

Key Points
mRNA decapping gene EDC4 is a novel fusion partner of MLL in AML.
Genes functioning in mRNA decapping may compose a distinct group of MLL fusion partners that links MLL function with mRNA decapping in AML.
Introduction
Translocations involving MLL (aka KMT2A) located on chromosome 11q23 occur in acute myeloid leukemia (AML) and lymphoblastic leukemia. In AML, they generally confer an adverse prognosis, unless the MLLT3 (aka AF9) gene is involved.1 More than 130 different translocation partner genes (TPGs) have been identified, forming the MLL recombinome.2
Recently, the scavenger messenger RNA (mRNA) decapping enzyme DCPS has been identified to be required for survival of AML cells, but not normal hematopoietic cells, and a DCPS inhibitor showed antileukemic activity.3,4 DCPS is also 1 of 2 genes (the other being DCP1A) involved in mRNA decapping and having been described as TPG of MLL in single leukemia cases.5-7
Here, we describe a novel MLL fusion with another mRNA decapping component, ie, the enhancer of mRNA decapping 4 gene (EDC4; also known as GE1 or HEDLS), in AML. EDC4 is part of a multiprotein complex in the cytoplasmic P bodies. It is required for the interaction of DCP2 and its cofactor DCP1 to remove the 5′-cap that is generated during transcription and crucial for protection from degradation.8,9 As recently shown, EDC4 has an additional, nuclear activity in DNA double strand break repair; its deficiency induces a phenotype comparable to BRCA1 deficiency.10 Mutations in EDC4 occur in various cancers, but rearrangements have not yet been described.11
Methods
Sample processing
Samples, collected at various time points as indicated in supplemental Figure 1, were enriched for mononuclear cells (MNCs) via Ficoll-Hypaque and depleted from CD3+ cells via autoMACS (Miltenyi Biotec), as previously described.12 CD3+ cells served as germline control. The patient provided written informed consent for the research use of the clinical data and biomaterial in accordance to the Declaration of Helsinki.
RNA sequencing
RNA sequencing libraries were prepared using the NEBNext Ultra RNA Library Prep Kit (New England Biolabs); sequencing was performed on a HiSeq 2500 system (Illumina). Fusion transcripts were detected by Genomon-fusion pipeline (https://github.com/Genomon-Project/), as previously described.13 Reference transcript sequences were for MLL NM_005933.1 and for EDC4 NM_014329.4. The primers used for polymerase chain reaction and sequencing of reverse-transcribed RNA had the following the sequences: MLL, ccagctggaaaattggtgtt; EDC4, gatgatcctgcgaaagtggt.
Animal model
NOD.Cg-PrkdcscidIl2rgtm1Sug/JicTac (NOG) were obtained from Taconic, Denmark. At 6 to 8 weeks of age, mice received 3 × 106 primary blasts from peripheral blood to establish patient-derived xenograft (PDX) models. Cells were injected into the tibia, and animals were observed for clinical signs of leukemia (hind limb paresis, weight loss, bad overall condition). When moribund, mice were euthanized and human leukemic cells harvested from bone marrow (BM) and spleen. Subsequently, 3 × 106 human leukemic cells were implanted into naive recipient mice. The study was carried out in accordance with the recommendations by the Society of Laboratory Animal Science. The animal experiments were approved by the regional council (Regierungspräsidium Freiburg, ref. 35, permit no. G-12/86).
Exome sequencing
The SureSelect Human All Exon v5 Kit (Agilent Technologies) was used for exome capturing from genomic DNA, and sequencing was performed on a HiSeq 2500 system (Illumina). Sequence alignment and mutation calling were performed using the Genomon pipeline (https://github.com/Genomon-Project/), as previously described,13,14 with minor modifications. Candidate mutations with (1) Fisher’s exact test, P < .01; and (2) a variant allele frequency in matched normal samples <0.2 was adopted and further filtered by excluding (1) known variants listed in the 1000 Genomes Project (May 2011 release), NCBI dbSNP build 131, National Heart, Lung, and Blood Institute Exome Sequencing Project 5400, the Human Genome Variation Database (October 2013 release), or an in-house single-nucleotide polymorphism database; and (2) variants present in unidirectional reads only.
Single-cell RNA sequencing
Single-cell RNA sequencing was performed on the MNC fraction of an available peripheral blood sample collected shortly before time point t2 using the chromium system (10X Genomics). Approximately 8000 cells were loaded on the chip, and library preparation was done according to the manufacturer´s instructions. The library was sequenced on 1 lane 26+74 bp paired-end on a HiSeq4000 machine (Illumina). Reads were processed with cell ranger software using an annotation that contained protein coding genes only to yield a unique molecular identifier (UMI) count table that was then further processed with Seurat.15 Low-quality cells were excluded from the analysis and regression was done using UMI count and cell-cycle stage. The normalized data were clustered using 7 PCA components as determined by an elbow plot. Marker genes were calculated by comparing a single cluster within the same cell type to all other clusters of the same cell type.
Results and discussion
Identification of EDC4 as a fusion partner of MLL
A 55-year-old patient presented with a myelodysplastic syndrome (MDS; timepoint [t] −1) that progressed to AML (t0). The patient refused treatment beyond supportive care. Six months later, blast expansion occurred (t1). The patient received 5 courses of decitabine (t2), followed by 3 months of hydroxyurea (t3) (supplemental Figure 1). Cytogenetics at t0 revealed a previously undescribed translocation involving the chromosomal location of MLL: 46,XX,t(11;16)(q23;q12)[12]/46,XX[8]. The MLL rearrangement was detectable via interphase fluorescence in situ hybridization in 84 of 100 cells.
RNA sequencing identified the fusion partner to be EDC4 on chromosome 16q22 (Figure 1A). The translocation led to the in-frame fusion of MLL exon 13 to EDC4 exon 6, linked by 19 nucleotides of EDC4 intron 5. The predicted amino acid sequence of this linker was ALNTLLR (Figure 1A). MLL-EDC4 was present at diagnosis and during the course of the AML, but not during MDS (Figure 1B).
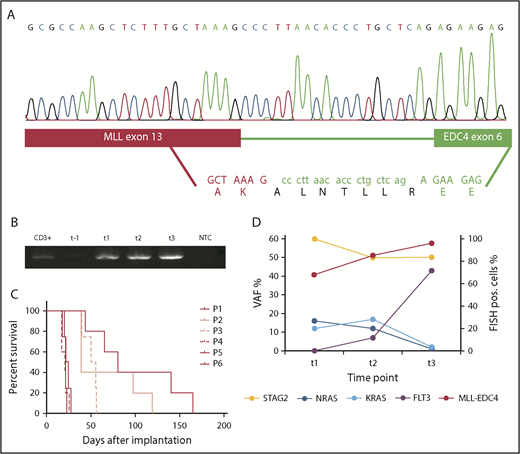
Identification of the MLL-EDC4 fusion and accompanying mutations. (A) In-frame fusion of MLL exon 13 (red bar) to EDC4 exon 6 (green bar) linked by 19 nucleotides from EDC4 intron 5, resulting in the predicted amino acid sequence ALNTLLR inserted between MLL p.K1565 and EDC4 p.E215. (B) Gel electrophoresis of the polymerase chain reaction on reverse-transcribed MLL-EDC4 mRNA at the indicated time points. (C) Patient-derived xenograft model of the MLL-EDC4+ AML. Survival of NOG mice after injection of primary patient blasts (P1) collected at t2 and after serial transplantation (P2-P6). (D) VAFs (derived from exome sequencing) of the mutations in STAG2 (NM_006603: c.463-1G>C), NRAS (p.G12C), KRAS (p.G13D), and FLT3 (p.D835V) at t1, t2, and t3, displaying the presence of the NRAS and KRAS mutations before the start of decitabine and the increase of the FLT3-mutated clone toward t3. FISH, fluorescence in situ hybridization; NTC, no template control; P, passage; pos., positive; VAF, variant allele frequency.
Identification of the MLL-EDC4 fusion and accompanying mutations. (A) In-frame fusion of MLL exon 13 (red bar) to EDC4 exon 6 (green bar) linked by 19 nucleotides from EDC4 intron 5, resulting in the predicted amino acid sequence ALNTLLR inserted between MLL p.K1565 and EDC4 p.E215. (B) Gel electrophoresis of the polymerase chain reaction on reverse-transcribed MLL-EDC4 mRNA at the indicated time points. (C) Patient-derived xenograft model of the MLL-EDC4+ AML. Survival of NOG mice after injection of primary patient blasts (P1) collected at t2 and after serial transplantation (P2-P6). (D) VAFs (derived from exome sequencing) of the mutations in STAG2 (NM_006603: c.463-1G>C), NRAS (p.G12C), KRAS (p.G13D), and FLT3 (p.D835V) at t1, t2, and t3, displaying the presence of the NRAS and KRAS mutations before the start of decitabine and the increase of the FLT3-mutated clone toward t3. FISH, fluorescence in situ hybridization; NTC, no template control; P, passage; pos., positive; VAF, variant allele frequency.
The MLL-EDC4 protein has a predicted molecular weight of >200 kD and contains most of EDC4, including the α-helical domain at the C terminus, which mediates binding to DCP2 and the exonuclease XRN1. However, it lacks ∼80% of the WD40 domain of EDC4, which is required for the interaction with DCP1.9,16
MLL-EDC4 stands out because of its fusion of MLL exon 13; <0.5% of MLL-rearranged cases harbor a breakpoint in exon 13 or farther downstream.2 However, as in most MLL rearrangements, the plant homology domain (PHD)/bromodomain (BD) function is likely impaired in MLL-EDC4 (PHD1 and PHD2 are retained; PHD3, BD, and PHD4 are missing). An intact PHD/BD domain is critical for determining whether MLL acts as transcriptional activator or repressor; moreover, PHD2 and PHD3 are important for maintaining the stability of MLL.2 Thus, the MLL-EDC4 fusion could lead to perturbation of epigenetic gene regulation by MLL. Moreover, it is tempting to speculate that MLL-EDC4 may lead to EDC4 haploinsufficiency, as the heterozygous knockout of EDC4 in a mouse model induces a myeloid phenotype featuring increased granulocyte counts.17
To study the potential oncogenic function of MLL-EDC4, we attempted to express it in mammalian cells; however, we were not able to successfully clone the full-length MLL-EDC4 because of its high molecular weight. A truncated construct including only the conserved WD40 region of EDC4 fused to MLL did not result in transformation of normal BM progenitor cells, whereas MLL-AF9 and E2A-PBX1 did (data not shown). Self-renewal capacity of the MLL-EDC4+ AML could be demonstrated through serial transplantation using a PDX model established from primary patient blasts in NOG mice (Figure 1C).
Identification of accompanying gene mutations and clonal evolution
To identify cooperating mutations and characterize clonal evolution, we performed exome sequencing. We identified a STAG2 mutation as potential founder mutation during MDS (t1), which persisted throughout the disease course (Figure 1D). At the time of acquisition of MLL-EDC4 (t1), 1 mutation each in KRAS (p.G13D) and NRAS (p.G12C) was detectable. Toward the terminal phase (t3), these mutations disappeared and a FLT3 mutation (p.D835V) emerged (Figure 1D).
Mutations in STAG2 impair sister chromatid cohesion after DNA replication and thereby segregation of chromosomes into daughter cells.18 Aberrant chromosome segregation has also been associated with alterations in the splicing machinery,19 and the recent report on DCPS function in AML indicated a direct link between mRNA decapping and splicing regulation.3 Hence, one may speculate that it was the combined effect of mutated STAG2 and MLL-EDC4 on chromatid cohesion, splicing, and mRNA degradation that conferred a clonal advantage.
NRAS or KRAS mutations each occur in ∼20% and FLT3 mutations in 10% to 15% of patients with MLL-rearranged AML.20 Similar mutation patterns of MLL-EDC4+ and other MLL-rearranged AMLs suggest that they share biological features. Of note, the previously described MLL-DCPS+ AML harbored no mutations in FLT3 or NRAS (K. H. Metzeler, Ludwig-Maximilians University, Munich, Germany, written communication, 12 October 2018).4
Characterization of the phenotype of the MLL-EDC4+ AML
As assessed by flow cytometry of the BM, the blasts at t0 were CD117+ with partial coexpression of CD33 (65%), CD34 (65%) and MPO (20%).
To further characterize the phenotype associated with MLL-EDC4, we performed single-cell RNA sequencing on CD3+ cell-depleted MNCs. Of the cells, 86% were identified to be leukemic blasts. The remaining cells were monocytes or residual erythroblasts (Figure 2A). Focusing on a set of candidate genes with potential biological relevance,21,22 the blasts featured strong coexpression of MYB, HOXA9, and HOXA10 and lower expression of EDC4, HOXA4, and HOXA5. In contrast, HOXB5, HOXB6 and HOXC4 were not detected (Figure 2B). Because one consequence of MLL-EDC4 could be impaired DNA repair resulting from EDC4 deficiency,10 it appears noteworthy that the AML displayed marked upregulation of PARP1 and genes of the MRN complex (ie, MRE11, RAD50, and NBN) (data not shown).
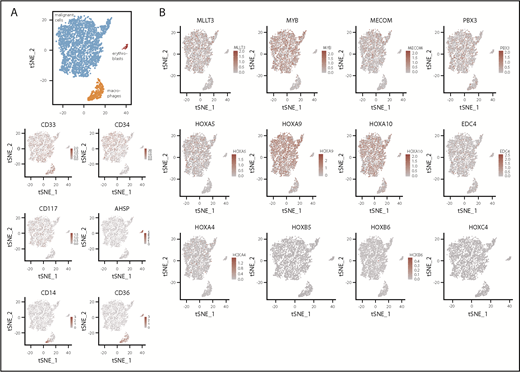
Single-cell RNA expression analysis. (A) tSNE plot with coloring according to cell type as determined by marker gene expression and as depicted in the heatmaps. Expression level is provided as UMI counts. (B) Expression of selected genes across all cells. tSNE, t-distributed stochastic neighbor embedding.
Single-cell RNA expression analysis. (A) tSNE plot with coloring according to cell type as determined by marker gene expression and as depicted in the heatmaps. Expression level is provided as UMI counts. (B) Expression of selected genes across all cells. tSNE, t-distributed stochastic neighbor embedding.
We next determined clusters of cells based on the differences in gene expression (clusters 0-7) and identified marker genes for a given cluster (supplemental Figure 2). These analyses confirmed the phenotypical heterogeneity among the leukemic blasts and identified the transcription factor HEMGN (aka EDAG, which regulates myelopoiesis)23 and the GTPase GIMAP7 to be enriched among CD34+ cells.
In conclusion, the fusion of MLL with EDC4 characterized here complements the recently established role of mRNA decapping in AML. In conjunction with the previously described MLL fusions to DCP1A and DCPS, our findings raise the possibility that mRNA decapping enzymes compose a distinct group of recurrent TPGs of MLL, emphasizing a possible link between MLL functions and mRNA decapping. In our case, MLL-EDC4 likely cooperated with aberrant chromatid cohesion and proliferation signaling; self-renewal capacity was demonstrated in a PDX model.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank J. Surrallés Calonge (Universitat Autònoma de Barcelona, Bellaterra, Spain) for insightful discussions on EDC4, and C. Meyer (University of Frankfurt, Frankfurt, Germany) and K. H. Metzeler (Ludwig-Maximilians University, Munich, Germany) for background information on the MLL-DCPS positive AML case.
This research was supported by the Translational Research Training in Hematology of the European Hematology Association and American Society of Hematology (H.B.); the German Research Foundation (SPP 1463 LU 429/8-2 [M.L.]; CRC992 MEDEP C04 [M.L.]; FOR 2674 BE 6461/1-1 [H.B.], LU 429/16-1 [M.L.], MA 7792/1-1 [J.-P.M.], RI 1283/15-1 [K.R.]; and DU 1287/2-1 [J.D.-A.]); and German Cancer Aid (111210) (H.B.).
Authorship
Contribution: H.B. undertook conception and design; patient care; specimen acquisition; data acquisition, analyses, and interpretation; analysis coordination; and manuscript preparation; G.G. undertook specimen processing; data acquisition, analyses, and interpretation; and analysis coordination; K.K. provided RNA sequencing and exome sequencing data acquisition, analyses, and interpretation; J.-P.M. managed single-cell RNA sequencing data acquisition, analyses, and interpretation; J.D.-A. undertook data acquisition, analyses, and interpretation; T.M. provided specimen processing, data acquisition and analyses, and analysis coordination; C.N. managed specimen processing, data acquisition, and analyses; M.P. managed fluorescence in situ hybridization data acquisition, analyses, and interpretation; J.D. undertook conception and design and data interpretation; M.L.C. undertook conception and design and data interpretation; J.S. provided patient-derived xenograft model generation, and data analyses and interpretation; K.R. managed single-cell RNA sequencing experimental design and interpretation; S.O. undertook RNA sequencing and exome sequencing data acquisition, analyses, and interpretation; and M.L. undertook conception and design; patient care; specimen acquisition; data acquisition, analyses, and interpretation; analysis coordination; and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Michael Lübbert, Department of Medicine I, Medical Center, University of Freiburg, Hugstetter Str 55, 79106 Freiburg, Germany; e-mail: michael.luebbert@uniklinik-freiburg.de.

