

Key Points
The exocyst component EXOC3 controls platelet granule secretion and glycoprotein receptor trafficking in platelets.
Deletion of platelet EXOC3 exacerbates arterial thrombosis and enhances hemostatic function.

Abstract
The exocyst is an octameric complex comprising 8 distinct protein subunits, exocyst complex components (EXOC) 1 to 8. It has an established role in tethering secretory vesicles to the plasma membrane, but its relevance to platelet granule secretion and function remains to be determined. Here, EXOC3 conditional knockout (KO) mice in the megakaryocyte/platelet lineage were generated to assess exocyst function in platelets. Significant defects in platelet aggregation, integrin activation, α-granule (P-selectin and platelet factor 4), dense granule, and lysosomal granule secretion were detected in EXOC3 KO platelets after treatment with a glycoprotein VI (GPVI)-selective agonist, collagen-related peptide (CRP). Except for P-selectin exposure, these defects were completely recovered by maximal CRP concentrations. GPVI surface levels were also significantly decreased by 14.5% in KO platelets, whereas defects in proximal GPVI signaling responses, Syk and LAT phosphorylation, and calcium mobilization were also detected, implying an indirect mechanism for these recoverable defects due to decreased surface GPVI. Paradoxically, dense granule secretion, integrin activation, and changes in surface expression of integrin αIIb (CD41) were significantly increased in KO platelets after protease-activated receptor 4 activation, but calcium responses were unaltered. Elevated integrin activation responses were completely suppressed with a P2Y12 receptor antagonist, suggesting enhanced dense granule secretion of adenosine 5′-diphosphate as a critical mediator of these responses. Finally, arterial thrombosis was significantly accelerated in KO mice, which also displayed improved hemostasis determined by reduced tail bleeding times. These findings reveal a regulatory role for the exocyst in controlling critical aspects of platelet function pertinent to thrombosis and hemostasis.
Introduction
Platelet granule secretion controls multiple aspects of platelet function extending beyond the classical paradigm of thrombosis and hemostasis. Therefore, the identity of target genes that can control platelet secretion, and control of specific granule subtypes, remains an attractive therapeutic avenue. Currently, there are multiple genes implicated as regulators of granule secretion. Clinically, the evidence is principally derived from patients with familial hemophagocytic lymphohistiocytosis (FHL), including FHL-3 (Munc13-4), FHL-4 (syntaxin 11), and FHL-5 (Munc18b), whose clinical symptoms often include a bleeding diathesis.1-3 Further studies on gene knockout (KO)/transgenic mice corroborate these findings but have also uncovered other secretory genes, including Rab27B, the v-SNARE VAMP8, and the t-SNARE SNAP23, whose absence causes a complete ablation of secretion from α-, dense, and lysosomal granules.4-7
The exocyst is a highly conserved octameric complex comprising 8 protein subunits, exocyst complex components (EXOC) 1 to 8. Each EXOC protein contains an extended coiled-coil domain, termed the “CorEx” motif, which mediates antiparallel pairing between components (EXOC1-2, EXOC3-4, EXOC5-6, and EXOC7-8) forming 4 heterodimers, providing the structural basis for assembly of the holo-complex.8 Historically, research on the identification and characterization of this complex has been inferred from yeast, with more recent studies in mammalian cells implicating this complex in a wide range of biological processes, including cell cycle progression, morphogenesis, migration, autophagy, and tumorigenesis.9-13 The best-characterized role for the exocyst is to regulate polarized secretion by trafficking and tethering secretory vesicles to the plasma membrane before SNARE-mediated fusion, which occurs through multiple interactions of exocyst components with t- and v-SNAREs and SNARE regulatory proteins, and directly with the plasma membrane.14-17
The targeting of the exocyst to the plasma membrane is regulated by the Ral guanosine triphosphatases (GTPases), in which EXOC2 and EXOC8 competitively bind GTP-loaded Ral.18,19 Recently, our group identified a redundant role for the Ral GTPases RalA and RalB as selective regulators of P-selectin exposure from α-granules.20 RalAB double KO (DKO) platelets exhibit normal dense and lysosomal granule release, whereas secretion of “soluble” α-granule cargo was also preserved. Proteomic studies have confirmed the expression of all EXOC proteins in human and mouse platelets, and phospho-proteomic analysis of platelet proteins identified EXOC1, EXOC4, and EXOC7 to be phosphorylated upon platelet activation.21-23 However, studies assessing functional roles for the exocyst in platelets are limited. Using permeabilized human platelets, Kawato et al24 showed that blocking the Ral–EXOC2 interaction with an antibody against the Ral-binding domain of EXOC2 could inhibit GppNHp-induced dense granule secretion. Here, platelet activation also induced an association between RalA and EXOC4, which is consistent with our observations showing constitutively active RalA and RalB bait proteins pulldown EXOC4. However, our functional analysis of RalAB DKO murine platelets revealed no apparent defect in dense granule secretion.20
Herein, we generated the first platelet-specific conditional mouse KO of an exocyst component, EXOC3, as a proxy to characterize and investigate the role of this complex in murine platelets. Studies from yeast and mammalian cells are supportive of a lack of functional redundancy between EXOC proteins regulating exocytosis, such that targeting an individual component ablates complex function.14,16,25 We hypothesized that loss of platelet EXOC3 would result in a relatively similar platelet phenotype to RalAB DKO mice, in which thrombosis and hemostasis are preserved but a selective secretion defect of P-selectin surface expression exists.
Materials and methods
Materials
Details of materials are provided in the supplemental Materials and methods.
Mice
EXOC3tm2a(EUCOMM)Hmgu mice were from the Wellcome Trust Sanger Institute. These were initially crossed with Flp recombinase transgenic mice, with subsequent crossing against PF4-Cre transgenic mice to generate wild-type (WT, PF4–) and conditional KO (PF4+) of EXOC3 in platelets. All animal studies were approved by the University of Bristol Research Ethics Committee, with mice bred and maintained in accordance with the UK Home Office regulations and Animals (Scientific Procedures) Act of 1986 (PPL No: 300/3445).
Washed platelet isolation
Age-matched (8-20 weeks) and sex-matched mice were killed by a gradual rise in carbon dioxide, with blood immediately drawn from the inferior vena cava into 4% (w/v) trisodium citrate. Anticoagulated blood was mixed with 150 µL acid citrate dextrose (71 mM citric acid, 85 mM sodium citrate, 111 mM glucose) and 800 µL modified N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-Tyrode’s buffer (135 mM NaCl, 3 mM KCl, 10 mM HEPES, 5 mM glucose, and 1 mM MgCl2, pH 7.3) and centrifuged at 180g for 8 minutes. Platelet-rich plasma was removed, with 2× further centrifugations and added HEPES-Tyrode’s buffer to give a total platelet-rich plasma volume of 2 mL. Platelets were pelleted at 580g for 10 minutes containing indomethacin (10 µM) and apyrase (0.02 U/mL), adjusted to 4 × 108/mL with apyrase (0.02 U/mL) and allowed to recover for at least 30 minutes at 30°C. For human platelet protein analysis, whole blood was drawn, after receipt of informed consent, from healthy volunteers in accordance with the Declaration of Helsinki and prepared as previously described.26
Hematology
Whole blood was analyzed on a Pentra ES 60 analyzer (Horiba).
Lumi-aggregometry
Simultaneous assessment of platelet aggregation and dense granule secretion (adenosine triphosphate [ATP] release) was performed as previously described.27
Platelet treatment with Naja kaouthia protease
Naja kaouthia (Nk) protease (from the venom of the cobra Naja kaouthia), which has also been previously shown to cleave murine glycoprotein (GP) Ibα,28 was purified as described elsewhere.29 Pelleted platelets were resuspended in HEPES-Tyrode’s buffer (4 × 108/mL) and treated with or without 5 µg/mL Nk protease containing 1 mM CaCl2 for 30 minutes at 37°C. Next, a 2× volume of CGS buffer (120 mM NaCl, 25.8 mM trisodium citrate, 5.5 mM glucose, pH 6.5) was added, and platelets were pelleted and resuspended as described.
Calcium signaling
Pelleted platelets were resuspended in CGS buffer (4 × 108/mL) and incubated for 45 minutes at 30°C with 0.1% (w/v) Pluronic F-127, 0.35% (w/v) bovine serum albumin, and 4 µM Fura-2 LR/AM. Dye-loaded platelets were pelleted and resuspended in HEPES-Tyrode’s buffer (containing 1 mM CaCl2) and allowed to recover. Platelets (1 × 108/mL) were stimulated at 37°C under nonstirring conditions on an Infinite M200 Pro plate reader (Tecan) with indicated agonist concentrations for a total of 30 cycles.
Generation of soluble and particulate platelet fractions
A modified method described by Holden and Horton30 was used. In brief, platelets (1 × 109/mL) were treated as described and mixed with an equal volume of 2× permeabilization buffer (final concentration, 1× phosphate-buffered saline [PBS], 0.1% [w/v] digitonin, protease and phosphatase inhibitors) for 5 minutes on ice, then centrifuged at 10 000g for 10 minutes (4°C) to collect the “soluble” fraction. The remaining pellet was carefully washed 1× with PBS, then treated with an equal volume of 1× lysis buffer (1× PBS, 1% [v/v] Triton X-100, protease and phosphatase inhibitors), relative to the soluble fraction volume, for 10 minutes. Samples were centrifuged and the supernatant containing the “particulate” fraction removed. Fractions were mixed with 4× sodium dodecyl sulfate sample buffer, with 20 µL volumes loaded for western blotting analysis.
In vitro thrombosis
Anticoagulated whole blood thrombus formation at arterial shear was performed as previously reported.31
Arterial thrombosis
WT or EXOC3 KO mice were weighed and anesthetized by intraperitoneal injection (100 mg/kg ketamine and 10 mg/kg xylazine). Platelets were labeled with a Dylight-488–conjugated anti-GPIbβ antibody (0.1 µg/g body weight; Emfret Analytics) following IV infusion into the jugular vein. The carotid artery was exposed, and a 2 × 1-mm piece of filter paper, saturated with 12% ferric chloride, was applied for 1 minute. Thrombus formation was monitored for 15 minutes by fluorescence time-lapsed microscopy using a BX51-WI microscope (Olympus) with a Rolera-XR digital camera (QImaging) and recorded by using StreamPix 4.24.0 (NorPix). Images were analyzed by using ImageJ software 1.46 as previously reported.32
Tail vein bleeding
Anesthetized mice had a 5-mm segment of the mouse tail excised and immediately immersed in a tube containing prewarmed saline (37°C). Total bleed time was recorded up to an experimental end point of 10 minutes (600 seconds).
Statistical analysis
Statistical analysis was performed by using GraphPad Prism 8. Data are representative of >3 experiments, presented as mean ± standard error of the mean, with exact numbers of independent experiments detailed in each figure legend. Statistical differences between samples were determined by using 2-tailed Student t tests and 2-way analyses of variance where appropriate, with Bonferroni’s post hoc test. A value of P < .05 was considered statistically significant.
Results
Characterization of the exocyst complex in platelets, hematology, and platelet surface receptor analysis from EXOC3 KO mice
Initial experiments set out to characterize the exocyst and our KO model in platelets. Loss of EXOC3 in KO platelets was confirmed, and densitometric analysis revealed a significant reduction in expression of other exocyst components compared with WT platelets (Figure 1A). Expression of α-granule proteins (PF4 and P-selectin/CD62P) and previously characterized platelet secretion regulators (RalA, Munc13-4, VAMP-8, and SNAP-23) were comparable between WT and KO platelets. Experiments in resting or CRP-stimulated WT platelets revealed that EXOC3, EXOC4, and EXOC7 could coimmunoprecipitate with EXOC2 to comparable levels. Similarly, in KO platelets, exocyst components could coimmunoprecipitate irrespective of activation, although at much lower levels compared with WT platelets (Figure 1B). Phos-tag acrylamide experiments were used to assess EXOC phosphorylation changes in response to CRP. In WT platelets, time-dependent phosphorylation and dephosphorylation changes were observed with EXOC4 and EXOC7, respectively, whereas no phosphorylation changes were detected with EXOC3. Lambda phosphatase treatment of lysates confirmed that the upper bands for pEXOC4 and pEXOC7 were specific phosphorylated forms of the protein (supplemental Figure 1). A similar pattern was observed in KO platelets, although under resting conditions, a significant enhancement of phosphorylated EXOC4 was observed (Figure 1C).
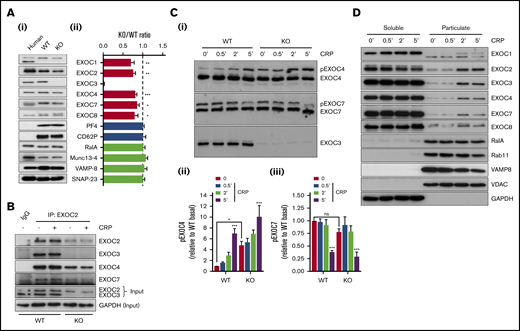
Characterization of exocyst expression, association, posttranslational modification, and movement in EXOC3 KO platelets. (Ai) Representative immunoblots of exocyst complex components and platelet secretory markers from human and murine (WT and EXOC3 KO) platelets are shown. (Aii) Densitometric analysis of relative murine protein abundance expressed as a KO:WT ratio after loading adjustment. (B) Washed platelets (5 × 108/mL) from WT and KO platelets were rested or stimulated with 5 µg/mL CRP for 5 minutes, lysed, and subjected to coimmunoprecipitation analysis of exocyst components with an EXOC2 antibody. Immunoprecipitation inputs were monitored for exocyst expression and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as loading control. (Ci) Lysates from WT and KO platelets (5 × 108/mL) treated with 5 µg/mL CRP for indicated times (minutes) were subject to phos-tag immunoblot analysis of EXOC3, 4 and 7. Densitometric analysis of changes in phosphorylated EXOC4 (Cii) and EXOC7 (Ciii) (pEXOC4/pEXOC7) were measured. (D) After stimulation of washed platelets (1 × 109/mL) with 5 µg/mL CRP for indicated times (minutes), platelet proteins were separated into soluble and particulate fractions and analyzed for exocyst expression and various “control” proteins for the respective fractions. Data are presented as the mean ± standard error of the mean. n = 5 (A); n = 3 (B-D). *P < .05, **P < .01, ***P < .001 vs WT (A), unstimulated sample (Cii-iii), or as indicated. IgG, immunoglobulin G; ns, not significant.
Characterization of exocyst expression, association, posttranslational modification, and movement in EXOC3 KO platelets. (Ai) Representative immunoblots of exocyst complex components and platelet secretory markers from human and murine (WT and EXOC3 KO) platelets are shown. (Aii) Densitometric analysis of relative murine protein abundance expressed as a KO:WT ratio after loading adjustment. (B) Washed platelets (5 × 108/mL) from WT and KO platelets were rested or stimulated with 5 µg/mL CRP for 5 minutes, lysed, and subjected to coimmunoprecipitation analysis of exocyst components with an EXOC2 antibody. Immunoprecipitation inputs were monitored for exocyst expression and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as loading control. (Ci) Lysates from WT and KO platelets (5 × 108/mL) treated with 5 µg/mL CRP for indicated times (minutes) were subject to phos-tag immunoblot analysis of EXOC3, 4 and 7. Densitometric analysis of changes in phosphorylated EXOC4 (Cii) and EXOC7 (Ciii) (pEXOC4/pEXOC7) were measured. (D) After stimulation of washed platelets (1 × 109/mL) with 5 µg/mL CRP for indicated times (minutes), platelet proteins were separated into soluble and particulate fractions and analyzed for exocyst expression and various “control” proteins for the respective fractions. Data are presented as the mean ± standard error of the mean. n = 5 (A); n = 3 (B-D). *P < .05, **P < .01, ***P < .001 vs WT (A), unstimulated sample (Cii-iii), or as indicated. IgG, immunoglobulin G; ns, not significant.
To assess exocyst localization, WT platelets were untreated or activated with CRP at defined time points and separated into “soluble” or “particulate” fractions. In resting platelets, the exocyst complex was predominantly localized to the soluble fraction, consistent with a cytosolic localization as verified by glyceraldehyde-3-phosphate dehydrogenase enrichment. However, upon stimulation, a portion of the complex mobilized to the particulate fraction, enriched with platelet organelles, indicated by markers of secretory vesicles (RalA and VAMP8), late/recycling endosomes (Rab11), and mitochondria (VDAC) (Figure 1D). Identical movement of the exocyst was also observed in human platelets (data not shown).
Hematologic analysis of KO blood revealed no significant changes in blood cell counts, but there was a small, yet significant increase in mean platelet volume (8.3% vs WT) (Table 1). Analysis of platelet GP receptors in KO platelets revealed significant alterations in surface expression, with CD42b/GPIbα and CD49b/GPIa levels increased by 20.3% and 8.4%, respectively, whereas GPVI levels were reduced by 14.5%. CD41/GPIIb levels were not altered.
Analysis of whole blood and platelet receptor levels in EXOC3 KO mice
| Parameter | WT | KO | N | P | ||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| WBC, ×103/μL | 6.34 | 1.37 | 7.569 | 2.88 | 13 | .19 |
| RBC, ×106/μL | 9.63 | 1.21 | 10.42 | 1.81 | 13 | .49 |
| Plt, ×103/μL | 827 | 155 | 735 | 160 | 13 | .09 |
| MPV, µm3 | 5.27 | 0.23 | 5.71 | 0.21 | 13 | .001 |
| PCT, % | 0.34 | 0.08 | 0.33 | 0.06 | 13 | .42 |
| CD41, MFI | 6904 | 1645 | 7012 | 1428 | 13 | .67 |
| CD42b, MFI | 4194 | 1352 | 5049 | 1266 | 13 | .001 |
| GPVI, MFI | 734 | 186 | 628 | 127 | 13 | .002 |
| CD49b, MFI | 589 | 107 | 639 | 104 | 13 | .01 |
| Parameter | WT | KO | N | P | ||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| WBC, ×103/μL | 6.34 | 1.37 | 7.569 | 2.88 | 13 | .19 |
| RBC, ×106/μL | 9.63 | 1.21 | 10.42 | 1.81 | 13 | .49 |
| Plt, ×103/μL | 827 | 155 | 735 | 160 | 13 | .09 |
| MPV, µm3 | 5.27 | 0.23 | 5.71 | 0.21 | 13 | .001 |
| PCT, % | 0.34 | 0.08 | 0.33 | 0.06 | 13 | .42 |
| CD41, MFI | 6904 | 1645 | 7012 | 1428 | 13 | .67 |
| CD42b, MFI | 4194 | 1352 | 5049 | 1266 | 13 | .001 |
| GPVI, MFI | 734 | 186 | 628 | 127 | 13 | .002 |
| CD49b, MFI | 589 | 107 | 639 | 104 | 13 | .01 |
Whole blood from WT and EXOC3 KO mice was analyzed on a Pentra ES 60 hematologic analyzer (Horiba), with cell counts corrected for citrate dilution. Washed platelets (2 × 107/mL) from WT and EXOC3 KO platelets were analyzed for surface expression of CD41 (GPIIb), CD42b (GPIbα), GPVI, and CD49b (GPIa) by using flow cytometry. Median fluorescent intensity (MFI) values are reported. Data are presented as the mean ± standard deviation (SD). n = 13. An unpaired, 2-tailed Student t test was used to determine P values, with statistically significant differences in bold.
MPV, mean platelet volume; PCT, plateletcrit; Plt, platelet; RBC, red blood cell; WBC, white blood cell.
Defective GPVI-mediated responses from EXOC3 KO platelets
Platelet responses to GPVI stimulation revealed significant defects in KO platelet aggregation (Figure 2A-Bi), dense granule secretion (Figure 2A-Bii), integrin GPIIbIIIa (αIIbβ3) activation (Figure 2Ci), and α-granule secretion involving P-selectin surface exposure (Figure 2Cii). Apart from P-selectin exposure, all defective responses were fully recovered with increasing CRP concentrations. Thrombus formation over collagen was also significantly reduced in KO blood (Figure 2D). To understand these functional defects further, proximal signaling responses downstream of GPVI receptor ligation were assessed. Activation of Syk and LAT after CRP treatment of 0.5 minute was significantly reduced in KO platelets (Figure 3A), whereas GPVI-mediated calcium responses were also significantly attenuated. Analysis of serine phosphorylated PKC substrates in KO platelets also revealed defects in response to CRP (supplemental Figure 2). Further downstream of GPVI signaling, “rescue” experiments with exogenous adenosine 5′-diphosphate (ADP) fully recovered the defect in CRP-mediated αIIbβ3 activation in KO platelets (Figure 3Ci), but the defect in P-selectin exposure was not fully rescued (Figure 3Cii). Similarly, combined stimulation of KO platelets with ADP and U46619 displayed normal integrin activation but a significant defect in P-selectin exposure.

Defective functional responses in EXOC3 KO platelets after GPVI stimulation and whole blood thrombus formation over collagen. (A-B) Washed platelets (2 × 108/mL) from WT and EXOC3 KO platelets were assessed for CRP-induced platelet aggregation and dense granule secretion responses by lumi-aggregometry. Representative aggregation (Ai-iii) and secretion (Aiv-vi) traces in response to increasing concentrations of CRP are shown. Quantified area under the curve (AUC) (Bi) and peak ATP (Bii) values were calculated to determine changes in aggregation and secretion, respectively. (C) Washed platelets (2 × 107/mL) from WT and EXOC3 KO mice were treated with indicated concentrations of CRP (0.1-30 µg/mL) for 10 minutes, fixed, and analyzed for integrin αIIbβ3 activation (i) and α-granule secretion of P-selectin (CD62P) (ii) by flow cytometry using a JON/A and P-selectin antibody, respectively. (D) Anticoagulated whole blood from WT and EXOC3−/− mice was loaded with 2 µM DiOC6 for 10 minutes and subsequently perfused over a collagen-coated surface (50 µg/mL) at 1000 s−1 for 3 minutes. Representative images of thrombus accumulation (i) and quantification of change in percent surface coverage over time (ii) and end point at 3 minutes (iii) are provided (scale bar, 200 µm). Data are presented as the mean ± standard error of the mean. n = 5-7 (A-B); n = 7 (C); n = 5 (D). *P < .05, **P < .01, ***P < .001, ****P < .0001 vs indicated sample.
Defective functional responses in EXOC3 KO platelets after GPVI stimulation and whole blood thrombus formation over collagen. (A-B) Washed platelets (2 × 108/mL) from WT and EXOC3 KO platelets were assessed for CRP-induced platelet aggregation and dense granule secretion responses by lumi-aggregometry. Representative aggregation (Ai-iii) and secretion (Aiv-vi) traces in response to increasing concentrations of CRP are shown. Quantified area under the curve (AUC) (Bi) and peak ATP (Bii) values were calculated to determine changes in aggregation and secretion, respectively. (C) Washed platelets (2 × 107/mL) from WT and EXOC3 KO mice were treated with indicated concentrations of CRP (0.1-30 µg/mL) for 10 minutes, fixed, and analyzed for integrin αIIbβ3 activation (i) and α-granule secretion of P-selectin (CD62P) (ii) by flow cytometry using a JON/A and P-selectin antibody, respectively. (D) Anticoagulated whole blood from WT and EXOC3−/− mice was loaded with 2 µM DiOC6 for 10 minutes and subsequently perfused over a collagen-coated surface (50 µg/mL) at 1000 s−1 for 3 minutes. Representative images of thrombus accumulation (i) and quantification of change in percent surface coverage over time (ii) and end point at 3 minutes (iii) are provided (scale bar, 200 µm). Data are presented as the mean ± standard error of the mean. n = 5-7 (A-B); n = 7 (C); n = 5 (D). *P < .05, **P < .01, ***P < .001, ****P < .0001 vs indicated sample.
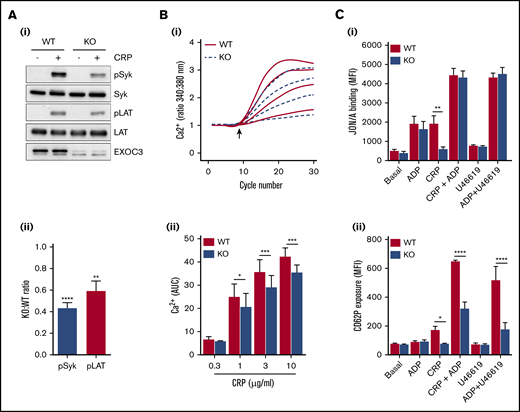
Alterations in proximal signaling events and dense granule secretion underlie defective GPVI responses in EXOC3 KO platelets. (A) Representative immunoblots of changes in phosphorylation of early GPVI signaling molecules, Syk and LAT, after CRP treatment (5 µg/mL) for 0.5 minute (i) with densitometric analysis of relative changes in Syk/LAT phosphorylation expressed as a KO:WT ratio after total protein loading adjustment (ii). (B) Washed platelets (1 × 108/mL) from WT and EXOC3 KO platelets were loaded with Fura-2AM before stimulation with increasing concentrations of CRP (0.3-10 µg/mL) and monitored for changes in cytosolic calcium levels. The kinetics of mean changes in calcium responses from 4 independent experiments (i) with quantified area under the curve (AUC) analysis (ii) are presented. The arrow in panel Bi denotes agonist addition. (C) Washed platelets (2 × 107/mL) were treated for 10 minutes with ADP (10 µM), CRP (0.3 µg/mL), U46619 (3 µM) alone, or combined as indicated, fixed, and monitored for changes in integrin αIIbβ3 activation (i) and α-granule secretion of P-selectin (CD62P) (ii) by using flow cytometry. Data are presented as the mean ± standard error of the mean. n = 7 (A); n = 4 (B); n = 3 (C). *P < .05, **P < .01, ***P < .001, ****P < .0001 vs indicated sample.
Alterations in proximal signaling events and dense granule secretion underlie defective GPVI responses in EXOC3 KO platelets. (A) Representative immunoblots of changes in phosphorylation of early GPVI signaling molecules, Syk and LAT, after CRP treatment (5 µg/mL) for 0.5 minute (i) with densitometric analysis of relative changes in Syk/LAT phosphorylation expressed as a KO:WT ratio after total protein loading adjustment (ii). (B) Washed platelets (1 × 108/mL) from WT and EXOC3 KO platelets were loaded with Fura-2AM before stimulation with increasing concentrations of CRP (0.3-10 µg/mL) and monitored for changes in cytosolic calcium levels. The kinetics of mean changes in calcium responses from 4 independent experiments (i) with quantified area under the curve (AUC) analysis (ii) are presented. The arrow in panel Bi denotes agonist addition. (C) Washed platelets (2 × 107/mL) were treated for 10 minutes with ADP (10 µM), CRP (0.3 µg/mL), U46619 (3 µM) alone, or combined as indicated, fixed, and monitored for changes in integrin αIIbβ3 activation (i) and α-granule secretion of P-selectin (CD62P) (ii) by using flow cytometry. Data are presented as the mean ± standard error of the mean. n = 7 (A); n = 4 (B); n = 3 (C). *P < .05, **P < .01, ***P < .001, ****P < .0001 vs indicated sample.
Dense granule secretion, integrin αIIbβ3 activation, and changes in CD41 surface expression are elevated in EXOC3 KO platelets after protease-activated receptor 4 activation
Assessment of protease-activated receptor 4–activating peptide (PAR4-AP)–induced platelet aggregation revealed no significant difference in WT and KO responses (Figure 4A-Bi). However, dense granule secretion analysis of the same sample sets showed significantly enhanced levels of ATP release in KO platelets (Figure 4Bii). Integrin αIIbβ3 activation was also significantly elevated in KO platelets (Figure 4Ci), but P-selectin exposure was unaltered (Figure 4Cii). Furthermore, KO platelets had significantly increased levels of changes in surface CD41 expression in response to PAR4-AP or thrombin stimulation (Figure 4D). To understand these enhanced responses, calcium levels were assessed as a readout of PAR4 receptor activity, which did not reveal any significant alteration in calcium mobilization in KO platelets (Figure 5A). Subsequent experiments in platelets pretreated with the P2Y12 inhibitor cangrelor (AR-C) showed that PAR4-mediated enhancement of αIIbβ3 activation was inhibited in KO platelets, confirming a critical role for enhanced dense granule secretion (Figure 5B). To try and understand a mechanism for this finding, platelets were pretreated with the αIIbβ3 antagonist tirofiban to determine if increased outside-in signaling was a contributing factor. However, enhanced dense granule secretion in KO platelets was preserved in the presence of tirofiban (Figure 5C), despite a complete ablation of aggregation responses (supplemental Figure 8). Platelet spreading on fibrinogen-coated surfaces was also unaltered in KO platelets (data not shown). It was further speculated that the increase in CD42b surface receptor levels in KO platelets could be facilitating PAR4 responses, analogous to a role played by CD42b in facilitating PAR4 responses in murine platelets.33 Cleavage of the major ligand-binding subunit of CD42b with purified Nk protease was verified (supplemental Figure 9A), but loss of this ectodomain fragment did not attenuate dense granule release in KO (or WT) platelets (Figure 5D).
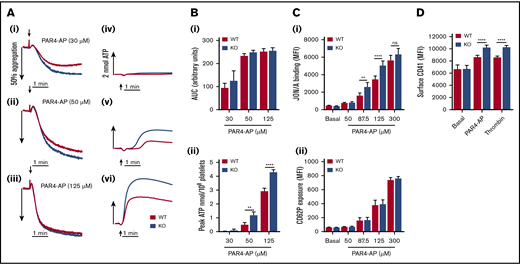
EXOC3 KO platelets elicit hyperactive responses to stimulation with PAR4-AP. (A-B) Washed platelets (2 × 108/mL) from WT and EXOC3 KO platelets were stimulated with increasing concentrations of PAR4-AP and monitored for changes in platelet aggregation and dense granule secretion. Representative aggregation (Ai-iii) and secretion (Aiv-vi) traces are shown. Quantified area under the curve (AUC) (Bi) and peak ATP (Bii) values were calculated to assess aggregation and secretion responses, respectively. (C) Washed platelets (2 × 107/mL) from WT and EXOC3 KO mice were treated with indicated concentrations of PAR4-AP (50-300 µM) for 10 minutes, fixed and analyzed for JON/A binding (i) and P-selectin surface exposure (CD62P) (ii) by using flow cytometry. (D) Similar to panel C, washed platelets were stimulated with PAR4-AP (300 µM) or thrombin (1 U/mL) for 10 minutes and monitored for changes in surface CD41 levels. Data are presented as the mean ± standard error of the mean. n = 4-5 (A-B); n = 8 (C); n = 9 (D). **P < .01, ****P < .0001 vs indicated sample.
EXOC3 KO platelets elicit hyperactive responses to stimulation with PAR4-AP. (A-B) Washed platelets (2 × 108/mL) from WT and EXOC3 KO platelets were stimulated with increasing concentrations of PAR4-AP and monitored for changes in platelet aggregation and dense granule secretion. Representative aggregation (Ai-iii) and secretion (Aiv-vi) traces are shown. Quantified area under the curve (AUC) (Bi) and peak ATP (Bii) values were calculated to assess aggregation and secretion responses, respectively. (C) Washed platelets (2 × 107/mL) from WT and EXOC3 KO mice were treated with indicated concentrations of PAR4-AP (50-300 µM) for 10 minutes, fixed and analyzed for JON/A binding (i) and P-selectin surface exposure (CD62P) (ii) by using flow cytometry. (D) Similar to panel C, washed platelets were stimulated with PAR4-AP (300 µM) or thrombin (1 U/mL) for 10 minutes and monitored for changes in surface CD41 levels. Data are presented as the mean ± standard error of the mean. n = 4-5 (A-B); n = 8 (C); n = 9 (D). **P < .01, ****P < .0001 vs indicated sample.

Enhanced dense granule secretion of ADP potentiates integrin αIIbβ3activation in PAR4-AP–stimulated EXOC3 KO platelets, which is independent of changes in calcium mobilization, integrin outside-in signaling, and GPIbα surface levels. (A) Washed platelets (1 × 108/mL) from WT and EXOC3 KO platelets were stimulated with increasing concentrations of PAR4-AP (50-300 µM) and monitored for changes in cytosolic calcium levels. The kinetics of mean changes in calcium responses from 4 independent experiments (i) with quantified area under the curve (AUC) analysis (ii) are presented. (B) Washed platelets (2 × 107/mL) were pretreated for 10 minutes with vehicle or 1 µM P2Y12 receptor antagonist cangrelor (AR-C) before stimulation with 125 µM PAR4-AP and monitored for changes in JON/A binding by using flow cytometry. Washed platelets (2 × 108/mL) from WT and EXOC3 KO platelets were pretreated with vehicle, tirofiban (50 µg/mL) (C) or Nk protease (5 µg/mL) (D), and assessed for changes in PAR4-AP (125 µM)–mediated dense granule secretion responses by lumi-aggregometry. Representative secretion traces (Ci,Di) and peak ATP secretion values (Cii,Dii) are displayed. Data are presented as the mean ± standard error of the mean. n = 4 (A); n = 5 (B); n = 3 (C); n = 4 (D). *P < .05, **P < .01, ****P < .0001 vs indicated sample. RFU, relative fluorescence units.
Enhanced dense granule secretion of ADP potentiates integrin αIIbβ3activation in PAR4-AP–stimulated EXOC3 KO platelets, which is independent of changes in calcium mobilization, integrin outside-in signaling, and GPIbα surface levels. (A) Washed platelets (1 × 108/mL) from WT and EXOC3 KO platelets were stimulated with increasing concentrations of PAR4-AP (50-300 µM) and monitored for changes in cytosolic calcium levels. The kinetics of mean changes in calcium responses from 4 independent experiments (i) with quantified area under the curve (AUC) analysis (ii) are presented. (B) Washed platelets (2 × 107/mL) were pretreated for 10 minutes with vehicle or 1 µM P2Y12 receptor antagonist cangrelor (AR-C) before stimulation with 125 µM PAR4-AP and monitored for changes in JON/A binding by using flow cytometry. Washed platelets (2 × 108/mL) from WT and EXOC3 KO platelets were pretreated with vehicle, tirofiban (50 µg/mL) (C) or Nk protease (5 µg/mL) (D), and assessed for changes in PAR4-AP (125 µM)–mediated dense granule secretion responses by lumi-aggregometry. Representative secretion traces (Ci,Di) and peak ATP secretion values (Cii,Dii) are displayed. Data are presented as the mean ± standard error of the mean. n = 4 (A); n = 5 (B); n = 3 (C); n = 4 (D). *P < .05, **P < .01, ****P < .0001 vs indicated sample. RFU, relative fluorescence units.
Accelerated thrombosis and improved hemostatic function in EXOC3 KO mice
In vivo parameters of thrombosis and hemostasis were investigated in mice deficient in platelet EXOC3. Thrombosis was monitored by using a ferric chloride injury model of the carotid artery. Representative images of arterial thrombi at distinct time points are presented (Figure 6Ai). Arterial thrombosis was significantly accelerated in KO mice after 7.5 minutes of injury, whereas end point analysis at 15 minutes revealed no significant difference compared with WT mice (Figure 6Aii-iii). Hemostasis was modeled by using tail tip excision and total bleed time monitored for up to 10 minutes (600 seconds). It was observed that KO mice had significantly lower bleed time levels (245 seconds) compared with WT mice (359 seconds), consistent with improved hemostatic function (Figure 6B).
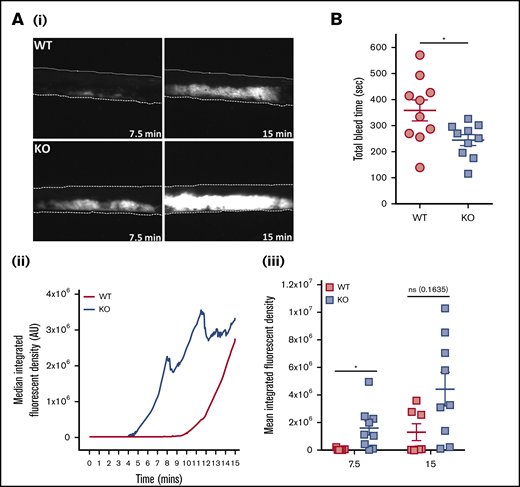
Thrombosis is accelerated and hemostasis improved in EXOC3 KO mice. (A) Anesthetized mice were infused with Dylight-488–conjugated anti-GPIbβ antibody before ferric chloride–mediated injury of the carotid artery up to a 15-minute experimental end point. Displayed are representative images of thrombus formation (i), with median fluorescence intensity values plotted over time (ii) and total median fluorescence intensity values compared at 7.5- and 15-minute time points (iii). (B) For tail bleeding analysis, anesthetized mice had tails excised 5 mm from the tip before immersion in 37°C saline, and total bleed time was monitored over a 10-minute experimental end point. Data are presented as the mean ± standard error of the mean. n = 7 for WT mice and n = 9 for KO mice (A); n = 10 (B). *P < .05 vs indicated sample. AU, arbitrary units.
Thrombosis is accelerated and hemostasis improved in EXOC3 KO mice. (A) Anesthetized mice were infused with Dylight-488–conjugated anti-GPIbβ antibody before ferric chloride–mediated injury of the carotid artery up to a 15-minute experimental end point. Displayed are representative images of thrombus formation (i), with median fluorescence intensity values plotted over time (ii) and total median fluorescence intensity values compared at 7.5- and 15-minute time points (iii). (B) For tail bleeding analysis, anesthetized mice had tails excised 5 mm from the tip before immersion in 37°C saline, and total bleed time was monitored over a 10-minute experimental end point. Data are presented as the mean ± standard error of the mean. n = 7 for WT mice and n = 9 for KO mice (A); n = 10 (B). *P < .05 vs indicated sample. AU, arbitrary units.
Discussion
To date, most studies investigating roles for the exocyst complex in human biology have adopted small interfering RNA, CRISPR/Cas-9 editing, or overexpression approaches in a cell culture environment to delineate roles regarding exocyst assembly, movement, and function.14,34-36 Here, we generated a conditional KO of platelet EXOC3, allowing both in vitro and in vivo functional assessment, with specific emphasis on platelet granule secretion and consequences for thrombosis and hemostasis.
EXOC3 KO platelets had significantly reduced expression of all other detected exocyst components, a finding aligned with other mammalian cells reporting decreased expression of EXOC proteins after selective targeting of an exocyst component (Figure 1A).36,37 It is possible that loss of EXOC3 causes an instability of the complex, and therefore unbound components are targeted for proteasomal degradation. Independent of activation status, our coimmunoprecipitation analysis shows the complex to be constitutively associated, even in the absence of EXOC3, albeit with reduced levels of intact complex (Figure 1B). It is difficult to understand whether this remaining intact complex is functional in KO platelets. Phos-tag analysis of lysates from CRP-stimulated platelets confirmed that posttranslational modifications of EXOC4 and EXOC7 persist in KO platelets, with a clear enhancement of pEXOC4 under resting conditions, but it is not clear what functional relevance these modifications have on exocyst function in platelets (Figure 1C). Phosphorylation of EXOC4 (Ser-32) has been reported in adipocytes, and despite an implicated role for the exocyst in regulating insulin-mediated GLUT4 trafficking in these cells, pEXOC4 has no apparent mechanistic involvement in this trafficking response.38,39 Interestingly, blocking pEXOC7 (Ser-250) inhibits matrix metalloproteinase secretion in tumor cells, whereas the same study also showed that pEXOC7 is important for increased exocyst complex assembly.40 Clearly, our phos-tag data on EXOC4 and EXOC7 do not discriminate between specific phosphorylation sites, whereas the exocyst coimmunoprecipitation and trafficking data imply the complex is fully assembled under resting/activated conditions. Considering the importance of pEXOC7 to secretory processes in other cell systems, it would be interesting to speculate that the time-dependent decrease in pEXOC7 in response to CRP reflects an “off” switch to limit/control granule tethering events involved in specific aspects of secretion. No phosphorylation changes of EXOC3 were observed, consistent with the study by Beck et al,21 but proteomic analysis of the platelet lysine “acetylome” confirmed acetylation of a lysine residue on EXOC3 under resting conditions.41 Cross talk between posttranslational modifications has been previously reported, such that acetylation of EXOC3 could regulate pEXOC4, but this is purely speculative.42 However, the enhanced basal level of pEXOC4 may also reflect a compensatory response due to loss of EXOC3, as is known to occur with gene KO approaches.43
Our exocyst trafficking data clearly show a “net movement” of the complex after activation from the “soluble” (cytosolic) to a “particulate” fraction, enriched with platelet organelles (Figure 1D). This includes platelet granules as evidenced by VAMP8 expression, which is constitutively bound to all granular (α-, dense, and lysosomal) membranes.4 Although these data do not definitively show which compartment the exocyst traffics to, it is highly probable it traffics to secretory granules consistent with its established role in exocytosis. Interestingly, loss of EXOC3 did not affect exocyst complex movement to the particulate fraction in response to CRP (supplemental Figure 3). It was further shown that the Ral GTPases RalA and RalB are partly required for this trafficking response, as the net movement of the exocyst was reduced in RalAB DKO platelets (supplemental Figure 4B). However, changes in pEXOC4 and pEXOC7 to CRP in RalAB DKO platelets was comparable to changes in WT platelets, suggesting a restricted role for the Ral GTPases in trafficking the exocyst to the target membranes (supplemental Figure 4A).
Key roles for the exocyst in controlling receptor trafficking have been established.38,44 In KO platelets, resting CD41 levels were comparable with those in WT platelets, but CD42b and CD49b levels were increased (Table 1). Considering the subtle, yet significant increase (8.3%) of mean platelet volume in KO platelets, this could explain, in part, the observed receptor level increase, and values have not been adjusted to account for this subtle change in volume. However, despite the larger cell surface area, GPVI surface levels were significantly decreased. Analysis of total CD42b and GPVI expression levels by western blotting did not reveal any significant differences between genotypes (supplemental Figure 5). It is unclear why directionality differences in terms of CD42b and GPVI surface level expression were observed but likely points to distinct mechanisms in which these platelet receptors are trafficked to and/or maintained on the platelet surface.
Analysis of the GPVI pathway revealed weak defects in CRP-mediated aggregation, ATP secretion, and αIIbβ3 activation in KO platelets, which were overcome by maximal CRP concentrations (Figure 2A-C). Similarly, weak defects in α-granule release (PF4) and lysosomal secretion (β-hexosaminidase) were also observed (supplemental Figure 6). Interestingly, defects in surface exposure of P-selectin were not fully restored in response to maximal CRP concentrations, despite integrin activation been fully recovered, implying a regulatory role for the exocyst in GPVI-mediated P-selectin release (Figure 2Cii). The other minor defects in GPVI responses could be interpreted in two ways: one that simply reflects a minor role for the exocyst in regulating GPVI responses, or the other, which involves an indirect effect, due to decreased surface expression. Defective CRP platelet responses have been reported in murine platelets with reduced (≤50%) GPVI levels.45 Analysis of proximal signaling nodes downstream of GPVI ligation revealed defects in activation of 2 critical GPVI components, Syk and LAT, in KO platelets (Figure 3A); calcium responses were also significantly reduced (Figure 3B).
It is difficult to appreciate how the exocyst could be directly affecting such early GPVI signaling responses. We therefore propose that defects in GPVI activation are an indirect consequence of decreased receptor levels, possibly affecting receptor clustering and avidity, causing decreased dense granule secretion and subsequent functional defects. In support of this, exogenous ADP fully recovered defects in CRP-mediated αIIbβ3 activation in KO platelets (Figure 3Ci). Notably, heterozygote FcR-γ chain mice, which have a 50% reduction in GPVI levels, exhibit normal shear-induced thrombus formation over collagen surfaces, suggesting that our observed defects in EXOC3 KO blood perfused over collagen (Figure 2D) may involve another undetermined mechanism.46
Assessment of the PAR4 pathway in KO platelets revealed several unexpected findings, notably, enhanced dense granule secretion and integrin activation (Figure 4A-Ci). Despite the clear increase in dense granule release, no significant enhancements of platelet aggregation were detected (Figure 4A). Under our experimental conditions, PAR4-mediated aggregation responses were insensitive to secreted ADP acting via P2Y12, as cangrelor did not alter WT or KO aggregation responses (supplemental Figure 7A). It was also confirmed that the enhanced dense granule release in EXOC3 KO platelets is preserved, despite an absence of ADP feedback via P2Y12 (supplemental Figure 7B). However, by fluorescence-activated cell sorting (FACS) analysis, we could clearly observe a critical functional role for secreted ADP in regulating PAR4-AP–induced integrin activation (Figure 5B). Unlike the clear defect in CRP-mediated P-selectin exposure in KO platelets, PAR4-AP–induced changes in surface P-selectin were unaltered, which may partly reflect compensatory input due to increased ADP release with PAR4 stimulation (Figure 4Cii). Changes in surface CD41 levels were significantly enhanced in KO platelets, possibly implying altered trafficking of CD41 reserves from the open canalicular system and not α-granules (Figure 4D). Similar surface enhancements of CD41 were observed with thrombin. It is important to note that the enhanced surface levels of CD41 could, in part, explain the increased levels of αIIbβ3 integrin activation observed in KO platelets.
Calcium mobilization in PAR4-AP–activated KO platelets was normal, suggesting that PAR4 surface levels are unaltered in EXOC3 KO platelets (Figure 5A). A possible role for enhanced outside-in signaling (due to increased CD41 levels) contributing to elevated dense granule release in KO platelets was excluded by lumi-aggregation experiments using tirofiban. Another critical component of PAR-mediated responses in platelets is CD42b, whose ectodomain contains multiple ligand-binding sites, and it has been well documented that thrombin responses require input from both PARs and CD42b.47,48 It has also been described that PAR4-mediated aggregation responses, with PAR4-AP, are reduced if the CD42b ectodomain is cleaved.33 Considering the significant increase in CD42b levels in EXOC3 KO platelets, we selectively cleaved its ectodomain to assess if it could, in part, facilitate increased PAR4-AP–induced KO responses. Cleavage of the CD42b ectodomain by Nk protease was verified by FACS analysis, and Nk-treated WT platelets had defective PAR4-AP and thrombin aggregation responses (supplemental Figure 9). However, enhanced dense granule secretion responses were preserved in Nk-treated KO platelets, excluding a cooperative role for CD42b in this response. Similarly, elevated CD41 surface levels and αIIbβ3 activation to PAR4-AP persisted in Nk-treated KO platelets (data not shown). Presently, there is no clear mechanism for the enhanced dense granule secretion response. Interestingly, a recent observation in human umbilical vein endothelial cells reported that depletion of EXOC4 or EXOC7 enhanced thrombin-induced release of von Willebrand factor from Weibel-Palade bodies and therefore corroborates our unexpected observations in platelets, suggesting the exocyst could serve as a “clamp” to specifically regulate dense granule secretion.49 However, it is also possible that alterations in platelet dense granule cargo sorting and/or biogenesis could be occurring at the level of the megakaryocyte. In this regard, dense granule maturation and differentiation in megakaryocytes is distinct from that in α-granules and requires cargo delivery from early/recycling endosomes where the exocyst complex resides, and could therefore explain the observed changes in platelet dense granule secretion.50-52
Considering the differential in vitro effects of GPVI- and PAR4-driven responses in EXOC3 KO platelets, it was interesting to assess the overall relevance of these within in vivo models. Gene KOs for both receptors display defects in thrombosis, with only bleeding defects reported for PAR4 KO mice.53,54 Here, arterial thrombosis revealed a significant acceleration of thrombosis in EXOC3 KO mice, with WT responses “catching up” toward the assay end point (Figure 6A). This would therefore be supportive of a dominant role for PAR4 responses in driving this response, presumably via enhanced dense granule secretion but also possibly via increased outside-in signaling (increased CD41 levels/activation) and input from changes in surface expression of other receptors, including CD42b. Hemostasis, quantified by total bleed time, was also significantly improved in EXOC3 KO mice and likely reflects a prominent role for PAR4 signaling in controlling blood loss, as reported in previous studies.54,55
In summary, the major and unexpected finding from EXOC3 KO platelets was increased PAR4-mediated dense granule secretion, which has potential consequences for the enhanced thrombotic and hemostatic responses observed. Also presented is sufficient evidence to support a key role for this complex in trafficking GPs to and from the platelet surface. Clearly, further mechanistic investigations on the exocyst complex should be directed toward early/recycling endosomal trafficking and granule biogenesis pathways in megakaryocytes, in particular dense granules. Notably, the exocyst appears to positively control specific α-granule secretion events (ie, P-selectin exposure but not PF4 release) in response to GPVI and secondary mediators of platelet activation, ADP and thromboxane A2. These observations suggest that in the absence of the exocyst, irreversible fusion of α-granule and plasma membranes in response to GPVI signaling is impeded by an inefficient tethering process. This hypothesis is directly aligned with our observations from RalAB DKO platelets, which is further supported by our exocyst trafficking data in these platelets suggesting a mechanistic link between Rals and the exocyst in platelets.20 Importantly, there are several other distinct upstream regulators controlling the exocyst, which likely explain the differences in platelet phenotypes between RalAB DKO and EXOC3 KO mice.25,56
It would be interesting to assess whether targeting other EXOC proteins phenocopies our EXOC3 KO observations. Although selectively removing an individual EXOC protein is generally favorable with implicating a role for the entire complex, a recent study suggests functional redundancy exists between exocyst components, and this redundancy is most likely due to paralogous gene expression.57 EXOC3 has 3 known paralogues, EXOC3-like protein 1 (EXOC3L1), EXOC2L2, and EXOC3L4, all of which (apart from EXOC3L1) are expressed at similar levels to EXOC3 in murine platelets.23 However, considering the striking phenotype observed in EXOC3 KO mice, a redundancy between EXOC3 paralogues, in platelets, seems unlikely.
Requests for data sharing may be submitted to the corresponding author (Alastair W. Poole; e-mail: a.poole@bristol.ac.uk).
Acknowledgment
This project was funded by a research grant from the British Heart Foundation (RG/15/16/31758) (A.W.P.).
Authorship
Contribution: T.G.W. designed, performed and analyzed experiments, and wrote the manuscript; Y.L, C.M.W., and E.W.A. performed and analyzed experiments; R.K.A. provided vital reagents and revised the manuscript; and A.W.P. designed and analyzed experiments and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alastair W. Poole, School of Physiology, Pharmacology & Neuroscience, Biomedical Sciences Building, University Walk, Bristol, BS8 1TD, United Kingdom; e-mail: a.poole@bristol.ac.uk.

