

Key Points
We unveil the distinct patterns of IGHV repertoire and discuss the correlation between IGHV and other genetic abnormalities in LPL/WM.
IGHV4 usage was a predictive marker of shorter progression-free survival in patients with LPL/WM.

Abstract
Lymphoplasmacytic lymphoma/Waldenström macroglobulinemia (LPL/WM) is a heterogeneous disease in which the role of immunoglobulin heavy-chain genes (IGHs) remains unknown. To determine the clinical relevance of the IGH repertoire in patients with LPL/WM, we performed immunoglobulin gene rearrangement and complementarity determining region 3 (CDR3) analysis. The IGH variable gene (IGHV) repertoire was remarkably biased in LPL/WM. IGHV3-23, IGHV4-34, IGHV3-30, IGHV3-7, and IGHV3-74 accounted for one-half of the cohort’s repertoire. Most cases (97.1%) were found to carry mutated IGHV genes, based on a 98% IGHV germline homology cutoff. IGHV3-30 was associated with long heavy chain CDR3, indicating there was specific antigen selection in LPL/WM. Patients with IGHV3-7 were significantly more likely to harbor the 6q deletion (P < .001) and an abnormal karyotype (P = .004). The IGHV hypermutation rate in patients with the MYD88 L265P mutation was significantly higher than that of wild-type patients (P = .050). IGHV3-23 and IGHV3-74 segments were more frequently detected in patients with MYD88-mutated LPL/WM (P = .050), whereas IGHV3-7 presented more frequently in MYD88 wild-type patients (P = .042). Patients with IGHV4, especially IGHV4-34, had higher levels of lactate dehydrogenase, and IGHV4 was a predictive marker of shorter progression-free survival. These results showed for the first time that the IGHV repertoire has clinical relevance in LPL/WM.
Introduction
Lymphoplasmacytic lymphoma/Waldenström macroglobulinemia (LPL/WM) is an uncommon, mature B-cell lymphoma characterized by the hypersecretion of immunoglobulin M (IgM) and bone marrow infiltration of clonal lymphoplasmacytic cells.1 Most patients with LPL/WM have an indolent clinical course with a median survival of 10 years,2 whereas ∼10% to 15% of patients present with a more rapidly progressive disease.3 Previously reported adverse prognostic factors are older age, B symptoms, anemia, low albumin serum, high lactate dehydrogenase (LDH), high β 2-microglobulin, low platelet count, high IgM levels, and an abnormal karyotype.4,5 However, the precise B-cell origin and molecular pathogenesis of LPL/WM remain poorly defined.
Immunoglobulin heavy-chain variable genes (IGHVs) are critical for defining epitope binding affinity and B-cell differentiation in B-cell non-Hodgkin lymphoma.6 The IGHV status is evidence of the origin of tumor B cells and reflects the status of tumor clones before and after transformation.7 For instance, diseases characterized by germline immunoglobulin genes may originate from naïve B cells, whereas diseases with mutated IGHV genes are derived from B cells that underwent a germinal center reaction in response to antigen stimulation.
The IGHV sequences of different B-cell tumors have been extensively studied in humans. It is known that those with chronic lymphocytic leukemia (CLL) exhibit a highly skewed IGHV repertoire, and patients with CLL somatic hypermutation statuses are understood to have a dependent prognostic factor.8 Patients with unmutated IGHV have a lower response rate to treatment and shorter progression-free survival (PFS) than patients with mutated IGHV, who responded better to fludarabine-based immune-chemotherapy. Some cases lie within an intermediate zone (eg, patients with the specific IGHV gene IGHV3-21 have a relatively poor prognosis independent of their mutational status).9
Similar to the gene patterns of CLL, distinct imprints of IGHV rearrangement and their clinical relevance have been reported previously for splenic marginal zone lymphoma (SMZL), hairy cell leukemia, mantle cell lymphoma, and follicular lymphoma.10-14 For instance, patients with SMZL had a predominant use of IGHV1-210 and IGHV4-34 was preferentially used in hairy cell leukemia-variant cases with unmutated IGHV.14 However, the IGHV repertoires in LPL/WM are different from those in CLL and other B-cell lymphoma and are characterized by high IGHV mutation rates, the overrepresentation of IGHV3-23 gene usage, and short CDR3 segments.15,16 These findings indicate that LPL/WM cells are derived from postgerminal center memory B cells subjected to T cell-dependent antigen selection. However, because of the rarity of LPL/WM, published studies on IGH repertoires have only been conducted on a limited number of samples. In addition, the clinical relevance of the IGH repertoire for LPL/WM remains largely unexplored. The aim of our study was to explore the IGHV repertoire of patients with LPL/WM in the largest series explored so far, and to evaluate the correlation between IGHV rearrangements, genetic aberrations, and the clinical characteristics of patients with LPL/WM.
Methods
Patients
A total of 162 patients received a diagnosis of LPL/WM between 2012 and 2019 from the Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Science, and Peking Union Medical College were included in this study. Diagnosis was established according to the consensus criteria from the Second International Workshop of WM.17 Clinical features including age, sex, diagnosis, biological parameters (platelet count, white blood cell count, and lactate dehydrogenase, β2-microglobulin, serum protein profile, and peak immunoglobulin levels), International Prognostic Scoring System (IPSS),18 and survival data were collected. All cases involved in this study were approved by the ethics committees of the Chinese Academy of Medical Sciences & Blood Disease Hospital, and patients’ informed consent was obtained in accordance with the Declaration of Helsinki.
Specimens and DNA extraction
Genomic DNA was isolated from samples at the time of diagnosis of LPL/WM. DNA was extracted from fresh frozen bone marrow (n = 151), peripheral blood mononuclear cell samples (n = 8), or formalin-fixed, paraffin-embedded lymph node specimens (n = 3) using the QIAmp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. Purity was checked using the NanoPhotometer spectrophotometer (Implen, CA). Total cellular RNA was isolated using Trizol Reagent (Thermo Fisher Scientific) from 18 bone marrow aspirates. Complementary DNA was amplified using the SuperScript II Reverse Transcriptase Kit (Invitrogen, Carlsbad, CA).
Sequencing and analysis of IGHV gene sequences
Polymerase chain reaction (PCR) amplification of IGHV-IGHD-IGHJ was performed on genomic DNA or complementary DNA samples using the IGH Somatic Hypermutation Assay v2.0 (Invivoscribe Technologies, San Diego, CA). PCR amplicons were purified with ExoASP-IT (Affymetrix, Santa Clara, CA) and then subjected to direct sequencing using the BigDye Terminator V3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). The sequencing reaction was performed as follows: 96°C for 1 minute, followed by 25 cycles of 96°C for 10 seconds, 50°C for 15 seconds, 60°C for 2 minutes, and then a final extension step of 3 minutes at 72°C. PCR products were sequenced for forward and reverse reads. Sequencing products were purified with the BigDye Terminator Purification Kit (Applied Biosystems) and analyzed using the 3730 Genetic Analyzer (Applied Biosystems). Sequences were aligned to homologous sequences in the IMGT (https://www.imgt.org/IMGT_vquest/vquest) and IGBLAST (https://www.ncbi.nlm.nih.gov/igblast/) databases. IGHV sequences with <98% homology to the germline sequence were considered mutated, whereas those with ≥98% homology to the germline sequence were considered unmutated. The Comprehensive R Archive Network software package (https://mirrors.tuna.tsinghua.edu.cn/CRAN/) was used to analyze IGHV segments combined with IGHD and IGHJ segments.
Stereotyped subset assignment
To identify possible distinct stereotyped subsets in patients with LPL/WM, the sequences we obtained were analyzed based on established criteria for CDR3 stereotypes in CLL19 : (1) 50% amino-acid identity and 70% amino-acid similarity, (2) sequence lengths of no more than 2 amino acids difference, and (3) use of the same IGHV gene.
Karyotype and fluorescence in situ hybridization analysis
Bone marrow samples were cultured at 37°C in RPM1 1640 (Gibco-BRL) for 24 hours. Colchicine was added at 0.2 μg/mL for 50 minutes and then hypotonic shock was performed with 0.4% KCL for 40 minutes. Cells were fixed in 3:1 (v/v) methanol-acetic acid reagent 3 times. G banding of chromosomes involved was treated by trypsin followed by Giemsa staining; G-banded metaphase karyotypes were centrally reviewed with an International System for Human Cytogenetic Nomenclature (ISCN 2020).
Fluorescence in situ hybridization analysis was performed on the interphase nuclei of uncultured BM cells. Commercial probes including 11q22.3(LSI ATM/CEP11), 17p13.1(LSI TP53/CEP17), and 13q14.2 (LSI RB-1) (Vysis, Abbott) were used for routine screening according to the manufacturer’s instructions. The threshold levels for del (11q22), del (17p13.1), and del (13q14.2) were 1.81%, 2.89%, and 6.17%. A total of 200 interphase nuclei were analyzed.
Screening for MYD88 L265P mutations
Amplification of MYD88 was performed with genomic DNA (forward primer: 5′-GGGATATGCTGAACTAAGTTGCCAC-3′, reverse primer: 5′-GACGTGTCTGTGAAGTTGGCATCTC-3′). The PCR reaction was performed as follows: 95°C for 5 minutes, followed by 35 cycles of 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 1 minute, and then a final extension step of 7 minutes at 72°C. PCR products were directly sequenced as described previously. The sensitive QX200 droplet digital PCR system from Bio-Rad (ddPCR, Bio-Rad, Hercules, CA, USA) was used to detect MYD88 L265P mutations when Sanger sequencing results were negative. All reactions were prepared using the ddPCR Supermix for probes (Bio-Rad, catalog no. 1863024). MYD88 L265P somatic mutations were detected using ddPCR Mut Assay MYD88 p.L265P (Bio-Rad, catalog nos. 10042964 and 10042967) according to the manufacturer’s introductions. Samples with a low tumor mutation burden (<1%) were not included due to low confidence.
Statistical analysis
A χ2 or Fisher exact test was used to compare categorical variables. t test was used to compare continuous variables. Pearson’s correlation test was used to determine the correlation between CDR3 amino acid length and IGHV mutation rate. PFS was calculated from diagnosis to any form of disease progression or death. Overall survival (OS) was defined as being from the time of diagnosis to death or last follow-up. Survival analyses were performed using the Kaplan-Meier method, and the log-rank test was used to determine differences between groups. For all comparisons, 2-sided tests were used, and P ≤ .05 was considered significant. All statistical analyses were performed using SPSS software (version 21.0; IBM, Chicago, IL).
Results
Patient characteristics
Productive IGHV-D-J rearrangements were obtained for 136 of 162 patients (84.0%). The clinical characteristics of the 136 patients are shown in Table 1. The study population consisted of 136 patients with LPL/WM and productive IGHV rearrangements. The median age was 62 years (range, 20-82 years), and 18.3%, 38.2%, and 43.5% of patients were assigned as low risk, intermediate risk, and high risk, respectively, according to their IPSS risk score.18
Clinical characteristics of patients with LPL/WM at diagnosis
| Characteristics | Median (range) |
|---|---|
| Age, y | 62 (20-82) |
| Sex, n (%) | |
| Male | 92 (67.6) |
| Female | 44 (32.4) |
| IPSS stage, n (%) | |
| Low risk | 24 (18.3) |
| Intermediate risk | 50 (38.2) |
| High risk | 57 (43.5) |
| WBC, ×109/L | 5.5 (0.9-118.3) |
| PLT, ×109/L | 154.0 (14.0-971.0) |
| HGB, g/L | 84.0 (44.0-153.0) |
| ALB, g/L | 33.0 (20.5-49.8) |
| High LDH, n (%) | 20 (15.2) |
| IgM, g/L | 29.9 (0.3-144.0) |
| Serum β2-MG , g/L | 3.9 (1.1-23.7) |
| IGHV mutation status, n (%) | |
| Mutated | 132 (97.1) |
| Nonmutated | 4 (2.9) |
| With 6q deletion | 8 (7.5) |
| Characteristics | Median (range) |
|---|---|
| Age, y | 62 (20-82) |
| Sex, n (%) | |
| Male | 92 (67.6) |
| Female | 44 (32.4) |
| IPSS stage, n (%) | |
| Low risk | 24 (18.3) |
| Intermediate risk | 50 (38.2) |
| High risk | 57 (43.5) |
| WBC, ×109/L | 5.5 (0.9-118.3) |
| PLT, ×109/L | 154.0 (14.0-971.0) |
| HGB, g/L | 84.0 (44.0-153.0) |
| ALB, g/L | 33.0 (20.5-49.8) |
| High LDH, n (%) | 20 (15.2) |
| IgM, g/L | 29.9 (0.3-144.0) |
| Serum β2-MG , g/L | 3.9 (1.1-23.7) |
| IGHV mutation status, n (%) | |
| Mutated | 132 (97.1) |
| Nonmutated | 4 (2.9) |
| With 6q deletion | 8 (7.5) |
ALB, albumin; HGB, hemoglobin; PLT, platelets; serum β2-MG, serum β2-microglobulin; WBC, white blood cell.
VH, DH, and JH gene segment usage
In total, 136 productive IGHV-IGHD-IGHJ rearrangements were amplified. Data on VH, DH, and JH chain gene segment usage, as well as the rate of somatic hypermutation (SHM), for all patients with LPL/WM were collected. IGHV gene repertoire analysis showed that IGHV3 usage was the predominant subgroup (82/136, 60.3%), followed by IGHV4 (33/136, 24.3%), IGHV1 (13/136, 9.6%), IGHV6 (5/136, 3.7%), IGHV2 (2/136, 1.5%), and IGHV5 (1/136, 0.7%) (Figure 1A). Consistent with previous studies,16 IGHV3-23 (15.4%), IGHV4-34 (10.3%), and IGHV3-7 (8.1%) were overrepresented in patients with LPL/WM. The distribution of IGHV segments is shown in Figure 1B. Notably, 5 IGHV segments, including IGHV3-23, IGHV4-34, IGHV3-7, IGHV3-30, and IGHV3-74, accounted for almost one-half of the series (48.5%).
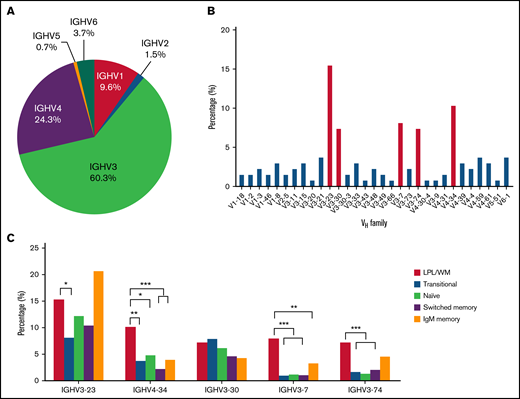
Distribution of IGHV gene segments in patients with LPL/WM. (A) IGHV gene usage in patients with LPL/WM. (B) Top 31 IGHV genes expressed in our series are shown along the x-axis. (C) IGHV distribution in LPL/WM and normal B cells. *P < .05, **P < .01, ***P < .001.
Distribution of IGHV gene segments in patients with LPL/WM. (A) IGHV gene usage in patients with LPL/WM. (B) Top 31 IGHV genes expressed in our series are shown along the x-axis. (C) IGHV distribution in LPL/WM and normal B cells. *P < .05, **P < .01, ***P < .001.
When we compared the use of individual IGHV genes in patients with LPL/WM to that of normal human B-cell Ig heavy chain repertoires, including transitional, naïve, switched memory, and IgM memory B cells,20 IGHV3-7 and IGHV4-34 were found to be significantly overrepresented in LPL/WM compared with normal B cells (P < .05). IGHV3-74 was significantly overrepresented in LPL/WM compared with transitional, naïve, and switched memory B cells (P < .001). Interestingly, IGHV3-23, IGHV3-7, and IGHV3-74 genes were reported to be significantly overrepresented in IgM memory cells compared with the other 3 normal B subsets.20 Moreover, the frequency of IGHV3 usage was highest in IgM memory cells.20 Thus, LPL/WM cells have repertoires that are closer to those of IgM memory cells than other stages of normal B cells (Figure 1C). The skewed IGHV rearrangement in LPL/WM patients reflects the developmental antigen selection induced by a different type of exogenous antigen.
The DH gene usage was detected in 134 patients. Among the IGHD family, IGHD3 was the most frequent subgroup (45/134, 33.6%), followed by IGHD6 (30/134, 22.4%) and IGHD2 (23/134, 17.2%). According to the IGHD analyses, the most frequently expressed segment was IGHD3-10 (21/134, 15.7%), followed by IGHD6-13 (18/134, 13.4%) (Figure 2A).

Distribution and associations of IGH gene segments in patients with LPL/WM. (A) Distribution of IGHD gene repertoire in patients with LPL/WM. (B). Distribution of IGHJ gene repertoire in patients with LPL/WM. Circular graphs of the associations between (C) IGHV and IGHD segments and (D) IGHV and IGHJ segments in patients with LPL/WM. The Comprehensive R Archive Network software package (https://mirrors.tuna.tsinghua.edu.cn/CRAN/) was used analyze IGHV segment combinations with IGHD and IGHJ segments.
Distribution and associations of IGH gene segments in patients with LPL/WM. (A) Distribution of IGHD gene repertoire in patients with LPL/WM. (B). Distribution of IGHJ gene repertoire in patients with LPL/WM. Circular graphs of the associations between (C) IGHV and IGHD segments and (D) IGHV and IGHJ segments in patients with LPL/WM. The Comprehensive R Archive Network software package (https://mirrors.tuna.tsinghua.edu.cn/CRAN/) was used analyze IGHV segment combinations with IGHD and IGHJ segments.
JH was identified in 136 patients, and the IGHJ4 segment was selected in more than one-half of these rearrangements (70/136; 51.5%), followed by IGHJ6 (23/136; 16.9%) and IGHJ5 (21/136; 15.4%) (Figure 2B).
Moreover, biased associations among selected IGHV and IGHD genes in LPL/WM were identified. Patients with IGHV3-30 and IGHV3-74 segments had a strong tendency toward the use of IGHD6-13 (3/10; 30.0%) and IGHD3-10 (3/10; 30.0%). Patients with IGHV4-34 segments had a bias toward usage of the IGHD6-13 segment (4/14; 28.6%). IGHV3-23 was associated with IGHD2-2 gene usage (3/20; 15.0%). No such propensities were found for IGHV3-7 segments (Figure 2C). Different combinations of IGHV and IGHJ genes were also identified. Of the 5 predominant segments, rearrangements using IGHV3-74 and IGHV3-23 showed biased recombination with IGHJ4, at percentages of 70.0% (7/10) and 65.0% (13/20), respectively. Moreover, IGHV4-34 segments were accompanied by roughly identical frequencies of IGHJ4 (4/14; 28.6%), IGHJ1 (3/14, 21.4%), IGHJ5 (3/14, 21.4%), and IGHJ6 (3/14, 21.4%) (Figure 2D).
Somatic hypermutation analysis
Following the classification of somatic hypermutation of 2% deviation from germline, 132 (97.1%) of the cohort were defined as having mutated IGHV genes. The percentage identity to the germline IGHV gene ranged from 81.4% to 99.3%, with a median of 93.2%. There was no significant difference in the percentages of SHM in the 5 predominantly used IGHV genes (with a median of 92.5% for IGHV3-23, 94.3% for IGHV4-34, 93.9% for IGHV3-30, 92.4% for IGHV3-7, and 92.0% for IGHV3-74, respectively). We divided all patients in the cohort into 4 groups according to IGHV hypermutation rate: unmutated (<2%, n = 4), minimally mutated (2%-4.9%, n = 32), moderately mutated (5.0%-9.9%, n = 81), and highly mutated (≥10.0%, n = 19). Then, we explored the distribution of germline identity subgroups among the different IGHV gene repertoires. We found IGHV3-23 was overrepresented in the moderately mutated subgroup, whereas IGHV4-34 had a higher proportion of minimally mutated rearrangements (P < .05) (Figure 3A).

Distinct mutation rates and CDR3 lengths of patients with IGHV4-34, IGHV3-23, IGHV3-30, IGHV3-7, and IGHV3-74 genes. (A) IGHV mutation rate in patients with different IGHV segments. (B) Correlation between CDR3 amino acid length and IGHV mutation rate. Linear regression fit for all data is shown by the blue line. (C) Those with the IGHV3-30 gene had longer VH CDR3 lengths than other subgroups. (D) Distribution of CDR3 lengths in patients with IGHV3-23 and IGHV3-30 segments. *P < .05.
Distinct mutation rates and CDR3 lengths of patients with IGHV4-34, IGHV3-23, IGHV3-30, IGHV3-7, and IGHV3-74 genes. (A) IGHV mutation rate in patients with different IGHV segments. (B) Correlation between CDR3 amino acid length and IGHV mutation rate. Linear regression fit for all data is shown by the blue line. (C) Those with the IGHV3-30 gene had longer VH CDR3 lengths than other subgroups. (D) Distribution of CDR3 lengths in patients with IGHV3-23 and IGHV3-30 segments. *P < .05.
CDR3 analysis
The median length of the VH CDR3 region in the 136 sequences at diagnosis was 14 amino acids (range, 6-25). Our analysis showed that VH CDR3 length was inversely correlated with mutation frequency (r = −0.21, P = .016; Figure 3B). The less mutated IGHV genes tended to be associated with a longer CDR3 length. There was a significant difference in the distribution of VH CDR3 lengths between the IGHV mutation subgroups. Among the 5 predominant segments, IGHV3-30 had longer VH CDR3 lengths compared with the other subgroups (mean lengths, 16.8 vs 13.9 amino acids, respectively, P < .05; Figure 3C). Patients with IGHV3-30 usage had significantly longer CDR3 than those with IGHV3-23 (P < .01) (Figure 3D). There was no statistically significant difference in mean length among the IGHV4-34, IGHV3-7, and IGHV3-74 subgroups. When all the cases were assigned to stereotyped subsets, 2 cases’ VH CDH3 sequences shared the patterns of CLL subsets 220 and 235,21 respectively (supplemental Table 1). No LPL/WM-specific stereotyped CDR3 sequences were found in our cohort.
Cytogenetic aberrations
Conventional cytogenetics was performed for 124 (124/136, 91.2%) patients at the time of diagnosis. Eighteen of them had poor proliferative abilities with fewer than 10 mitotic phases. Among the 106 remaining patients with a qualifying mitotic phase (>10), 28 patients (28/106, 26.4%) had cytogenetic abnormalities. Patients using IGHV3-7 segment had a high proportion of abnormal karyotypes than those using other IGHV segment (21.4% vs 2.5%, P = .004; Figure 4A). All 28 cases with cytogenetic abnormalities were IGHV mutated, and deletions of 6q were the most frequent abnormalities in LPL/WM (8/28, 28.6%). It was noted that patients with 6q deletions were characterized by more frequent usage of the IGHV3-7 gene (37.5% vs 4.3%, P < .001; Figure 4B). Complex karyotypes with at least 3 chromosomal aberrations were detected in 7 patients (7/28, 25.0%); however, no significant correlation was identified between karyotype complexity and CDR3 length (P = .881). Notably, 6 of the 8 patients (75.0%) with the IGHV3-7 gene had abnormal karyotypes, and complex karyotypes were detected in 3 of the 6 patients.
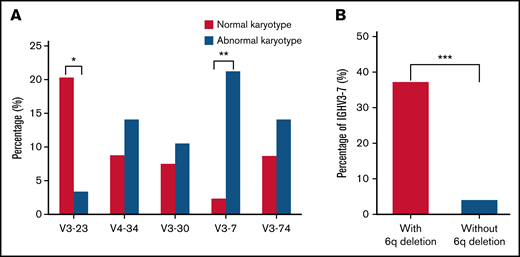
Links between cytogenetic aberrations and IGH segments. (A) Distribution of major IGHV gene segments in patients with normal or abnormal karyotypes. (B) Distribution of IGHV3-7 gene segments in patients with and without 6q deletion karyotype. *P < .05, **P < .01, ***P < .001.
Links between cytogenetic aberrations and IGH segments. (A) Distribution of major IGHV gene segments in patients with normal or abnormal karyotypes. (B) Distribution of IGHV3-7 gene segments in patients with and without 6q deletion karyotype. *P < .05, **P < .01, ***P < .001.
Mutations in MYD88 L265P
Because MYD88 has been reported to be mutated in most patients with LPL/WM,22 we performed MYD88 L265P mutation detection in 120 patients by Sanger sequencing and subsequent confirmation by ddPCR with a sensitivity of 0.1%. MYD88 mutations were observed in 105 of the 120 patients (87.5%). The IGHV SHM rate in patients with the MYD88 L265P mutation was higher than patients with wild-type MYD88 (mean rate 7.3% vs 5.8%, P = .050; Figure 5A). The CDR3 amino acid length of patients with wild-type MYD88 was significantly longer than that of patients with MYD88 L265P (mean length, 17.5 vs 14.6, P = .008; Figure 5B). When we detected the 5 predominant IGHV segments mentioned previously, we found that IGHV3-23 and IGHV3-74 segments were more frequently detected in mutated MYD88 patients with LPL/WM (26.7% vs 0.0%, P = .050). In addition, IGHV3-7 presented more frequently in MYD88 wild-type patients compared with mutated MYD88 patients (26.7% vs 6.7%, P = .042) (supplemental Table 2). Patients with IGHV3-23, IGHV3-74, and IGHV3-7 were characterized by a high mutation rate (range, 3.70%-16.07%; mean rate, 7.80%).
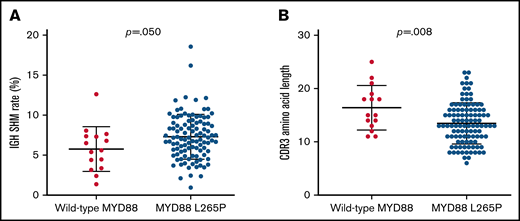
Mutations in MYD88 L265P with IGH segments. (A) MYD88 L265P patients had higher SHM rate than wild-type MYD88 patients. (B) Chart showing CDR3 amino acid lengths in patients with either mutated or wild-type MYD88.
Mutations in MYD88 L265P with IGH segments. (A) MYD88 L265P patients had higher SHM rate than wild-type MYD88 patients. (B) Chart showing CDR3 amino acid lengths in patients with either mutated or wild-type MYD88.
Clinical correlations
The median follow-up for all patients was 26.6 months (range, 0.5-137 months), and most treated patients received bortezomib-based therapy or rituximab-based therapy (91/124, 73.4%). We then explored the association between IGHV gene usage and clinical outcome. We first evaluated whether patients with distinct IGHV genes had different clinical characteristics. IGHV3 and IGHV4 were the top 2 most frequently presented gene rearrangements and accounted for 85% of the patients in our series. Thus, we studied the clinical features of patients with IGHV3 and IGHV4 genes. The detailed clinical features, treatment options, lines of therapy, cytogenetic abnormalities, and MYD88 mutation status between patients with IGHV4+ and IGHV4−, IGHV3+, and IGHV3− are shown in supplemental Table 3. IGHV3 segments were frequently associated with moderate to severe anemia (62.5% vs 37.5%, P = .039) and hypoalbuminemia (66.3% vs 33.7%, P = .007). Elevated serum LDH levels were more frequently observed in patients with IGHV4 segments (27.3% vs 11.1%, P = .050), especially in those with IGHV4-34 (50.0% vs 11.0%, P = .001). IGHV4 was correlated with adverse outcomes (median PFS 29.4 vs 61.4 months, P = .044; Figure 6A). The 3-year PFS rate of patients with IGHV4 usage was lower than patients without (46.3% vs 73.3%). However, no statistically significant differences were found between patients with or without IGHV4 in terms of OS (3-year PFS, 65.5% vs. 93.8%, P = .101) (Figure 6B). When we only included the patients with elevated LDH level into survival analysis, we found IGHV4 lost its prognostic significance. In addition, the presence of IGHV3-7 had no statistically significant effect on PFS or OS (supplemental Figure 1). There were no significant survival associations for the other IGHV-D-J segment usage patterns.
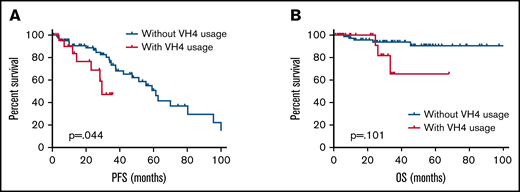
Survival analysis of patients with IGHV4 gene. (A) PFS and (B) OS estimates for patients with LPL/WM according to IGHV4 gene usage.
Survival analysis of patients with IGHV4 gene. (A) PFS and (B) OS estimates for patients with LPL/WM according to IGHV4 gene usage.
Discussion
IGH rearrangements have often been used as molecular signs of lymphoid malignancy clonality. In our study, we analyzed the IGH gene repertoire of a cohort of 136 patients with LPL/WM with monoclonal spikes to find correlations with clinical characteristics. LPL/WM has a biased IGHV repertoire. It provides useful clues in the maturation status of specific B-cell entities and origin of B-tumor cells. Our study confirmed a previous hypothesis that the biased usage of IGHV genes in LPL/WM is completely different from other B-cell lymphoproliferative disorders.16,23,24 Although we observed a predominance of IGHV3 usage, with a percentage of 60.3% in LPL/WM, the percentage of IGHV3 usage was lower than that of other LPL/WM populations (eg, France, 77%; Spain, 76%; Greece, 74.3%; and Italy, 87%).15,16,23,25 The distribution of IGHV repertoire in LPL/WM in several representative studies is listed in supplemental Table 4. Most notably, IGHV4-34 (10.3%) was frequently used in our patients, but is relatively rare in the Western LPL/WM cohort, with an incidence of 2%25 or less.23 Interestingly, our previous studies indicated that the IGHV4-34 usage was more frequently detected in Chinese patients with CLL and SMZL than those reported in other series.11,26,27 The incidence of lymphoma and the distribution of subtypes vary among different ethnic and geographic populations. Besides, our previous study indicated that Chinese patients with CLL have a higher frequency of mutated IGHV, distinct IGHV segments usage, and a different gene mutation spectrum compared with the Western cohort.26 Therefore, the different IGHV patterns, especially the higher frequency of IGHV4 gene usage in Chinese patients with LPL/WM, might be attributed to their different ethnic background and antigen stimulation. Further studies are expected to demonstrate the phenotypic and genotypic differences between the IGHV gene utility of Chinese and Western people with LPL/WM.
Patients with LPL/WM display biased immunogenetic signatures, indicating their distinct antigen exposure histories. LPL/WM cells are characterized by a lack of class switch recombination and IGHV mutation.23,28 The immunophenotype of LPL/WM cells matches that of memory B cells (smIgM+/CD10–/CD20+/CD23−/CD27+/CD38low+/CD81+).29 The transcriptome profile of WM cells also supports the hypothesis that the normal counterparts of LPL/WM cells are CD25+CD22low memory B cells.30 A recent study undertook genome-wide methylation analyses of flow-sorted LPL/WM tumor cells and revealed that patients with LPL/WM naturally segregate into 2 groups related to either their normal memory B cell or plasma cell profiles.31 Accordingly, immunophenotyping and molecular analysis support a B-memory cell origin for WM. In this study, we compared the IGHV repertoire of LPL/WM cells with that of normal B cells at different developmental stages. The results revealed that LPL/WM cells have repertoires closer to those of IgM memory cells than other stages of normal B cells. These results further support the hypothesis that IgM memory B cells are the primary candidates for the origin of LPL/WM cancer cells, as formulated according to IGHV arrangement analysis.
Both genetic lesions and B-cell receptor signaling are oncogenic drivers in LPL/WM. We explored the correlation between IGHV and cytogenetic abnormalities in patients with LPL/WM, and cytogenetic data obtained at the time of diagnosis. The deletion of 6q was the most common chromosome abnormality in LPL/WM cells, as has been described previously.32 We first reported that the IGHV3-7 gene was more highly expressed in patients with LPL/WM with abnormal karyotypes. More importantly, the gene more frequently presented in cases with 6q deletion than those without. These results indicated that distinct IGHV repertoires in patients with LPL/WM may contribute to their secondary genetic changes. The presence of 6q deletions has been suggested to discern patients with LPL/WM from those with IgM monoclonal gammopathy of unknown significance, provide sights to the transformation of LPL/WM33 and tend to serve as a prognostic marker, although the latter conclusion remains controversial.22 There is no prognostic significance of 6q deletion in our cohort. That is the reason we found a significant higher usage of IGHV3-7 segment in patients with 6q deletions, but no prognostic value of IGHV3-7 usage was identified in our patients. However, conventional cytogenetics were performed to detect 6q deletion in our cohort. We had a lower detectable rate of 6q deletion than that of another study.34 Further research with more precise cytogenetic detection method is needed to verify the conclusion.
In addition to the cytogenetic aberrations, we further investigated the relationship between the IGHV repertoire and MYD88 mutation status. Previous studies indicated that patients with wild-type MYD88 had an older age, higher β2MG levels, and worse survival and higher risk of disease transformation.35-39 Therefore, we made a detailed comparison of preferential IGHV usage in MYD88-mutated patients among different cohorts (supplemental Table 4).15,16,25 We found the median IGHV SHM was higher in patients with MYD88 L265P. The usage of IGHV3-23 and IGHV3-74 were decreased in MYD88 wild-type patients, consistent with the study of Petrikkos et al.16 IGHV3-7 usage was more frequently observed in MYD88 wild-type patients than that in MYD88-mutated patients (26.7% vs 6.7%, P = .042). Inconsistent with these results, a moderately elevated proportion of IGHV3-7 usage was reported in MYD88-mutated patients in Gachard’s study with no statistical significance (28% vs 11%, P = .623).15 The cohort size of these studies has curtailed the confidence of these conclusions. Although part of the results was inconsistent with previous studies, these existing results could support that patients with different MYD88 mutation status experience different antigenic stimulation and have different biological characteristics.
The biased usage of IGHV genes and a stereotyped B-cell receptor support the role of antigen-driven mechanisms in the pathogenesis of B-cell lymphoproliferative disorders. Previous studies have reported on the clinical relevance and prognostic significance of IGHV mutational status and repertoires in CLL, FL, mantle cell lymphoma, and MZL.40-43 One of our study purposes was to discuss the effects of IGHV gene usage and mutation rates on the clinical features and survival of the patients with LPL/WM. Clinical characteristics such as age, hemoglobin, platelets, β2-microglobulin, and serum monoclonal immunoglobulin are predictive of outcomes in patients with LPL/WM,18 and some studies found that the addition of elevated serum LDH further improved the ability of IPSS to identify a group of patients with significantly worse outcomes.44,45 In our current cohort, we analyzed the prognostic value of IGHV rearrangement for the first time in LPL/WM. We suggested that IGHV4 had an adverse effect on PFS in LPL/WM. To clarify whether the adverse prognostic significance of IGHV4 usage depend on other known prognostic factors, we compared the patient population, clinical features, treatment regimens, lines of therapy, cytogenetic abnormalities, and MYD88 mutation status between patients with IGHV4+ and IGHV4− (supplemental Table 3). The only significant difference we found between patients with and without IGHV4 was LDH level. Elevated serum LDH levels were more frequently observed in patients with IGHV4 segments, especially in those with IGHV4-34. Disappointingly, we found IGHV4 lost its prognostic significance when we only included the patients with elevated LDH level. This indicated that the prognostic value of IGHV4 might depend on LDH level. However, LPL/WM is a highly heterogeneous and indolent disease. The conclusion needs to be confirmed by further study with larger cohort size and long-term follow-up.
In conclusion, we unveiled distinct patterns of IGH gene usage, specific mutational statuses, and restricted VH CDR3 lengths in patients with LPL/WM. The IGHV segment usage were found associated to the presence of cytogenetic alterations and MYD88 mutations. In addition, IGHV4 segment usage proved to be a potential prognostic risk factor in LPL/WM. These findings provide evidence for the concept that specific IGH gene repertoires may promote or select genetic lesions and have an impact on the clinical outcomes of LPL/WM.
Acknowledgments
This work was supported by grants from the National Nature Science Foundation of China (81970187, 82170193, 81920108006, 81600181, and 81900203), Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2021-I2M-C&T-B-081), The Nature Science Foundation of Tianjin (18JCQNJC12100), State Key Laboratory of Experimental Hematology Research (Z20-07, Z21-09), and Special Support Program for the HighTech Leader and Team of Tianjin (TJTZJH-GCCCXCYTD-2-18).
Authorship
Contribution: S.Y. and L.Q. designed the study; J.W. and Y.Y. wrote the manuscript; S.Y. conceived the project and provided leadership; J.W. and Y.Y. performed the experiments, analyzed data, and prepared figures; G.S. performed computational analysis; S.Y., W.X., Z.Y., T.W., R.L., W.L., D.Z., and G.A. managed patients and collected samples; Y.M. performed Comprehensive R Archive Network analysis; Y.W., Y.Y., J.C., and Y.J. performed clinical data annotation; J.Z., Y.J., Q.L., C.L., Q.S., and H.W. were responsible for pathologic diagnosis; Z.X. and J.W. suggested revisions; and all authors reviewed the manuscript and provided final approval for submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shuhua Yi, State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Haihe Laboratory of Cell Ecosystem, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300020, China; e-mail: yishuhua@ihcams.ac.cn; or Lugui Qiu, State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Haihe Laboratory of Cell Ecosystem, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300020, China; e-mail: qiulg@ihcams.ac.cn.

