

Key Points
AITL-like tumors in mice continuously require key Tfh factors, such as Bcl6 and SAP.
Molecular pathways crucial for Tfh identity and T-B collaboration could be promising therapeutic targets for AITL.

Abstract
Angioimmunoblastic T-cell lymphoma (AITL) is an aggressive peripheral T-cell lymphoma driven by a pool of neoplastic cells originating from T follicular helper (Tfh) cells and concomitant expansion of B cells. Conventional chemotherapies for AITL have shown limited efficacy, and as such, there is a need for improved therapeutic options. Because AITL originates from Tfh cells, we hypothesized that AITL tumors continue to rely on essential Tfh components and intimate T-cell–B-cell (T-B) interactions. Using a spontaneous AITL mouse model (Roquinsan/+ mice), we found that acute loss of Bcl6 activity in growing tumors drastically reduced tumor size, demonstrating that AITL-like tumors critically depend on the Tfh lineage–defining transcription factor Bcl6. Because Bcl6 can upregulate expression of signaling lymphocytic activation molecule–associated protein (SAP), which is known to promote T-B conjugation, we next targeted the SAP-encoding Sh2d1a gene. We observed that Sh2d1a deletion from CD4+ T cells in fully developed tumors also led to tumor regression. Further, we provide evidence that tumor progression depends on T-B cross talk facilitated by SAP and high-affinity LFA-1. In our study, AITL-like tumors relied heavily on molecular pathways that support Tfh cell identity and T-B collaboration, revealing potential therapeutic targets for AITL.
Introduction
Angioimmunoblastic T-cell lymphoma (AITL) is an aggressive non-Hodgkin lymphoma representing 15% to 20% of peripheral T-cell lymphomas.1 Patients have generalized lymphadenopathy, hypergammaglobulinemia, and autoimmune hemolytic anemia with poor prognosis (5-year survival rate, ∼33%).2,3 Typically, tumors display oligoclonal expansion of T cells and an effacement of lymph node architecture with prominent arborization of endothelial venules.1,4,5 Gene expression profiling, immunohistochemical studies, and xenograft experiments established that neoplastic cells in AITL are derived from CD4+ T follicular helper (Tfh) cells.6-9 However, the actual Tfh tumor cell content in the AITL tumor mass is kept low throughout disease progression, with concomitant expansion of bystander B cells and other reactive immune cells.5,6,10 Currently, chemotherapy is the most common treatment of AITL, but its limited efficacy demands more effective therapeutic options.3
The etiology of AITL has not been fully elucidated; however, genome sequencing of AITL tumor samples has uncovered heterogeneous somatic mutations with a few recurrent genes. The most frequently mutated genes include epigenetic modifiers (IDH2, TET2, and DNMT3A), genes encoding the small GTPase RhoA, and components of T-cell receptor/costimulatory signaling, including the Src-related protein tyrosine kinase Fyn.10-13 Importantly, activating mutations in T-cell signaling components were found in ∼50% of AITL patients, suggesting that hyperactivation of T cells during germinal center (GC) reactions may play an important role in the malignant transformation of Tfh cells and tumor growth.12,13
Tfh cells are a subset of CD4+ T cells whose differentiation is driven by the transcription factor Bcl6.14,15 Excessive Tfh cell activity can lead to pathologies such as autoimmunity, as well as Tfh-driven lymphomas.15,16 Directly or indirectly, Bcl6 suppresses other lineage-committing transcription factors and promotes the expression of Tfh signature genes, such as those encoding CXCR5, PD-1, and SAP, as well as key cytokines such as IL-21.17-19 Tfh cells have a unique ability to migrate into B-cell follicles, forming transient yet stable T-cell–B-cell (T-B) conjugates during GC reactions. These processes are supported by signaling lymphocytic activation molecule (SLAM) family receptors and their adaptor protein, SLAM-associated protein (SAP),20-24 as well as LFA-1.25 After repeated interaction with T cells, GC B cells that have received sufficient T-cell help and B-cell receptor signaling downregulate Bcl6 and upregulate IRF4 to differentiate into antibody-secreting plasma cells.26,27
Mice possessing 1 copy of the sanroque allele of Roquin (Roquinsan), a dominant negative point mutation, spontaneously develop AITL-like disease.16 The sanroque mutation increases the stability of an array of mRNA species, such as those coding for ICOS, OX40, and IFN-γ in CD4+ T cells, because of the disruption of Roquin-mediated mRNA degradation machinery.28 Mice homozygous for this sanroque mutation (Roquinsan/san) have hyperactive Tfh cells, spontaneous GC reactions, and lupus-like autoimmune disease, combined with AITL-like disease within 4-months of age.29 Heterozygous Roquinsan/+ mice do not develop lupus-like disease, but still have hyperactive GC reactions and present with AITL-like disease (∼50% incidence rates at 6 months of age).16 Although ROQUIN gene mutations have not been discovered in AITL patients,30 hyperactivation of T cells and Tfh cells are shared features of tumors arising in Roquinsan/+ mice and AITL patients.
In this study, the AITL-like tumors in Roquinsan/+ mice accumulated B cells with features of early-stage plasma cells. This B-cell expansion coincided with highly proliferative CD4+CXCR5+PD-1+ Tfh-like cells equipped with Bcl6, SAP, and high-affinity LFA-1. Importantly, acute abrogation of Bcl6 or SAP function or inhibition of high-affinity LFA-1 led to partial or full tumor regression. Taken together, these data suggest that AITL-like tumors in Roquinsan/+ mice are driven by Tfh-like CD4+ cells that continuously interact with B cells in a manner resembling GC T-B interactions.
Methods
Mice
Mice with the Roquinsan allele16,29 were provided by C. Vinuesa (Australian National University) and bred with other lines of mice to make composite mouse lines. UBC-CreERT2 (Jax 008085)31 and CD4-CreERT2 mice32 were used for tamoxifen-inducible ubiquitous or CD4+ cell–specific knockout models. Bcl6 conditional knockout mice were provided by T. Takemori (RIKEN, Japan).33 SAP conditional knockout (Sh2d1af)34 and SAPR78A mice (Sh2d1aR78A)35 were previously described. All mice had been backcrossed on the C57BL/6J background for more than 10 generations before the composite lines were generated. Mice were maintained in the animal facility at the Institut de Recherches Cliniques de Montréal (IRCM) in a specific pathogen-free environment, and all experiments were performed in compliance with animal use protocols approved by the IRCM Animal Care Committee.
Antibodies and chemicals
All antibodies, streptavidin conjugates, and reagents used for flow cytometry were from ThermoFisher unless otherwise stated: B220 (RA3-6B2), Bcl6 (K112-91; BD Biosciences), CD4 (GK1.5), CD16/CD32 (2.4G2; BioXCell), CD95 (BD Biosciences), CD138 (281-2; BioLegend or BD Biosciences), CXCR5 (SPRCL5), GL7, ICAM-1 (YN1/1.7.4), IRF4 (3E4), Ki-67 (SolA15), PD-1 (J43; BD Biosciences), SAP (1A9; BD Biosciences or produced by the André Veillette laboratory), streptavidin-APC, and streptavidin-PE-Cy. Dead cells were stained with 7-aminoactinomycin D (BD Biosciences) or a fixable viability dye. Fixation and permeabilization kits (BD Biosciences and ThermoFisher) were used to perform intracellular staining. Tamoxifen was purchased from Millipore Sigma, isoflurane from CDMV, and lovastatin from Selleck Chemicals. Anti-LFA1 (CD11a; M17/A) was purchased from BioXCell.
Flow cytometry
Single-cell suspensions of lymph nodes were prepared by mechanical dissociation via a 70-μm nylon mesh filter (BD Biosciences), in phosphate-buffered saline or staining buffer with 1% bovine serum albumin (Wisent). Cells were first blocked with anti-CD16/CD32 and then stained with primary antibodies, followed by streptavidin conjugates. For intracellular stains, cells were fixed and permeabilized with Cytofix/Cytoperm solution (BD Biosciences) or Fix/Perm Buffer (ThermoFisher), according to the manufacturer’s instructions. ICAM-1 binding assay was performed as described in the supplemental Methods. Samples were acquired using LSR Fortessa (BD Biosciences) and analyzed by using FlowJo, version 10 (Treestar).
Tumor monitoring and treatment
Tumors in cervical, axillary, brachial, and inguinal lymph nodes were palpated and monitored by in vivo sonographic imaging (VEVO 770; Visual Sonics), using the RMV 707B scanhead. A minimum of 5 cross-sectional ultrasound images were taken, and the largest diameter and cross-sectional surface area were chosen (∼5- to 12-mm diameter; ∼13- to 77-mm2 cross-sectional surface area). To induce Cre recombinase activity, oral gavage was performed for 5 consecutive days (200 μg/g of body weight per day in corn oil). Lovastatin was administered through intraperitoneal injections (2 mg/kg, every other day for 2 weeks). ELISA, human AITL histology, as well as genomic DNA preparation and PCR are described further in the supplemental Methods.
Statistical analysis
Data were analyzed using Prism 7.0 (GraphPad Software). When comparing 2 groups, an unpaired Student t test was performed. When comparing 3 groups, 1-way analysis of variance was used. For time courses of tumor regression, 2-way analysis of variance was used. For comparing tumor incidence rates, Fisher’s exact test was used. P < .05 was considered statistically significant.
Results
Signs of elevated helper T-cell activities in Roquinsan/+ tumor-bearing mice
A longitudinal study of AITL patient biopsy samples revealed that B-cell follicles in tumorous lymph nodes gradually disappear, leading to a complete disruption of T-B-cell boundaries.5,36 This hallmark feature of AITL has been recapitulated in lymph node tumors spontaneously developing in Roquinsan /+ mice.16 As with human AITL, rather than a disproportionate expansion of T cells, it is an expansion of B cells that contributes to most of the tumor mass.16 Although it has been presumed that this B-cell expansion is driven by the hyperactive helper functions of neoplastic CD4+ cells, the different B-cell populations in Roquinsan /+ tumor-bearing mice during progression have not been well characterized.16 Thus, we phenotyped B-cell populations in tumor-free, nontumorous, and tumorous lymph nodes of Roquinsan /+ mice (6-13 months old; Figure 1A).
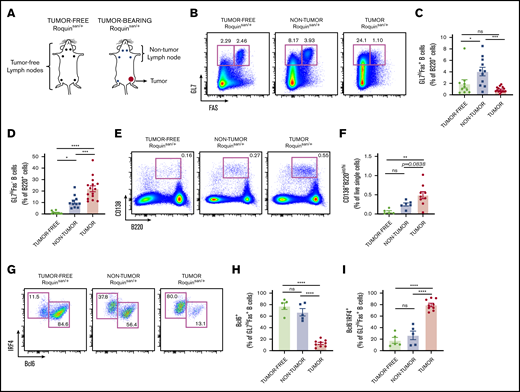
An increase in plasmablasts in tumor lymph nodes of Roquinsan/+mice. (A) Pictogram illustrating differences between tumor-free and -bearing mice, as well as providing a pictorial explanation of tumor-free, nontumorous (non-tumor), and tumorous (tumor) lymph nodes. (B-D) Representative flow cytometric analyses and frequencies of B-cell populations using the surface markers Fas and GL7 (n = 10 tumor-free; n = 11 nontumorous; and n = 15 tumorous samples). (E-F) Representative flow cytometry plots and histograms comparing the frequency of plasmablasts (n = 5 tumor-free, n = 6 nontumorous; and n = 9 tumorous samples). (G-I) Expression profile of Bcl6 and IRF4 in GL7hiFas+ B cells as depicted through representative flow cytometry plots and frequencies (n = 5 tumor-free; n = 5 nontumorous; and n = 9 tumorous samples). Error bars in panels C-D,F,H-I represent the standard error of the mean (SEM). Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not significant.
An increase in plasmablasts in tumor lymph nodes of Roquinsan/+mice. (A) Pictogram illustrating differences between tumor-free and -bearing mice, as well as providing a pictorial explanation of tumor-free, nontumorous (non-tumor), and tumorous (tumor) lymph nodes. (B-D) Representative flow cytometric analyses and frequencies of B-cell populations using the surface markers Fas and GL7 (n = 10 tumor-free; n = 11 nontumorous; and n = 15 tumorous samples). (E-F) Representative flow cytometry plots and histograms comparing the frequency of plasmablasts (n = 5 tumor-free, n = 6 nontumorous; and n = 9 tumorous samples). (G-I) Expression profile of Bcl6 and IRF4 in GL7hiFas+ B cells as depicted through representative flow cytometry plots and frequencies (n = 5 tumor-free; n = 5 nontumorous; and n = 9 tumorous samples). Error bars in panels C-D,F,H-I represent the standard error of the mean (SEM). Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not significant.
Consistent with mild spontaneous GC reactions, there was a greater frequency of GC B cells (B220+GL7hiFas+) in nontumorous than in tumor-free lymph nodes (Figure 1B-C). Interestingly, in tumors, the frequency of typical GC B cells was reduced, as confirmed by flow cytometric analysis of B220+GL7hiFas+ (Figure 1B-C) and B220+Bcl6+Fas+ (supplemental Figure 1B) cell populations. We reasoned that this reduction could be related to a rapid transition of GC B cells into the plasma cell lineage, reflecting hyperactive helper T-cell activities during tumor growth. Consistent with this, we observed an increase of CD138+B220int plasmablasts in tumors (Figure 1E-F). Furthermore, we found that B220+GL7hiFas+ cells in tumors had an increased subset of cells that express high levels of IRF4 and reduced Bcl6 (Figure 1G-I), a population reported to be the immediate precursor of plasma cells.26,27 The B220+GL7hiFas− cells that accumulated in tumorous lymph nodes were almost exclusively IRF4hi Bcl6−. We also confirmed that tumor-bearing mice have elevated IgG2c and IgG1 titers (supplemental Figure 2A-B), consistent with previous data.16 This hypergammaglobulinemia is a shared feature between human AITL and our mouse model.2 Thus, tumor-bearing Roquinsan /+ mice have signs of ongoing T-B-cell cross talk within tumors, despite a lack of conventional GCs.
Persistent activity of Bcl6 is essential for tumor progression
When heterogeneous human AITL tumor cell populations were xenografted into immunodeficient mice, after repeated rounds of serial transplantation, putative neoplastic CD4+ T cells persisted, whereas CD8+ T and B cells gradually disappeared.8 Importantly, the enriched T-lineage cells were mostly Bcl6+, suggesting that sustained expression of Bcl6 in CD4+ tumor cells may be necessary for AITL tumor progression. Similar to this observation, we found that consecutive sections of AITL patient biopsy samples had staining patterns where CD3 and Bcl6 cells overlapped (Figure 2A). Multicolor immunofluorescence imaging confirmed a predominant expression of Bcl6 in CD4+ cells in AITL, whereas Bcl6 expression was restricted within the GC in human tonsils (supplemental Figure 3A-B). In fact, Bcl6 positivity is now considered a reliable diagnostic tool, especially for AITL patients with bone marrow involvement.37,38 In Roquinsan /+ tumors, the conventional CD4+CXCR5+PD-1+ Tfh cell population was decreased with reduced PD-1 expression (Figure 2B-D).16 However, this difference was less pronounced when we gated on CD4+CXCR5+Bcl6+ cells (supplemental Figure 4A-B) and high levels of Bcl6 in CD4+CXCR5+PD-1+ cells were maintained, regardless of tumor status (Figure 2E-F). Because this CD4+CXCR5+PD-1+ population expresses the highest levels of Bcl6 and is also highly proliferative (Figure 2G), we suspect that these cells represent the neoplastic cells driving tumor growth by interacting with B cells. Based on these, we predicted that acute ablation of Bcl6 activity halts AITL-like tumor growth. To test this idea, we generated mice expressing a CD4-driven tamoxifen-inducible Cre recombinase and conditioned Bcl6 genes on the Roquinsan/+ background (Roquinsan/+; Cd4-CreERT2+/−; Bcl6f/f; Figure 2H). Upon exposure to Cre recombinase activity, the conditioned Bcl6 gene loses exons 7 to 9 (encoding Zn-finger domains) and thereby its capacity to drive GC B or Tfh differentiation.33,39 We confirmed that our Cd4-CreERT2 system efficiently removed the floxed Bcl6 DNA segment specifically in CD4-expressing cells (supplemental Figure 5A). Under the same conditions, the Bcl6 antibody (clone K112-91) detected intact levels of Bcl6 in Cre-experienced Tfh-like cells (supplemental Figure 5B), suggesting that the deleted allele still produced a nonfunctional truncated version of Bcl6. Once these mice developed full-size tumors, we followed tumor growth by palpation and sonographic imaging after administration of tamoxifen. Notably, tamoxifen treatment alone in control tumor-bearing mice (Roquinsan/+; Cd4-CreERT2+/−) reduced tumor size within the first week after treatment (Figure 2I), perhaps because of a general anticancer effect of tamoxifen.40 However, tumors grew back after the initial shrinkage at 2 weeks after treatment. In contrast, Roquinsan/+; Cd4-CreERT2+/−; Bcl6f/f mice showed continued tumor regression (Figure 2I-J). Mice that showed tumor regression had no recurrence in primary or distal lymph nodes up to 12 weeks after tamoxifen treatment (Figure 2I). Therefore, we concluded that persistent Bcl6 activity is indispensable for the continued growth of AITL-like tumors in Roquinsan/+ mice.
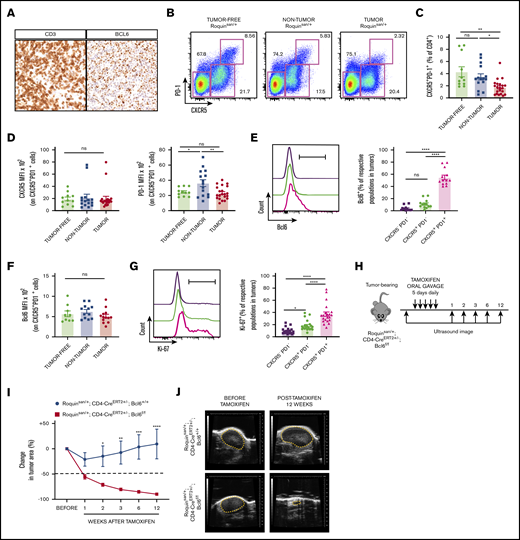
Disruption of functional Bcl6 gene in growing AITL-like tumors leads to tumor regression. (A) Exemplary immunohistochemistry sections of AITL patient samples depicting CD3 and Bcl6 expression. (B) Representative flow cytometric plots showing subsets of CD4 cells based on expression of PD-1 and CXCR5. (C) Comparing frequencies of CD4+CXCR5+PD-1+ cells between tumor-free, nontumorous (non-tumor), and tumorous (tumor) samples (n = 11 tumor-free; n = 15 nontumorous; and n = 21 tumorous samples). (D) MFI of CXCR5 and PD-1 on CD4+CXCR5+PD-1+ cells (n = 11 tumor-free; n = 15 nontumorous; and n = 21 tumorous samples). Bcl6 expression in CXCR5−PD1−, CXCR5+PD-1−, and CXCR5+PD-1+ cells from tumor samples (E) and Bcl6 MFI of CD4+CXCR5+PD-1+ cells (F) (n = 9 tumor-free; n = 12 nontumorous; and n = 13 tumorous). (G) Representative histogram and frequency of Ki-67 levels in tumor CD4+ subsets (n = 21 tumorous). (H) Roquinsan/+ mice were bred with a CD4-specific, tamoxifen-inducible Cre-recombinase to target the Bcl6 gene (Roquinsan/+; Cd4-CreERT2+/−; Bcl6f/f). Representative time course (I) and sonograms (J) demonstrating tumor regression in mice with Bcl6 gene deletion (n = 5 Roquinsan/+; Cd4-CreERT2+/−; Bcl6+/+; n = 6 Roquinsan/+; Cd4-CreERT2+/−; Bcl6f/f). Error bars in panels C-G,I represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Disruption of functional Bcl6 gene in growing AITL-like tumors leads to tumor regression. (A) Exemplary immunohistochemistry sections of AITL patient samples depicting CD3 and Bcl6 expression. (B) Representative flow cytometric plots showing subsets of CD4 cells based on expression of PD-1 and CXCR5. (C) Comparing frequencies of CD4+CXCR5+PD-1+ cells between tumor-free, nontumorous (non-tumor), and tumorous (tumor) samples (n = 11 tumor-free; n = 15 nontumorous; and n = 21 tumorous samples). (D) MFI of CXCR5 and PD-1 on CD4+CXCR5+PD-1+ cells (n = 11 tumor-free; n = 15 nontumorous; and n = 21 tumorous samples). Bcl6 expression in CXCR5−PD1−, CXCR5+PD-1−, and CXCR5+PD-1+ cells from tumor samples (E) and Bcl6 MFI of CD4+CXCR5+PD-1+ cells (F) (n = 9 tumor-free; n = 12 nontumorous; and n = 13 tumorous). (G) Representative histogram and frequency of Ki-67 levels in tumor CD4+ subsets (n = 21 tumorous). (H) Roquinsan/+ mice were bred with a CD4-specific, tamoxifen-inducible Cre-recombinase to target the Bcl6 gene (Roquinsan/+; Cd4-CreERT2+/−; Bcl6f/f). Representative time course (I) and sonograms (J) demonstrating tumor regression in mice with Bcl6 gene deletion (n = 5 Roquinsan/+; Cd4-CreERT2+/−; Bcl6+/+; n = 6 Roquinsan/+; Cd4-CreERT2+/−; Bcl6f/f). Error bars in panels C-G,I represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
SAP is critical for tumor progression
Next, we sought to identify key mechanisms downstream of the Bcl6-dependent Tfh program that support AITL-like tumor growth in Roquinsan/+ mice. As a master regulator of Tfh cell differentiation, Bcl6 has many direct or indirect target genes, ∼50% of which are distinct from those identified in B cells.17,19 Relevant to its role in Tfh differentiation, Bcl6 overexpression in human CD4+ T cells leads to upregulation of SAP, along with CXCR5 and PD-1.18 SAP has been shown to be crucial for the formation of T-B conjugates during early and late stages of the GC reaction.20,22 This is the likely reason that SAP deficiency drastically reduced tumor incidence in Roquinsan/+ mice.16 Consequently, we predicted that sustained expression of SAP is critical for AITL-like tumor growth. We found increased frequencies of SAPhi cells among CD4+ T cells (Figure 3A) and Tfh-like cells from tumors in Roquinsan/+ mice, although we observed no differences in SAP median fluorescence intensity (MFI) among Tfh-like cells (Figure 3B). Notably, SAPhi CD4+ T cells from tumors had more Ki-67+ cells when compared with SAPhi CD4+ T cells from nontumorous or tumor-free lymph nodes (Figure 3C). Within tumors, SAPhi CD4+ T cells were more proliferative than SAPlo CD4+ T cells (Figure 3D). These results suggest that tumor cells require high levels of SAP expression for their growth. To directly test this idea, we generated Roquinsan/+; Cd4-CreERT2+/−; Sh2d1af/for Sh2d1af/y mice (the Sh2d1a gene is X-linked; Figure 3E). As expected, abrogation of SAP protein expression in CD4+ T cells by administration of tamoxifen (supplemental Figure 5E) led to durable tumor regression until 12 weeks after treatment (Figure 3F-G). Similar results were obtained when the Sh2d1a gene was deleted ubiquitously by using a UBC-CreERT2 system in growing tumors (supplemental Figure 6). Despite similar tumor regression kinetics caused by abrogation of functional Bcl6 or SAP, the 2 Tfh signature proteins appear to have distinct roles in AITL-like tumor maintenance. First, acute disruption of Bcl6 gene function does not cause immediate loss of SAP protein (supplemental Figure 5C). Second, Bcl6 disruption leads to a small but clear reduction of CXCR5 expression levels among Tfh-like cells without grossly affecting their total frequency (supplemental Figure 5D). In contrast, abrogation of SAP expression leads to a clear twofold reduction in the percentage of Tfh-like cells without affecting CXCR5 levels (supplemental Figure 5F).
![Abrogation of SAP expression from AITL-like tumors leads to tumor regression. (A) Increased expression of the SLAM adaptor protein, SAP in CD4 cells from AITL-like tumors (n = 5 tumor-free; n = 8 nontumorous [non-tumor]; and n = 12 tumorous [tumor] samples). (B) Frequency and MFI of SAP in CXCR5+PD-1+ cells from tumor-free, nontumorous, and tumor samples (n = 5 tumor-free; n = 8 nontumorous; and n = 12 tumorous samples). (C) SAPhi CD4 cells from tumors are more proliferative, as depicted through increased frequencies of Ki-67+ cells (n = 4 tumor-free; n = 7 nontumorous; and n = 11 tumorous samples). (D) SAPhi CD4+ tumor cells are more proliferative than SAPlo as shown in representative histogram and frequencies. (E) Roquinsan/+ mice were bred with a CD4 specific tamoxifen-inducible Cre recombinase and conditional Sh2d1a allele (Roquinsan/+; Cd4-CreERT2+/−; Sh2d1af/y or f/f). Representative time course (F) and sonograms (G) demonstrating tumor regression in mice with CD4-specific abrogation of SAP (n = 5 Roquinsan/+; Cd4-CreERT2+/−; Sh2d1a+/+; n = 9 Roquinsan/+; Cd4-CreERT2+/−; Sh2d1af/y or f/f). In panel F, we reused CD4-CreERT2 control data shown in Figure 2I to perform statistical analysis for different time points. Error bars in panels A-D,F represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/5/10.1182_bloodadvances.2019001114/2/m_advancesadv2019001114f3.png?Expires=1764968443&Signature=EW7Nxjnj-8QgEq0zebZIC00~~-pUnl04~KTGealEvMskGQ3R8iJXxiVGP7x5OeC6a0XpNRMbVdCN6pbbqUeZCCqrWnC45bvEPLQxmhPkYvJR2~ArBsug4vPaMFmatknOV6UpRAFr82H~RXiQGqMwOL~9QjljANy9X7mjxLXGx~I5zfL85QTkkK6wwCvizbEkSht06RjkafTDeGgdK1wUCcK5o8xlzc1f~o2EJp1c-noCqCjQClehugoiB1cVwfFYeTAM~Tp3UDA4GtxddgNSWTGk3-cyqcBi29m9N3ifS4doWbp6XkCMVimwGva5rX79uZCsEo2k8-B9H0V~9GCMMw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Abrogation of SAP expression from AITL-like tumors leads to tumor regression. (A) Increased expression of the SLAM adaptor protein, SAP in CD4 cells from AITL-like tumors (n = 5 tumor-free; n = 8 nontumorous [non-tumor]; and n = 12 tumorous [tumor] samples). (B) Frequency and MFI of SAP in CXCR5+PD-1+ cells from tumor-free, nontumorous, and tumor samples (n = 5 tumor-free; n = 8 nontumorous; and n = 12 tumorous samples). (C) SAPhi CD4 cells from tumors are more proliferative, as depicted through increased frequencies of Ki-67+ cells (n = 4 tumor-free; n = 7 nontumorous; and n = 11 tumorous samples). (D) SAPhi CD4+ tumor cells are more proliferative than SAPlo as shown in representative histogram and frequencies. (E) Roquinsan/+ mice were bred with a CD4 specific tamoxifen-inducible Cre recombinase and conditional Sh2d1a allele (Roquinsan/+; Cd4-CreERT2+/−; Sh2d1af/y or f/f). Representative time course (F) and sonograms (G) demonstrating tumor regression in mice with CD4-specific abrogation of SAP (n = 5 Roquinsan/+; Cd4-CreERT2+/−; Sh2d1a+/+; n = 9 Roquinsan/+; Cd4-CreERT2+/−; Sh2d1af/y or f/f). In panel F, we reused CD4-CreERT2 control data shown in Figure 2I to perform statistical analysis for different time points. Error bars in panels A-D,F represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Abrogation of SAP expression from AITL-like tumors leads to tumor regression. (A) Increased expression of the SLAM adaptor protein, SAP in CD4 cells from AITL-like tumors (n = 5 tumor-free; n = 8 nontumorous [non-tumor]; and n = 12 tumorous [tumor] samples). (B) Frequency and MFI of SAP in CXCR5+PD-1+ cells from tumor-free, nontumorous, and tumor samples (n = 5 tumor-free; n = 8 nontumorous; and n = 12 tumorous samples). (C) SAPhi CD4 cells from tumors are more proliferative, as depicted through increased frequencies of Ki-67+ cells (n = 4 tumor-free; n = 7 nontumorous; and n = 11 tumorous samples). (D) SAPhi CD4+ tumor cells are more proliferative than SAPlo as shown in representative histogram and frequencies. (E) Roquinsan/+ mice were bred with a CD4 specific tamoxifen-inducible Cre recombinase and conditional Sh2d1a allele (Roquinsan/+; Cd4-CreERT2+/−; Sh2d1af/y or f/f). Representative time course (F) and sonograms (G) demonstrating tumor regression in mice with CD4-specific abrogation of SAP (n = 5 Roquinsan/+; Cd4-CreERT2+/−; Sh2d1a+/+; n = 9 Roquinsan/+; Cd4-CreERT2+/−; Sh2d1af/y or f/f). In panel F, we reused CD4-CreERT2 control data shown in Figure 2I to perform statistical analysis for different time points. Error bars in panels A-D,F represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Selective abrogation of SAP-Fyn signaling pathway does not greatly alter AITL-like disease
SAP promotes the production of cytokines such as IL-4 by activating the Src family protein kinase Fyn.35 When SAP-Fyn interactions were abrogated by the SAP R78A point mutation, the ability of SAP to activate Fyn was abolished.35 However, this SAP R78A mutant retained the ability to promote Tfh maturation and GC reactions.24,41 Interestingly, a few Fyn mutations with disrupted autoinhibitory mechanisms have been discovered in human AITL tumors, suggesting a positive role for Fyn kinase in AITL tumorigenesis.12,13 Although SAP-deficient Roquinsan/+ mice had dramatically reduced tumor incidence (no tumors developed in the 16 mice monitored),16 the contributions of the SAP-Fyn signaling pathway to disease incidence and severity have not been evaluated. To address these questions, we generated cohorts of Roquinsan/+ mice possessing the Sh2d1a+/y or Sh2d1aR78A/y genotype. Although there was some trend toward reduced tumor incidence, a substantial portion (9 of 29) of SAP R78A mutant mice still developed tumors (Figure 4A). Also, there was no difference in the number and size of tumors. Furthermore, the percentages of CD4+CXCR5+PD-1+ Tfh-like cells in tumors within wild-type or mutant SAP mice were similar (Figure 4B). We observed no differences in Bcl6 levels but found increased Ki-67+ populations in SAP-R78A tumors, compared with the number in SAP-wild-type tumors (Figure 4C). SAP-R78A tumors also recapitulated the phenotype of B-cell populations in SAP-wild-type tumors (Figure 4D-E). Last, there was no difference in the adhesive capacities of CD4+ T cells derived from the 2 groups of tumors, as judged by an ICAM-1 binding assay (supplemental Figure 7). These results indicate that SAP mostly uses Fyn-independent pathways, such as T-B-cell conjugation, to promote AITL-like disease.
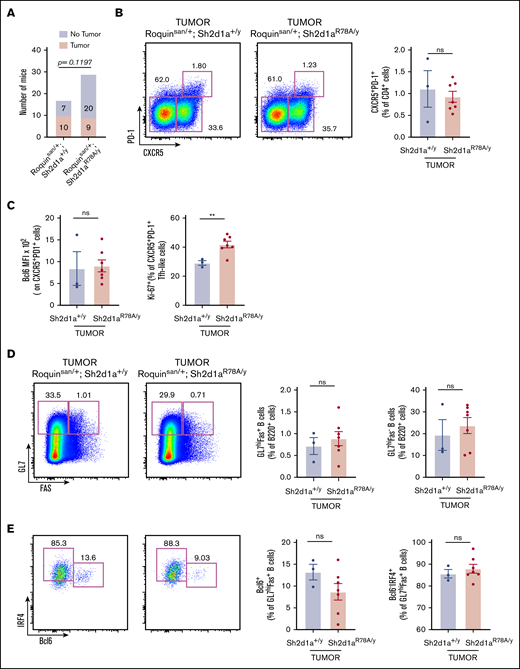
Loss of SAP-Fyn signaling pathway does not greatly alter AITL-like disease. (A) Abrogation of downstream signaling between SAP and Fyn kinase does not prevent tumor incidence, but may have a partial effect (n = 17 Roquinsan/+; Sh2d1a+/y and n = 29 Roquinsan/+; Sh2d1aR78A/y). (B-E) Tumors from Roquinsan/+; Sh2d1a+/y or Roquinsan/+; Sh2d1aR78A/y show comparable T-B-cell expression patterns and frequencies (n = 3 Roquinsan/+; Sh2d1a+/y tumors and n = 7 from Roquinsan/+; Sh2d1aR78A/y tumors). Error bars in panels B-E represent the SEM. Data are pooled from at least 2 independent experiments. **P < .01.
Loss of SAP-Fyn signaling pathway does not greatly alter AITL-like disease. (A) Abrogation of downstream signaling between SAP and Fyn kinase does not prevent tumor incidence, but may have a partial effect (n = 17 Roquinsan/+; Sh2d1a+/y and n = 29 Roquinsan/+; Sh2d1aR78A/y). (B-E) Tumors from Roquinsan/+; Sh2d1a+/y or Roquinsan/+; Sh2d1aR78A/y show comparable T-B-cell expression patterns and frequencies (n = 3 Roquinsan/+; Sh2d1a+/y tumors and n = 7 from Roquinsan/+; Sh2d1aR78A/y tumors). Error bars in panels B-E represent the SEM. Data are pooled from at least 2 independent experiments. **P < .01.
Evidence of LFA-1–dependent T-B-cell cross talk in AITL-like tumor progression
Because ongoing T-B-cell cross talk was evident in growing AITL-like tumors, we wanted to test for upregulation of adhesion molecules on CD4+ and B220+ cells in tumors. It has been reported that Tfh cells express high levels of LFA-1 and that disruption of LFA-1 activity severely compromises Tfh cell survival and GC reactions.25 Interestingly, using an ICAM-1 binding assay, we found that CD4+ T cells from tumors had elevated levels of functional or high-affinity LFA-1 compared with tumor-free Roquinsan/+ mice, whereas nontumor CD4+ T cells had intermediate levels of active LFA-1 (Figure 5A). In addition, tumor samples contained B cells that expressed relatively higher levels of ICAM-1 compared with their counterparts in tumor-free mice, although the difference in absolute MFI was not very strong (Figure 5B), suggesting that B cells in tumor-bearing mice have a better capacity to engage with T cells. Among B-cell subsets, regardless of tumor progression, ICAM-1 levels were highest in B220+GL7hiFas+ GC B-like cells, the known partner of Tfh cells in GCs (Figure 5C). Accordingly, we tested whether repeated injection of LFA-1–blocking antibodies could reduce tumor size. However, we could not detect clear regression of AITL-like tumors. We reasoned that macromolecules, such as antibodies, may not infiltrate tumor lymph nodes because of their highly disorganized lymph node architecture. Therefore, we decided to test small-molecule inhibitors for LFA-1. A cholesterol-lowering drug, lovastatin, has been reported to inhibit LFA-1 allosterically.42,43 We validated this inhibitory effect of lovastatin in vitro using Tfh cells isolated from Peyer’s patches, with minimal change to cell viability (Figure 5D). Importantly, when we administered lovastatin to tumor-bearing Roquinsan/+ mice, we observed a full regression (>60% reduction in area) or partial regression (25% to 60% reduction in area) in 6 of 13 tumors (Figure 5E-F). However, the impact of lovastatin on tumor growth may involve mechanisms other than LFA-1 inhibition, such as alteration of cholesterol metabolism, warranting further study. Because absence of SAP in murine natural killer cells diminishes high-affinity LFA-1,44 we tested whether acute loss of SAP expression would lead to a drastic disappearance of high-affinity LFA-1 in CD4+ T cells. However, both in vitro and in vivo tamoxifen-induced abrogation of SAP expression did not affect active LFA-1 levels (supplemental Figure 8). Together, these data suggest that LFA-1 and ICAM-1 serve as adhesion molecules that promote T-B-cell cross talk and growth of AITL-like tumors in Roquinsan/+ mice.
![Elevated high-affinity LFA-1 and ICAM-1 levels in Roquinsan/+tumors. (A) Tumor samples have increased frequency of high-affinity LFA-1. Representative flow cytometric analyses and frequency of high-affinity LFA-1 expression on CD4+ cells from recombinant ICAM-1 binding assay (10 μg/mL of murine recombinant ICAM-1; n = 5 tumor-free; n = 4 nontumorous [non-tumor]; and n = 4 tumorous [tumor] samples). (B) Concomitant increased expression of ICAM-1 on B220+ cells in Roquinsan/+ tumors as shown in representative flow cytometric analyses and frequencies. (B-C) MFI of ICAM-1 on B-cell subsets (n = 5 tumor-free; n = 5 nontumorous; and n = 7 tumorous samples). (D) Incubating wild-type Peyer’s patch cells with Lovastatin during ICAM-1 binding assay verifies reduction in high-affinity LFA-1 levels on CD4+ T cells with minimal differences in cell viability (n = 5, wild type). (E) Left graph shows a time course of change in tumor size after treatment with lovastatin (2 mg/kg, intraperitoneal, every other day for 2 weeks) compared with the untreated control group. Lovastatin-treated mice were further subgrouped based on the following criteria: no response (<25% reduction in area or growth), partial response (25% to 60% reduction in area), or full response (>60% reduction in area) at the final time point (week 6). The right graph further compares each response type with the untreated control group. (F) Exemplary regressed tumor is shown in sonograms. Error bars in panels A-E represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/5/10.1182_bloodadvances.2019001114/2/m_advancesadv2019001114f5.png?Expires=1764968443&Signature=oPf23DuBmsLdDsABwndOAncY-Xuvd8yMz2FNDlF2JICtWEokW3dsQOKXzYQiSTshIs6rVuueHIs6H1xL8TZmDGQqiEEpi5iOuCHaN5LmOWyGcLT6tdJOgp4K9z6PR4olfH3ls5fFRgGTf~xsodi76zogJ4f-Rwri1KNH5IWWcUakrebKePmQM4uwIOqCRq6Y18AFB2v0VIw~PZF77i~QasD8ZmWXV8DlM~JiuXCDRFVV~UOF8Y3i1dNygN1AfQnNFyVN36HrrffhQAT45jeNCncVCypl9Rpgyy1mC6nZND8ICUz2f~o3LzlNl01iTdjHaYKGZWYyF0Id4jz8gnGADQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Elevated high-affinity LFA-1 and ICAM-1 levels in Roquinsan/+tumors. (A) Tumor samples have increased frequency of high-affinity LFA-1. Representative flow cytometric analyses and frequency of high-affinity LFA-1 expression on CD4+ cells from recombinant ICAM-1 binding assay (10 μg/mL of murine recombinant ICAM-1; n = 5 tumor-free; n = 4 nontumorous [non-tumor]; and n = 4 tumorous [tumor] samples). (B) Concomitant increased expression of ICAM-1 on B220+ cells in Roquinsan/+ tumors as shown in representative flow cytometric analyses and frequencies. (B-C) MFI of ICAM-1 on B-cell subsets (n = 5 tumor-free; n = 5 nontumorous; and n = 7 tumorous samples). (D) Incubating wild-type Peyer’s patch cells with Lovastatin during ICAM-1 binding assay verifies reduction in high-affinity LFA-1 levels on CD4+ T cells with minimal differences in cell viability (n = 5, wild type). (E) Left graph shows a time course of change in tumor size after treatment with lovastatin (2 mg/kg, intraperitoneal, every other day for 2 weeks) compared with the untreated control group. Lovastatin-treated mice were further subgrouped based on the following criteria: no response (<25% reduction in area or growth), partial response (25% to 60% reduction in area), or full response (>60% reduction in area) at the final time point (week 6). The right graph further compares each response type with the untreated control group. (F) Exemplary regressed tumor is shown in sonograms. Error bars in panels A-E represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Elevated high-affinity LFA-1 and ICAM-1 levels in Roquinsan/+tumors. (A) Tumor samples have increased frequency of high-affinity LFA-1. Representative flow cytometric analyses and frequency of high-affinity LFA-1 expression on CD4+ cells from recombinant ICAM-1 binding assay (10 μg/mL of murine recombinant ICAM-1; n = 5 tumor-free; n = 4 nontumorous [non-tumor]; and n = 4 tumorous [tumor] samples). (B) Concomitant increased expression of ICAM-1 on B220+ cells in Roquinsan/+ tumors as shown in representative flow cytometric analyses and frequencies. (B-C) MFI of ICAM-1 on B-cell subsets (n = 5 tumor-free; n = 5 nontumorous; and n = 7 tumorous samples). (D) Incubating wild-type Peyer’s patch cells with Lovastatin during ICAM-1 binding assay verifies reduction in high-affinity LFA-1 levels on CD4+ T cells with minimal differences in cell viability (n = 5, wild type). (E) Left graph shows a time course of change in tumor size after treatment with lovastatin (2 mg/kg, intraperitoneal, every other day for 2 weeks) compared with the untreated control group. Lovastatin-treated mice were further subgrouped based on the following criteria: no response (<25% reduction in area or growth), partial response (25% to 60% reduction in area), or full response (>60% reduction in area) at the final time point (week 6). The right graph further compares each response type with the untreated control group. (F) Exemplary regressed tumor is shown in sonograms. Error bars in panels A-E represent the SEM. Data are pooled from at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Discussion
We report evidence that CD4+ and B220+ cells in AITL-like tumors intimately influence each other and resemble Tfh-GC B-cell interactions in physiological GCs. First, there is an accumulation of B220+GL7hi Fas+ populations with signs of plasma cell commitment (Bcl6loIRF4hi), a product of successful Tfh-GC B collaboration. Second, AITL-like tumor progression relies heavily on continued expression of the Tfh lineage–defining transcription factor Bcl6 and high levels of SAP, a key element of T-B-cell conjugation. In addition, CD4+ T cells from tumor-bearing mice had increased active LFA-1, an adhesion molecule highly expressed on Tfh cells, as well as elevated levels of its binding partner ICAM-1 on B cells. Last, we found that tumor CD4+ T cells can drive tumor growth without Fyn-mediated SAP signaling mechanisms, consistent with previous studies that T-B-cell collaboration during GC reactions is SAP-Fyn independent.
It is intriguing that Bcl6 plays critical roles in both GC-derived T- and B-cell malignancies. Dysregulated Bcl6 expression is important in the development and progression of GC-derived B-cell lymphomas.45-48 In B-cell lymphomas, the oncogenic effects of persistent Bcl6 is most likely mediated by its ability to suppress cell cycle inhibitor p21 and p53.46 In contrast, BCL6 mutations have not been discovered in human AITL, but sustained expression of Bcl6 appears to be critical for maintaining malignant CD4+ cell populations in xenografted human AITL samples8 and our mouse model. Considering that ∼50% of Bcl6 target genes in Tfh cells are distinct from those regulated in GC B cells,17 Bcl6 may play different roles in T-cell lymphoma generation and progression than in B-cell lymphomas.
The association between Bcl6 and SAP has been well established in Tfh cells. Both in mouse and human Tfh cells, Bcl6 expression levels correlate highly with SAP levels and B-cell helper functions.18,49,50 High levels of SAP in Tfh cells appear to promote T-B-cell conjugation by inhibiting negative signals provided by SLAMF6.23 Analogous to this, Bcl6-expressing Tfh-like cells in tumor lymph nodes had higher levels of SAP expression, along with an expansion of B cells progressing into the plasma cell lineage. However, we found that acute disruption of Bcl6 activity does not lead to a drastic change in SAP expression level in CD4+ T cells. Because tumor regression becomes evident 2 weeks after tamoxifen treatment, we predict that these differences may be greater at later stages. However, we have not pursued this possibility, as newly generated T cells from the thymus may complicate the interpretation. Despite this, our data suggest that SAP is not an immediate downstream component of Bcl6. Further mechanistic studies are needed to identify proximal components of Bcl6-regulated genes relevant to AITL tumor progression.
There have been a handful of human AITL patients reported with somatic mutations in the FYN gene. These Fyn mutations disrupt autoinhibitory mechanisms and suggest that hyperactive Fyn could drive AITL tumor initiation and progression. Previous work has shown that Roquinsan/+ mice cannot develop tumors in SAP-deficient mice.16 However, specific roles for SAP-Fyn signaling in AITL-like tumor incidence and progression have not been clarified. In this study, we could not find clear differences in tumor incidence rates, severity, or characteristics, indicating that SAP-Fyn signaling has a very limited role in tumorigenesis and maintenance. Although it remains possible that persistently hyperactive Fyn activity through other upstream signals (such as T-cell receptor [TCR]) may drive tumor progression, it is not the main mechanism of how SAP drives AITL-like disease in our mouse model.
There are several mouse models of AITL that have provided insights into the pathogenesis of human AITL. An early study using transplantation of human AITL cells into immunodeficient mice confirmed that neoplastic CD4+ T cells express high levels of Bcl6.8 Studies using genetically engineered mouse models confirmed the relevance of the most prevalent somatic mutations discovered in human AITL. A recurrent RHOA G17V mutation is present in ∼60% to 70% of AITL patients and is often associated with loss-of-function mutations in TET2.10,12,13,51 Several recent mouse models support a 2-hit model of AITL tumorigenesis that requires both TET2 deficiency and expression of RhoA G17V. Loss of TET2 in hematopoietic progenitor cells is shown to predispose mice to both lymphoid and myeloid malignancies, but an additional RhoA G17V mutation in T cells is essential to initiate Tfh-driven lymphomas.51-54 These studies also showed that combining TET2-deficiency with the RhoA G17V mutation synergistically elevates Tfh differentiation, as well as PI3K-mTORC1 and TCR signaling pathways, which leads to AITL-like disease. Although spleen and swollen lymph nodes (<2 mm in diameter) in these mouse models share histological features with human AITL, they do not phenocopy the hypergammaglobulinemia and palpable lymphadenopathy seen in humans. In contrast, Roquinsan/+ mice give rise to easily detectable tumors with most of the pathological features of human AITL. The gross change in the size of tumor lymph nodes provides a unique opportunity to test the impact of gene deletion or drug treatment in a preclinical setting. Despite these advantages, it should be noted that ROQUIN gene mutations have not been detected in human AITL.30 Also, it remains unclear whether T cells in Roquinsan/+ mice transform into tumor cells in response to elevated TCR or cytokine signaling, as is often shown in peripheral T-cell lymphomas.13,55 Therefore, our findings made in Roquinsan/+ mice should be further validated in more relevant disease models. In this context, patient-derived xenograft models using human AITL cell lines with defined causative mutations may provide further insights.56,57
In summary, our data suggest that ongoing T-B-cell cross talk is a driving force for the progression of Tfh-derived T-cell lymphomas. Importantly, this may apply not only to AITL but also to other peripheral T-cell lymphomas with features of Tfh origin. Therefore, we propose that future therapies for AITL focus on T-cell signaling components that mediate T-B-cell interactions.
The authors will share renewable materials, data sets, and protocols upon e-mail request to the corresponding author.
Acknowledgment
The authors thank C. Vinuesa (Australian National University, Canberra, Australia) for the Roquinsan/+ mice, T. Takemori (RIKEN, Yokohama City, Japan) for Bcl6 cko, I. King (McGill University) for assistance with the ICAM-1 binding assay, and the IRCM Flow Cytometry Core Facility and IRCM Animal Facilities for their services.
This work was supported by operating grants from the Cancer Research Society (W.-K.S.) and Canadian Institutes of Health Research (CIHR) grants MT-14429, MOP-82906, and FDN-143338 (A.V.). A.V. holds the Canada Research Chair on Signaling in the Immune System. M.W. and J.L. received CIHR Master’s Awards, and M.W. and V.P. received Fonds de Recherche du Québec–Santé (FRQS) Doctoral Scholarships.
Authorship
Contribution: W.-K.S. conceptualized the study; M.W., J.C., M.-C.Z., Y.B., V.P., J.L., T.B., and A.V. developed experimental tools; M.W. performed the experiments; M.W. and W.-K.S. analyzed the data and prepared the figures; M.W. and W.-K.S. wrote the manuscript with input from J.C., Y.B., V.P., J.L., T.B., M.-C.Z., A.V., S.J.K., W.S.K., and Y.H.K.; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: A.V. has a contract with Bristol Myers-Squibb to study the mechanism of action of anti-SLAMF7 monoclonal antibody elotuzumab in multiple myeloma. The remaining authors declare no competing financial interests.
Correspondence: Woong-Kyung Suh, IRCM, 110 Ave des Pins Ouest, Montréal, QC H2W IR7, Canada; e-mail: woong-kyung.suh@ircm.qc.ca.

