (ANOVA). (B) Quantification of α granule constituents in mouse whole platelet lysates: PF4 (left) measured by ELISA, P-selectin (middle) and fibrinogen (right) measured by western blotting (mean ± SEM, n = 3; not significant [ns] using Student t test). (C) Quantification of β-hexosaminidase in mouse sera as a measure of lysosome content (mean ± SEM, n = 5, *P < .05 using Student t test).
Impact of Rab32 and/or Rab38 inactivation on platelet granule content. (A) Quantification of DG constituents in mouse whole platelet lysates: 5-HT measured by ELISA (left); ADP (middle) and ATP (right) measured by HPLC separation. Results are the mean ± SEM in at least 3 mice; **P < .01, ***P < .0001 using 1-way analysis of variance (ANOVA). (B) Quantification of α granule constituents in mouse whole platelet lysates: PF4 (left) measured by ELISA, P-selectin (middle) and fibrinogen (right) measured by western blotting (mean ± SEM, n = 3; not significant [ns] using Student t test). (C) Quantification of β-hexosaminidase in mouse sera as a measure of lysosome content (mean ± SEM, n = 5, *P < .05 using Student t test).
On the other hand, total platelet PF4, P-selectin and fibrinogen levels were normal, whatever the genotype of the mice (Figure 3B). VWF was likewise detected in DKO platelets by immunolabeling and found to be present in similar amounts as in WT platelets (data not shown), confirming that neither RAB protein plays a significant part in α granule biogenesis. Furthermore, β-hexosaminidase used as a marker of lysosomes was not decreased and was even slightly increased in DKO mice, indicating that lysosome biogenesis was not negatively impaired (Figure 3C). Finally, blood parameters were normal in the different combinations of genotypes (supplemental Table 1).
Altogether, we show that Rab32/38 DKO mice fully mimic HPS and that, unlike in the case of coat color and lung physiology, RAB32 and RAB38 seem to play equally redundant roles in the biogenesis of DGs.
RAB32 and RAB38 are required for dense granule formation
We next investigated whether the absence of DG contents was the result of the absence of DG compartments or reflected only their defective storage capacity. As seen on TEM images, DG formation in DKO mouse platelets was strongly impaired, with only a few granules being observed. Furthermore, these DGs presented an abnormal dense core having a very small, linear, or poorly condensed appearance (Figure 4A, right), in contrast to normal DGs having a typical “bull’s-eye” morphology35 (Figure 4A, left). No morphological defect in platelet DGs was observed in single knockout or in [Rab32+/−;Rab38−/−] or [Rab32−/−;Rab38+/−] mice (data not shown). Because empty or immature granules may not be identified by TEM, mepacrine uptake was quantified as a measure of DG amount.36,37 Mepacrine uptake was decreased by 45% in resting DKO platelets compared with WT (Figure 4B). As a control, we observed that thrombin preactivated platelets incorporated minor amount of mepacrine, irrespective of their genotype (Figure 4B). Altogether, these data indicate that DKO platelets combine a decreased DG number and an impaired granule content. We then used focused ion beam (FIB)/scanning electron microscopy analysis and 3-dimensional (3D) reconstruction38 to determine the exact number of DGs per platelet, even if empty. We counted 17.6 ± 1.4 DG (mean ± SEM) in WT platelets, consistent with our previous observations of a higher number of DGs in mouse than in human platelets (A.E., personal observations). A 70% decrease in the total number of DGs in DKO platelets (5.2 ± 0.5 DG/DKO platelet) was observed (Figure 4C). These morphological defects seemed to be restricted to DGs because α granules had a normal appearance (supplemental Figure 4A). A similar deficiency in DG biogenesis was already observed at the MK stage in the bone marrow by TEM analysis (supplemental Figure 4B). However, MVB were morphologically similar in DKO and WT MKs differentiated in vitro from Lin− cells (Figure 4D), suggesting that RAB32 and RAB38 intervene in DG biogenesis downstream of MVB compartmentalization. These observations indicate that RAB32 and RAB38 are both required to initiate the formation of DG compartments and for their maturation in terms of storage contents.
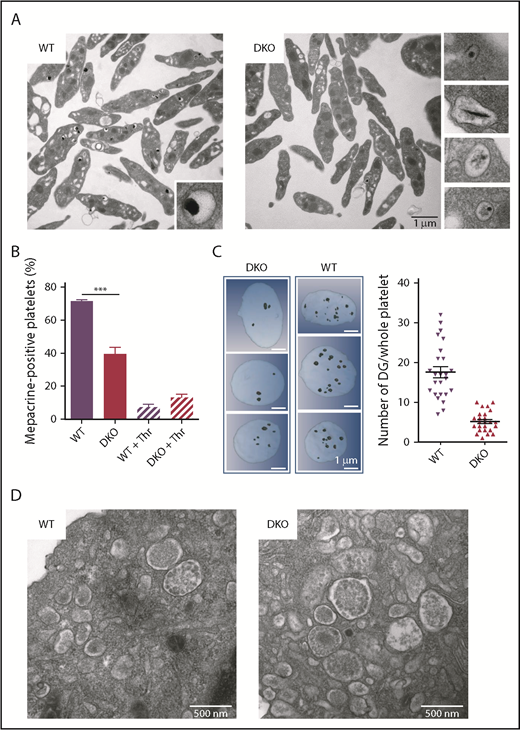
Decreased number and abnormal DGs in Rab32/Rab38 DKO mouse platelets. (A) TEM images showing the ultrastructure of WT and DKO platelets. Right, the close-up views illustrate the various abnormal DG morphologies in DKO mice. Images are representative of at least 3 independent platelet preparations. Scale bar, 1 µm. (B) Evaluation of mepacrine uptake by WT and DKO platelets. Bar graph represents the percentage of mepacrine-positive resting or degranulated platelets (prestimulation with 1 U/mL thrombin) following incubation with 10 µM mepacrine for 30 minutes (n = 3 independent experiments). ***P < .001 using 1-way ANOVA. (C) FIB/scanning electron microscopy images and 3D reconstructions of whole platelets. (Right panel) Dark spots represent DGs reconstructed by drawing the granule membranes on each slice. Note the heterogeneity of the size of the DGs in both genotypes. (Left panel) Quantification of the number of DGs per platelet using 3D FIB/scanning electron microscopy reconstructions (n = 25). (D) TEM images showing the presence of normal MVBs in DKO mouse MKs differentiated in culture from Lin− cells compared with WT. Images are representative of at least 20 MKs. Scale bars, 500 nm.
Decreased number and abnormal DGs in Rab32/Rab38 DKO mouse platelets. (A) TEM images showing the ultrastructure of WT and DKO platelets. Right, the close-up views illustrate the various abnormal DG morphologies in DKO mice. Images are representative of at least 3 independent platelet preparations. Scale bar, 1 µm. (B) Evaluation of mepacrine uptake by WT and DKO platelets. Bar graph represents the percentage of mepacrine-positive resting or degranulated platelets (prestimulation with 1 U/mL thrombin) following incubation with 10 µM mepacrine for 30 minutes (n = 3 independent experiments). ***P < .001 using 1-way ANOVA. (C) FIB/scanning electron microscopy images and 3D reconstructions of whole platelets. (Right panel) Dark spots represent DGs reconstructed by drawing the granule membranes on each slice. Note the heterogeneity of the size of the DGs in both genotypes. (Left panel) Quantification of the number of DGs per platelet using 3D FIB/scanning electron microscopy reconstructions (n = 25). (D) TEM images showing the presence of normal MVBs in DKO mouse MKs differentiated in culture from Lin− cells compared with WT. Images are representative of at least 20 MKs. Scale bars, 500 nm.
Severely impaired hemostasis and thrombosis in Rab32/Rab38-deficient mice
The main function of the secretion of DG content is to recruit other circulating platelets to the site of injury and amplify platelet activation. As patients with DG deficiencies have a compromised hemostatic capacity,1,7,39 we evaluated hemostasis and thrombosis. Hemostasis evaluated in a tail bleeding assay appeared unaffected in Rab32 or Rab38 single knockout mice, in accordance with data in cht mouse.21 In contrast, a marked increase in bleeding time was recorded in DKO animals with no cessation after 30 minutes (Figure 5A). No intermediate phenotype was observed in [Rab32−/−;Rab38+/−] or [Rab32+/−;Rab38−/−] mice, indicating that their partial 5-HT and ADP content was sufficient to prevent excessive bleeding.
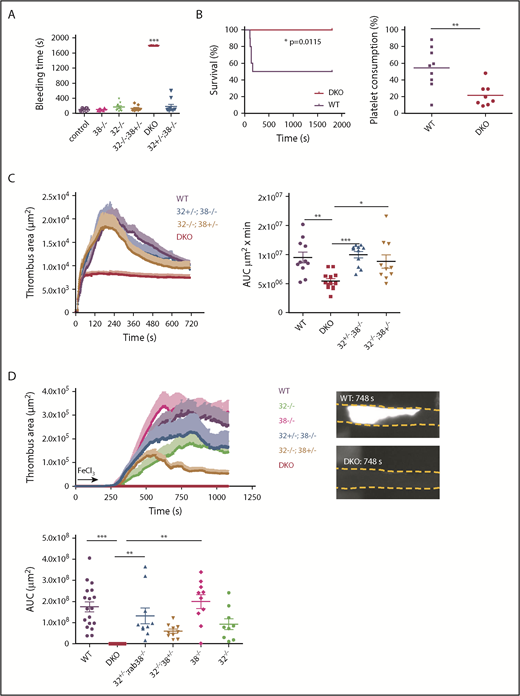
Defective hemostasis and thrombus growth in DKO mice. (A) Bleeding time measured as the time to the first cessation of bleeding. For DKO mice, bleeding was manually stopped at 1800 seconds; n = 10-12 mice, scatter plot with mean ± SEM, each dot corresponds to an individual mouse, ***P < .0001 with Kruskal-Wallis test and Dunn’s multiple comparison test. (B) TF-induced thromboembolism experiments. (Left panel) Percentage survival following tissue factor injection, n = 10 mice; statistics using a log-rank (Mantel-Cox) test. (Right panel) Individual and mean percentage of platelet consumption, n = 8-9 mice; **P < .01 using Student t test. (C) Laser-induced mesenteric arteriole injury. (Left panel) Curves representing the mean ± SEM thrombus area at each time point of 10 to 13 vessels observed from 7 to 9 mice. (Right panel) Bar graph of the area under the curve (AUC) of the individual curves corresponding to the graph shown in left. **P < .001, Kruskal-Wallis test. (D) FeCl3-induced carotid artery injury. FeCl3 was applied to the lateral side of the carotid for 150 seconds (arrow), after which thrombus growth was visualized from the top. (Upper left panel) Curves representing the mean ± SEM thrombus area at each time point, n = 10 mice. (Upper right panel) Representative top view showing the fluorescent platelet accumulation at the peak thrombus formation in WT mice (748 seconds) (upper image) and the absence of platelet accumulation in DKO mice (lower image). (Lower panel) Scatter bar graph of the AUC; **P < .001, ***P < .0001 using Kruskal-Wallis test and Dunn’s posttests.
Defective hemostasis and thrombus growth in DKO mice. (A) Bleeding time measured as the time to the first cessation of bleeding. For DKO mice, bleeding was manually stopped at 1800 seconds; n = 10-12 mice, scatter plot with mean ± SEM, each dot corresponds to an individual mouse, ***P < .0001 with Kruskal-Wallis test and Dunn’s multiple comparison test. (B) TF-induced thromboembolism experiments. (Left panel) Percentage survival following tissue factor injection, n = 10 mice; statistics using a log-rank (Mantel-Cox) test. (Right panel) Individual and mean percentage of platelet consumption, n = 8-9 mice; **P < .01 using Student t test. (C) Laser-induced mesenteric arteriole injury. (Left panel) Curves representing the mean ± SEM thrombus area at each time point of 10 to 13 vessels observed from 7 to 9 mice. (Right panel) Bar graph of the area under the curve (AUC) of the individual curves corresponding to the graph shown in left. **P < .001, Kruskal-Wallis test. (D) FeCl3-induced carotid artery injury. FeCl3 was applied to the lateral side of the carotid for 150 seconds (arrow), after which thrombus growth was visualized from the top. (Upper left panel) Curves representing the mean ± SEM thrombus area at each time point, n = 10 mice. (Upper right panel) Representative top view showing the fluorescent platelet accumulation at the peak thrombus formation in WT mice (748 seconds) (upper image) and the absence of platelet accumulation in DKO mice (lower image). (Lower panel) Scatter bar graph of the AUC; **P < .001, ***P < .0001 using Kruskal-Wallis test and Dunn’s posttests.
We next examined the Rab knockouts in in vivo thrombosis models. Mice inactivated for both Rab32 and Rab38 were resistant to thromboembolism induced by tissue factor administration (Figure 5B), in accordance with the important role of DG content in this model.32 Thrombosis resulting from vessel injury was also severely impaired in DKO mice, as observed in a model of localized deep laser-induced injury of the mesenteric arteriole vessel wall33 (Figure 5C). Finally, we also asked how DKO may behave in a model of FeCl3-induced injury in the carotid, generating a more severe lesion and hence a much larger thrombus.40 FeCl3 was applicated laterally, allowing to visualize platelet accumulation and thrombus growth into the vessel lumen34 (Figure 5D). There, thrombosis was profoundly impaired with absolutely no thrombus growth in DKO mice (Figure 5D, red line), whereas only the intermediate genotypes [Rab32−/−; Rab38+/−] and Rab32−/− also showed a tendency, though not significant compared with the WT, to reduced thrombus size. No difference was observed between WT and single KO mice. The mean ± SEM thrombus area at 600 seconds was 240 × 103 ± 42 × 103, 77 ± 17, 117 × 103, 326 × 103, 196 × 103 ± 56 × 103, and 114 × 103 ± 2 × 103 µm2 for WT, DKO, Rab32−/−, Rab38−/− [Rab32+/−;Rab38−/−], and [Rab32−/−;Rab38+/−] mice, respectively; only the differences between DKO and WT, [Rab32+/−;Rab38−/−] and Rab38−/− were significant (P < .05 using Kruskal-Wallis test with Dunn’s posttest).
Impaired platelet functions
To dissect the defective steps in thrombus formation, we next examined platelet aggregation under flow using hirudin-anticoagulated blood perfused through collagen-coated glass capillaries at an arterial shear rate of 1500 seconds−1. Platelets from WT mice adhered and formed compact aggregates as expected. By contrast, DKO mice formed only a layer over the surface with no growth in height (Figure 6A), reminiscent of the thrombosis behavior. This suggested that the initial phase of platelet attachment was normal, but followed by a major defect in thrombus growth, raising the hypothesis of a defect in αIIbβ3 integrin activation in the DKO mice resulting from defective DG content release. Similar results were observed by performing in vitro platelet aggregation. Although ADP response was fully normal, as expected because it is not dependent of DG secretion when using washed platelets (not shown), the responses to collagen or thrombin were decreased at low agonist concentrations and overcome when increasing concentrations (Figure 6B).
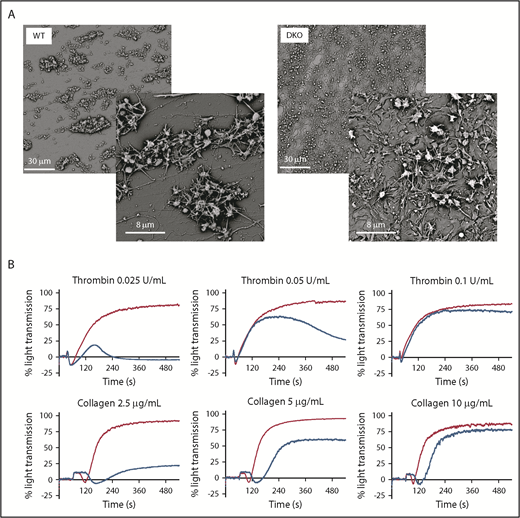
Defective platelet aggregate formation in DKO mice. (A) Whole blood anticoagulated with hirudin was perfused through glass capillaries coated with collagen I fibers for 5 minutes at a shear rate of 1500 seconds−1. Scanning electron microscopy images that are representative of at least 3 independent experiments. (B) Aggregation tracings of WT (red) and DKO (blue) washed platelets stimulated with various concentrations of agonists as indicated. Representative of at least 4 independent experiments.
Defective platelet aggregate formation in DKO mice. (A) Whole blood anticoagulated with hirudin was perfused through glass capillaries coated with collagen I fibers for 5 minutes at a shear rate of 1500 seconds−1. Scanning electron microscopy images that are representative of at least 3 independent experiments. (B) Aggregation tracings of WT (red) and DKO (blue) washed platelets stimulated with various concentrations of agonists as indicated. Representative of at least 4 independent experiments.
To further evaluate platelet functions, we measured JonA-PE labeling as a marker of αIIbβ3 integrin activation in thrombin-stimulated platelets over 300 s at concentrations ranging from 0.05 to 1 U/mL. We observed strongly reduced JonA-PE labeling at all-time points and concentrations in platelets from DKO mice compared with WT mice (Figure 7A). We next established that integrin activation could be rescued by exogenously added ADP at all-time points and for both thrombin concentrations (Figure 7B) (P < .001 between DKO and DKO+ADP). Under the same activation conditions, no critical defect in platelet P-selectin exposure was observed in DKO platelets (Figure 7C), and addition of ADP did not modify the response whatever the genotype (Figure 7D), suggesting that α granule release is not affected by DG defects as also indicated by the normal PF4 release (supplemental Figure 5). By contrast, lysosome exocytosis evaluated by LAMP1 externalization was found to be significantly decreased in DKO platelets compared with WT (Figure 7E) and only partially recovered by exogenously added ADP (Figure 7F).
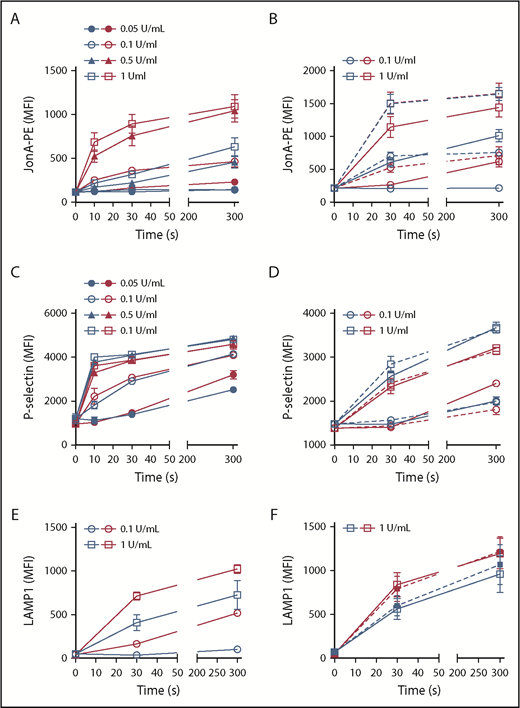
Thrombin-induced integrin αIIbβ3 activation, P-selectin exposure and LAMP1 exposure. (A-F) Washed platelets were stimulated with increasing thrombin concentrations and periods of time (numbers in graph legend indicate the final thrombin concentration) and analyzed by flow cytometry. Red symbols, WT; blue symbols, DKO. All graphs represent mean ± SEM for n = 3 independent experiments. Statistical analyses were performed using 2-way ANOVA, Bonferroni posttest, to compare differences between WT and DKO in similar stimulation conditions. (A) αIIbβ3 integrin activation as measured by JonA-PE labeling and represented as mean fluorescence intensity. At each time point and for all thrombin concentrations, DKO platelet JonA-PE labelings were significantly different from WT ones (P < .001), except after stimulation with 0.05 U/mL thrombin for 10 seconds, where there was no difference. (B) αIIbβ3 integrin activation in the presence (dotted lines) or absence (straight lines) of ADP (10 µM). The presence of ADP significantly increases JonA-PE labeling in DKO mice compared with absence of ADP for all conditions (P < .001). (C) P-selectin exposure at the surface of platelets. No significant difference was observed between WT and DKO platelets, except after stimulation with 0.05 U/mL thrombin for 300 seconds (P < .01). (D) P-selectin exposure in the presence (dotted lines) or absence (straight lines) of ADP (10 µM). The presence of ADP did not increase P-selectin exposure. No significant difference between WT and DKO platelets. (E, left) LAMP1 platelet surface exposure. LAMP1 exposure is significantly decreased in DKO platelets compared with WT (P < .001) except for 0.1 U/mL thrombin at 30 seconds. (F) Exogenous addition of 10 µM ADP (dotted lines) only partially rescued DKO platelet LAMP1 exposure in response to 1 U/mL thrombin.
Thrombin-induced integrin αIIbβ3 activation, P-selectin exposure and LAMP1 exposure. (A-F) Washed platelets were stimulated with increasing thrombin concentrations and periods of time (numbers in graph legend indicate the final thrombin concentration) and analyzed by flow cytometry. Red symbols, WT; blue symbols, DKO. All graphs represent mean ± SEM for n = 3 independent experiments. Statistical analyses were performed using 2-way ANOVA, Bonferroni posttest, to compare differences between WT and DKO in similar stimulation conditions. (A) αIIbβ3 integrin activation as measured by JonA-PE labeling and represented as mean fluorescence intensity. At each time point and for all thrombin concentrations, DKO platelet JonA-PE labelings were significantly different from WT ones (P < .001), except after stimulation with 0.05 U/mL thrombin for 10 seconds, where there was no difference. (B) αIIbβ3 integrin activation in the presence (dotted lines) or absence (straight lines) of ADP (10 µM). The presence of ADP significantly increases JonA-PE labeling in DKO mice compared with absence of ADP for all conditions (P < .001). (C) P-selectin exposure at the surface of platelets. No significant difference was observed between WT and DKO platelets, except after stimulation with 0.05 U/mL thrombin for 300 seconds (P < .01). (D) P-selectin exposure in the presence (dotted lines) or absence (straight lines) of ADP (10 µM). The presence of ADP did not increase P-selectin exposure. No significant difference between WT and DKO platelets. (E, left) LAMP1 platelet surface exposure. LAMP1 exposure is significantly decreased in DKO platelets compared with WT (P < .001) except for 0.1 U/mL thrombin at 30 seconds. (F) Exogenous addition of 10 µM ADP (dotted lines) only partially rescued DKO platelet LAMP1 exposure in response to 1 U/mL thrombin.
Discussion
The spontaneous Rab38 null mutation of the fawn-hooded rat, resulting in pigment dilution and defective DG biogenesis, was the first animal model of HPS available. Later, a Rab38 mutation in cht mouse was described, without DG anomalies.21 We provide an explanation for these discrepancies by showing that rat platelets do not express the RAB32 protein or mRNA. In contrast, human and mouse platelets express both RAB38 and RAB32. The absence of RAB32 in rat platelets has never been documented to date. One previous publication mentioned the presence in rat platelets of a putative immunoreactive long form of RAB32 having a molecular mass of 52 kDa instead of 25 kDa.41 This would be difficult to reconcile with the fact that RAB32 and RAB38 are paralogs.14,15 Using 2 different RAB32 antibodies, we found no evidence for this long form of the protein. Hence, DG biogenesis in rat platelets would appear to depend solely on RAB38, unlike in mouse and human platelets.
To investigate in vivo the respective roles of RAB32 and RAB38 in DG biogenesis and platelet functions in species expressing both proteins (in the platelet lineage), we generated mouse lines deficient in RAB32, RAB38, and both proteins. Our data support a full redundancy between the 2 RABs for DG biogenesis, which was unexpected from the reported higher expression of RAB32 over RAB38.42 This was not the case for other LROs because we observed that RAB38 plays a more important role in the biogenesis of melanosomes and multilamellar bodies compared with RAB32, in accordance with studies using cultured melanocytes9,11 and previous data in the cht mice and fawn-hooded rats, which both present pigmentation and lung defects.20,22,43
Up to now, 10 genes have been identified as causative for HPS in humans, whose mutation predicts the severity and diversity of symptoms.8,16,44,45 Yet, there is still a number of patients presenting with HPS of unknown origin. The identification of most of the genes involved in human HPS have been greatly facilitated by the availability of HPS mouse disease models.16,46 Our observations that, contrary to the rat, RAB38 can be compensated by RAB32 for DG biogenesis in mouse and probably also in human might explain why no mutation in the Rab38 gene has yet been reported in patients with HPS.47
Known mutations in HPS affect proteins involved in multisubunit protein complexes. Among these, the BLOC3, composed of HPS1 and HPS4 subunits, works as a guanine exchange factor for RAB32 and RAB38.25,48 It is thus tempting to suppose that the DKO mouse model could correspond to the HPS1 and HPS4 form of the human disease. HPS1 and HPS4 are typically characterized by varying oculocutaneous albinism, bleeding problems from defective DG biogenesis, and often lethal progressive lung fibrosis resulting from defective alveolar type II cells,49 which closely resembles the reported DKO phenotype. However, spontaneous mouse mutants for HPS1 (Hps1ep/ep) and HPS4 (Hps4le/le), do not exactly phenotypically mimic Rab32/Rab38 DKO mice. Although both Hps1ep/ep and Hps4le/le mutant mice display an important DG storage defect, they surprisingly have very mild coat color dilution almost exclusively visible on the ears and tail extremity.46,50 The observed discrepancy between Hps mutant mice and our DKO mice could suggest that RABs play roles independent of BLOC-3 that may account for the more severe pigmentation defect we observed. This is a likely possibility because, in melanocytes, RAB32 and RAB38 also interact with BLOC-2, AP-3, and AP-1, all proteins involved in the transport of cargos to melanosomes.9 In addition, we cannot exclude additional roles for RAB32 and RAB38, independent of protein trafficking.23 Of note, RAB32 has been described as an A-kinase anchoring protein acting at the surface of human mitochondria51 and xenopus melanosome.52 These differences indicate that care must be taken not to consider all HPS mouse models as identical. They also point to the utility of our DKO mice as a model mimicking severe HPS, which would be especially useful to study the mechanisms leading to the biogenesis of other LROs or to evaluate the effect of treatments.
Thus, the severe hemostasis and thrombosis phenotypes in RAB32- and RAB38-deficient mice appear in line with the DG biogenesis defect. Interestingly, we observed that in DKO mice, thrombi were totally unable to grow in height, although platelets were able to adhere to the site of lesion. This was confirmed in vitro where platelets from DKO mice did not form compact aggregates on collagen under flow conditions. Accordingly, we observed a strong decrease in αIIbβ3 integrin activation in DKO platelets, explaining the platelet behavior. The defect in integrin activation was fully recovered by exogenously added ADP, suggesting that it results essentially from the absence of amplification responses by DG-secreted agonists. In addition, we observed that lysosome secretion was affected in DKO, whereas no clear defect in α granule secretion was evidenced. These data are in accordance with works from Meng et al, who evaluated light-ear mice (Hps4le/le), showing defective platelet LAMP1 surface exposure but normal P-selectin exposure at thrombin concentration ≥0.05 U/mL.50 This indicates that α-granules exocytosis at these thrombin concentrations is independent of RAB32/38. LAMP1 exposure in DKO mice was hardly compensated by ADP suggesting that RAB32 and RAB38 are involved in their secretion machinery, or alternatively that some LAMP1 is also normally expressed at the DG membrane, and thus decreased in DKO-activated platelets.
In conclusion, we showed that unlike in the rat, RAB32 and RAB38 proteins are fully redundant and specific for DG biogenesis in the mouse. Deletion of both Rab32 and Rab38 genes reproduces a mouse HPS model with hypopigmentation of eyes and hair, associated with lung abnormalities and decreased DGs in platelets that are totally devoid of 5-HT and lack of nucleotides, mimicking severe human HPS disease. This animal model will be a useful tool to study the role of these 2 related RABs in other LROs and to address the role of DG content in the increasing diversity of platelet-mediated processes.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Juliette Mulvihill for reviewing the English of the manuscript.
This study was supported by the Association de Recherche et Développement en Médecine et Santé Publique and the European Union through the European Regional Development Fund. A.A. was supported by a French government fellowship.
Authorship
Contribution: A.A., F.L., C.G., and C.L. designed the research; A.A., J.W., J.B., C.Z., A.E., S.M., and C.B. conducted the experiments; M.F., B.H., and P.H.M. supervised experiments; and C.L., F.L., B.H., P.H.M., and C.G. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Catherine Léon, UMR_S1255 INSERM–Université de Strasbourg, Etablissement Français du Sang, 10 rue Spielmann, B.P. N 36, 67065 Strasbourg Cedex, France; e-mail: catherine.leon@efs.sante.fr.

