

Key Points
The present study demonstrates the dominant-negative impact of an in-frame large deletion on VWF biosynthesis and biogenesis of the WPBs.
The malformed WPBs/altered trafficking of its inflammatory cargos cause distresses in endothelial cell signaling pathways and phenotype.

Abstract
An in-frame heterozygous large deletion of exons 4 through 34 of the von Willebrand factor (VWF) gene was identified in a type 3 von Willebrand disease (VWD) index patient (IP), as the only VWF variant. The IP exhibited severe bleeding episodes despite prophylaxis treatment, with a short VWF half-life after infusion of VWF/factor VIII concentrates. Transcript analysis confirmed transcription of normal VWF messenger RNA besides an aberrant deleted transcript. The IP endothelial colony-forming cells (ECFCs) exhibited a defect in the VWF multimers and Weibel-Palade bodies (WPBs) biogenesis, although demonstrating normal VWF secretion compared with healthy cells. Immunostaining of IP-ECFCs revealed subcellular mislocalization of WPBs pro-inflammatory cargos angiopoietin-2 (Ang2, nuclear accumulation) and P-selectin. Besides, the RNA-sequencing (RNA-seq) analysis showed upregulation of pro-inflammatory and proangiogenic genes, P-selectin, interleukin 8 (IL-8), IL-6, and GROα, copackaged with VWF into WPBs. Further, whole-transcriptome RNA-seq and subsequent gene ontology (GO) enrichment analysis indicated the most enriched GO-biological process terms among the differentially expressed genes in IP-ECFCs were regulation of cell differentiation, cell adhesion, leukocyte adhesion to vascular endothelial, blood vessel morphogenesis, and angiogenesis, which resemble downstream signaling pathways associated with inflammatory stimuli and Ang2 priming. Accordingly, our functional experiments exhibited an increased endothelial cell adhesiveness and interruption in endothelial cell–cell junctions of the IP-ECFCs. In conclusion, the deleted VWF has a dominant-negative impact on multimer assembly and the biogenesis of WPBs, leading to altered trafficking of their pro-inflammatory cargos uniquely, which, in turn, causes changes in cellular signaling pathways, phenotype, and function of the endothelial cells.
Introduction
Deficient von Willebrand factor (VWF) causes von Willebrand disease (VWD), which is the most frequent inherited bleeding disorder.1,2 VWD is categorized as quantitative (type 1 and type 3) or qualitative (type 2). Type 3 VWD, the most severe form of the disease, is characterized by very low or no circulating VWF.3 In general, patients with type 3 VWD exhibit moderate to severe mucocutaneous bleeding as well as hemarthroses and muscle hematomas. VWD type 3 is inherited as an autosomal recessive trait either from homozygous or compound heterozygous mutation of the VWF gene (VWF).4
The VWF, comprising 52 exons, is transcribed into an 8.8-kb messenger RNA (mRNA) transcript that encodes a precursor VWF.5 The VWF precursor is composed of a signal peptide, a propeptide (D1-D2 domains), and a mature VWF subunit (domains of D′-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CK).6 The VWF is mainly synthesized in endothelial cells (ECs), and its biosynthesis involves dimerization (through CK domains), multimerization (by forming disulfide bonds between D3 domains), and proteolytic cleavage of the propeptide followed by tubular packing of multimers and targeting into Weibel-Palade bodies (WPBs).7,8 The WPBs are elongated secretory organelles specific to ECs that encompass VWF, a prerequisite for the existence of WPBs, and multiple pro-inflammatory and proangiogenic proteins such as P-selectin, angiopoietin-2 (Ang2), interleukin 6 (IL-6), and IL-8. VWF is released from ECs into the blood circulation via either a constitutive path or a regulated pathway upon stimulation of ECs by secretagogues.9-12 The half-life of VWF is reported to be about 12 hours, but the mechanisms involved in VWF clearance are still only partially understood.13-17 Nevertheless, it is clear that macrophages make a significant contribution to VWF removal from circulation.18,19
We previously reported a heterozygous de novo large deletion of exons 4 through 34 (del4-34) of VWF in an index patient (IP) diagnosed with type 3 VWD.20 The IP suffers from life-threatening bleeding despite prophylactic treatment with high doses of the plasma-derived VWF/factor VIII (FVIII) concentrates, and we have documented markedly accelerated elimination of VWF from the plasma. The single heterozygous large deletion could not provide a plausible explanation for the very low VWF levels and the clinical outcome observed in this patient. This study aimed to elucidate the underlying molecular mechanisms driving the unusual clinical manifestations in this VWD case by exploiting patient-derived endothelial colony-forming cells (ECFCs), heterologous expression of VWF in human embryonic kidney (HEK 293T) cells, and studies in the VWF knockout mouse model.
Materials and methods
Patients: phenotypic analysis
A 40-year-old male IP with severe VWD was recruited in the current study. The IP was diagnosed at the age of 8 years to have type 3 VWD resulting from very low VWF antigen (VWF:Ag) levels (Figure 1). International Society on Thrombosis and Haemostasis Bleeding Assessment Toll questionnaire was administered to record patient bleeding history.21 Moreover, the pharmacokinetic (PK) profiles of VWF/FVIII concentrates in the IP were investigated following administration of Voncento (CSL Behring, Germany), at a dose of 50 IU FVIII/kg body weight. The PK assessments were calculated following measurement of VWF:Ag, FVIII activity (FVIII:C), and platelet glycoprotein Ib binding activity of VWF (VWF:GPIbM), as described previously.22,23
![Phenotypic characteristics and genetic data of the index patient. (A) The phenotypic and genetic data of the index patient and his parents. No, no VWF gene large deletion was detected. (B) VWF:Ag, VWF:GPIb, and FVIII:C pharmacokinetics in the type 3 VWD index patient following administration of Voncento VWF/FVIII concentrate. Blood samples were obtained at multiple time points, before and at 15 and 30 minutes, and 1, 2, 4, 6, 8, 12, and 24 hours after infusion of Voncento (50 IU VWF:RCo/kg). The amount of VWF:Ag, VWF:GPIb, and FVIII:C was determined from collected plasma samples. The patient did not receive any VWF product 1 day before the PK study period. Data were analyzed by using GraphPad Prism version 8.0.1. The half-life (T1/2) of VWF:Ag, VWF:GPIb, and FVIII:C was calculated based on 1-phase exponential decay half-life of VWF:Ag, VWF:GPIb, and FVIII:C was determined as 5.2 hours (95% confidence intervals [CI], 3.6-7.7), 3.9 hours (95% CI, 3.2-4.7, and 14.9 hours (95% CI, 10.3-23.6), respectively. FVIII:C showed longer T1/2 compared with VWF markers apparently from the effect of increasing endogenous FVIII levels combined with decreasing levels of exogenous FVIII. VWF:RCo, von Willebrand factor ristocetin cofactor assay.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/3/10.1182_bloodadvances.2021005895/4/m_advancesadv2021005895f1.png?Expires=1769225859&Signature=ixpbjPmUemtzvVYcrA7AtJa8malz6H2SaFbxTC4SHqZ3HZqhNusNevEwOBUP9W9nI2fo6xhnbnJfmmfh7w63vc6dFSahnoO~9QhgEdGJgTK8tRm--~p0mwEjIJsINwH5BMxqYi3ZIbqqm5zg8MBFRvclgew1mvXlY75tOyvrxTz22sdJH62Z2qrFBGbV~9c69GgddSHRjjuO7LdN3bJxmDBMDNuFlqr~sfeILCNBEHEThtbX-GJbrUdBn8WB1pBmYD3ASP~VjMqTZZHR1X48TEUJ71xbiZd5Sx-45TrP-DSPp0UefFZa7tIP7XYx6LI2RdzJVe3js52nlpEbXCGq0w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Phenotypic characteristics and genetic data of the index patient. (A) The phenotypic and genetic data of the index patient and his parents. No, no VWF gene large deletion was detected. (B) VWF:Ag, VWF:GPIb, and FVIII:C pharmacokinetics in the type 3 VWD index patient following administration of Voncento VWF/FVIII concentrate. Blood samples were obtained at multiple time points, before and at 15 and 30 minutes, and 1, 2, 4, 6, 8, 12, and 24 hours after infusion of Voncento (50 IU VWF:RCo/kg). The amount of VWF:Ag, VWF:GPIb, and FVIII:C was determined from collected plasma samples. The patient did not receive any VWF product 1 day before the PK study period. Data were analyzed by using GraphPad Prism version 8.0.1. The half-life (T1/2) of VWF:Ag, VWF:GPIb, and FVIII:C was calculated based on 1-phase exponential decay half-life of VWF:Ag, VWF:GPIb, and FVIII:C was determined as 5.2 hours (95% confidence intervals [CI], 3.6-7.7), 3.9 hours (95% CI, 3.2-4.7, and 14.9 hours (95% CI, 10.3-23.6), respectively. FVIII:C showed longer T1/2 compared with VWF markers apparently from the effect of increasing endogenous FVIII levels combined with decreasing levels of exogenous FVIII. VWF:RCo, von Willebrand factor ristocetin cofactor assay.
Phenotypic characteristics and genetic data of the index patient. (A) The phenotypic and genetic data of the index patient and his parents. No, no VWF gene large deletion was detected. (B) VWF:Ag, VWF:GPIb, and FVIII:C pharmacokinetics in the type 3 VWD index patient following administration of Voncento VWF/FVIII concentrate. Blood samples were obtained at multiple time points, before and at 15 and 30 minutes, and 1, 2, 4, 6, 8, 12, and 24 hours after infusion of Voncento (50 IU VWF:RCo/kg). The amount of VWF:Ag, VWF:GPIb, and FVIII:C was determined from collected plasma samples. The patient did not receive any VWF product 1 day before the PK study period. Data were analyzed by using GraphPad Prism version 8.0.1. The half-life (T1/2) of VWF:Ag, VWF:GPIb, and FVIII:C was calculated based on 1-phase exponential decay half-life of VWF:Ag, VWF:GPIb, and FVIII:C was determined as 5.2 hours (95% confidence intervals [CI], 3.6-7.7), 3.9 hours (95% CI, 3.2-4.7, and 14.9 hours (95% CI, 10.3-23.6), respectively. FVIII:C showed longer T1/2 compared with VWF markers apparently from the effect of increasing endogenous FVIII levels combined with decreasing levels of exogenous FVIII. VWF:RCo, von Willebrand factor ristocetin cofactor assay.
Furthermore, plasma ADAMTS-13 antigen and activity levels were assessed by the enzyme-linked immunosorbent assay (ELISA)-based ADAMTS-13 screening kits (Technoclone, Austria). The development of anti-VWF alloantibodies was investigated using an ELISA-based approach for screening of antibodies (IgG, IgM, and IgA) against recombinant as well as ECFC-derived VWF.
The procedures using human samples were conducted according to the Declaration of Helsinki principles. This study was approved by the local ethics committee, and informed consent was obtained from the IP (vote 091/09).
Isolation of ECFCS and neutrophils
ECFCs were isolated from the blood of the patient and 6 healthy individuals based on the published standardized protocols.24-26 The isolation and culture of ECFCs from the IP were performed 3 times during the study (years 2014, 2018, and 2020). The characterization of the ECFCs was performed by immunofluorescence microscopy (detecting cell surface markers PECAM-1 and VE-cadherin) as well as flow cytometry (only done for the sampling in 2020) using fluorochrome-conjugated antibodies directed against endothelial markers CD31 (PECAM-1), CD309 (VEGFR2), CD201 (EPCR), CD141, CD34, and hematopoietic marker CD45, based on the standard approaches.27 ECFCs were used at passages between 6 and 10 in all experiments.
Neutrophils were isolated from peripheral blood of the IP and 6 controls using a standard procedure.28
RNA isolation and real-time PCR
Peripheral blood RNA and ECFCs RNA were isolated using the Tempus Spin RNA isolation kit (Applied Biosystems, UK) and RNeasy_Mini kit (Qiagen, Germany), respectively. The reverse transcription (RT) reactions and amplification of the full-length VWF complementary DNA (cDNA) were conducted in 10 overlapping fragments (supplemental Table 1) using Qiagen LongRange 2Step RT-polymerase chain reaction (PCR) kit (Qiagen, Germany). Additional multiplex RT-PCR reactions, exploiting primers across the junction of exons 2 and 3 (forward) and exons 52 (reverse) along with control internal primers, were performed to ascertain transcription of the del4-34 allele (supplemental Table 2). The RT-PCR products were separated on 1% agarose gel and sequenced.
Moreover, we quantified normal VWF transcripts in ECFCs using the TaqMan assay on an ABI 7500 real-time PCR system (Applied Biosystems, USA). The quantitative RT-PCR was conducted using TaqMan Reverse Transcription Reagents, TaqMan Universal PCR Master Mix (Applied Biosystems, USA), and exploiting 3 sets of a fluorogenic probe/forward and reverse primers combinations targeting 3 different sites in VWF (across exons 4-5, 11-12, and 43-45) cDNA (supplemental Table 3). Data were analyzed by 7500 software v2.0.6 based on the comparative CT (ΔΔ CT) method.
Whole-transcriptome analysis via RNA-seq
Total RNA was extracted from 2 healthy ECFCs (each 2 samples, n = 4) and IP-ECFCs (2 samples) as described previously. RNA-sequencing (RNA-seq) libraries were prepared using QuantSeq 3′-mRNA Library Prep kit (Lexogen, Austria), and sequenced on an Illumina HiSeq 2500 V4 platform (Illumina). After quality assessment and trimming tasks by Fastqc and Cutadapt, respectively, reads were aligned to the reference human genome GrCH38.8 (Ensemble) using the miARMa pipeline.29-31 Tophat2 and bowtie2 were used for mapping.30,32 The reads were converted to the fragments per kilobase million, and the second quality control step including normalization and filtering of transcripts was executed using Qlucore omics explorer 3.6 (Qlucore, Sweden). The genes with a P value ≤ .05 and fold change ≥2 (or absolute log2FC, mean difference, >1) were considered statistically significant differentially expressed genes (DEGs) and were investigated by Gene Ontology (GO) Consortium (released 09.10.2020) and Ingenuity Pathway Analysis ([IPA], QIAGEN, Germany).33,34
Assessment of VWF production in ECFCs
The amount of VWF:Ag secreted into media of 6 healthy ECFCs (each, 3 different samples from cell passages 6 to 10; N = 18) and IP-ECFCs (3 different ECFCs isolation in 2014, 2018, and 2020; each, 3 samples from cell passages of 6 to 10; N = 9) were measured. Seventy-two hours after seeding cells (at a density of 1.5 × 106 cells/10 mL per 75-cm2 flasks), the supernatant medium was collected and cells were lysed. Subsequently, the collected medium was concentrated on Amicon Centrifugal filter devices (Millipore, USA). Considering variations in cell proliferation rates (and its impact on doubling population) the VWF:Ag levels were normalized using total ECFCs lysates cellular protein content, after performing Bradford assay (Coomassie Plus Assay Kit; Thermo Scientific, USA). Furthermore, the secreted VWF multimers were analyzed by electrophoresis on 1.2% and 1.6% sodium dodecyl sulfate (SDS)-agarose gel.22,23
Immunofluorescence microscopy
Confluent healthy and IP ECFCs (all 3 ECFC isolation occasions) were fixed and immunostained as previously described.24 Polyclonal rabbit anti-human VWF (DAKO, Denmark) or sheep anti-VWF (Abcam, UK), anti-CD62P (MyBioSource, USA), anti-Ang2 (F-1; Santa Cruz Biotechnology, USA), as well as anti-CD31 (Life Technologies, USA) and anti-VE-cadherin (Santa Cruz Biotechnology, USA) were used for immunostaining of the ECFCs. Imaging of the cells was carried out using an Apotome.2 microscope (Carl Zeiss, Germany) or Olympus FluoView FV1000 confocal microscope. Three-dimensional (3D) images of piled-up z-stacks were generated by the ZEN 2.6 program (blue edition; Carl Zeiss, Germany). Quantification of colocalization was performed using ImageJ software, assessing Pearson correlation coefficients for a minimum of n = 30 regions of interest for each set of comparisons. The 3D image processing and morphometric assessments of the WPBs were performed by using the Vision4D 3.2 software (arivis AG, Imaging Science, Germany). The morphometric assessments of the WPBs were including measuring the length, depth, and width (by measuring the longest, middle, and shortest sides of the 3D-oriented bounds, respectively) plus the shape (sphericity factor, Ψ). The sphericity describes the roundness of the WPBs, represented as a value between 0 and 1.35
In-cell-based ELISA
To quantify ECFCs intracellular proteins PECAM-1 and Ang2, an in-cell ELISA assay was performed using an ICE kit (Abcam, UK). Cells of 3 healthy individuals and the IP were seeded into collagen precoated 96-well microplates. After 72 hours, cells were fixed and permeabilized followed by immunostaining (using the same primary antibodies as used for immunofluorescence imaging).
In vitro quantitation of endothelial adhesiveness
Confluent ECFCs (3 healthy individuals and the IP on 3 different occasions), on coverslips in 24-well plates, were incubated with Calcein AM-stained HL-60 cells (DSMZ, Germany) for 180 minutes. After a rinsing procedure, cells were fixed with 10% formalin.36 Using an inverted microscope, the images from at least 10 fields for each sample were acquired. Subsequently, adhesive ECs, which were surrounded by HL-60 clusters, were enumerated. Furthermore, fixed, labeled HL-60 cells were costained for VWF and were subjected to confocal immunofluorescence microscopy, as described previously.
Transient transfection experiments
A pMT2-VWF expression vector containing full-length wt human VWF (huVWF) cDNA was used and modified. Inverse PCR was used to remove the exons 4 through 34 from the VWF sequence (huVWF del4-34) (supplemental Table 4).37 HEK 293T (DSMZ, Germany) was transiently transfected with either huVWF or huVWF del4-34 constructs using either lipofectamine (Invitrogen, Germany) or calcium phosphate transfection techniques, as previously described.23 Conditioned media were collected and concentrated, VWF:Ag and VWF:GPIbM were quantified, and VWF multimer profile was analyzed (reference).23 The values (n = 9) were expressed as a percentage of the corresponding wt.
Clearance studies in mice
VWF-deficient mice on a C57BL/6 background were infused with 200U huVWF or huVWF del4-34/kg of body weight. At appropriate time points, mice were anesthetized, and blood from retro-orbital venipuncture was sampled. Percentage recovery, mean residence time, and plasma VWF half-life were determined.
Data analysis
All data are expressed as mean ± standard error of the mean (SEM). Statistical significance of the results was examined by unpaired Student t test using GraphPad Prism version 8.0.1 (GraphPad Software, USA). A P value < .05 was considered statistically significant.
Results
Patient characterization
As it was previously reported, the genetic investigations (including DNA sequencing of VWF as well as multiplex ligation-dependent probe amplification) revealed only a heterozygous VWF del4-34 in this type 3 VWD IP.20 No large deletion was found in the VWF of his mother and his father after multiplex ligation-dependent probe amplification analysis. Both his consanguineous parents had normal VWF:Ag levels, and they had no bleeding history (Figure 1A).
The IP is treated with high doses of a plasma-derived VWF/FVIII concentrate (1000-1500 IU every 12 hours). Nevertheless, he frequently suffers from spontaneous oral cavity bleeding. He additionally exhibited a long-term bleeding history of epistaxis, cutaneous bleeding, and bleeding from minor wounds. Furthermore, the IP is mentally impaired and he has chronic periodontitis. The latter 2 complications are not typical symptoms of VWD. The VWF PK assessments showed a short VWF half-life (5.2 hours) in the IP (Figure 1B). Alloantibodies against VWF were not detected in the IP’s plasma.
VWF RNA transcripts
The RT-PCR investigation proved the presence of a full-length normal VWF mRNA (excluding any deficiency in the transcription of the normal VWF allele) as well as a truncated transcript (del4-34) (Figure 2A). Real-time PCR exhibited normal VWF mRNA levels in the IP-ECFCs once primers/probe targeting sequences spanning exons 43-45 (outside of the deleted region) were applied (Figure 2B). Further, a substantial reduction in VWF mRNA levels was observed once the primers and probes directing sequences at exons 4 and 5 and 11 and 12 (sequences within deleted VWF del4-34 region) are exploited (Figure 2B). Complementarily, the truncated transcript resulting from the del4-34 allele was revealed after performing PCR reactions using allele-specific primers and subsequent sequencing analysis (Figure 2C).
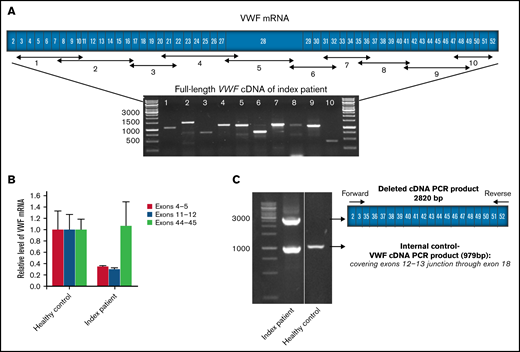
VWF mRNA transcription analysis. (A) The schematic scale of the VWF coding region (exons 2-52) with the position of designed primers for amplification of the full-length VWF cDNA and corresponding amplicons segments. Agarose gel electrophoresis image displays the 10 overlapping RT-PCR products of VWF using total RNA from the index patient (IP) as a template. (B) Comparative levels of IP-ECFCs VWF mRNA quantified by real-time PCR, using primer/probe combinations directing 3 different sites in VWF cDNA, across exons 4 and 5, 11 and 12, and 43 through 45. In the first set, the forward primer and the probe both were designed to target sequences in exon 4, and a reverse primer was designed across the exons 4-5 junction of VWF cDNA. In the second set, the forward and the reverse primers were directed at exons 11 and 12, respectively, and the fluorogenic probe targeted a sequence across the junction of the exons 11 and 12. In the third set, a fluorogenic probe as well as forward and reverse primers were targeting sequences in exon 44, exons 43 and 44, and 44 and 45 junctions, respectively. The measurements were performed based on the comparative CT (ΔΔ CT) method. Measurements of VWF mRNA levels were normalized to endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or actin β (ACTB) mRNA. (C) Agarose gel electrophoresis of multiplex RT-PCR products amplified using primers designed across the junction of exons 2-3 (forward) and exons 51-52 (reverse) along with control internal primers (forward and reverse primers targeting sites in exon 12-13 junctions and exon 18, respectively). RT-PCR products of RNA obtained from the IP demonstrate a larger product (2820 bp) corresponding to the abnormal deleted VWF transcript derived from deleted VWF allele (del4-34) and a smaller fragment (979 bp) relevant to the normal transcript (lane 1). However, RT-PCR using RNA from healthy control as the template shows only the smaller normal fragment (lane 2). Molecular weight marker: GeneRuler 1kb ladder (Thermo Scientific, Germany).
VWF mRNA transcription analysis. (A) The schematic scale of the VWF coding region (exons 2-52) with the position of designed primers for amplification of the full-length VWF cDNA and corresponding amplicons segments. Agarose gel electrophoresis image displays the 10 overlapping RT-PCR products of VWF using total RNA from the index patient (IP) as a template. (B) Comparative levels of IP-ECFCs VWF mRNA quantified by real-time PCR, using primer/probe combinations directing 3 different sites in VWF cDNA, across exons 4 and 5, 11 and 12, and 43 through 45. In the first set, the forward primer and the probe both were designed to target sequences in exon 4, and a reverse primer was designed across the exons 4-5 junction of VWF cDNA. In the second set, the forward and the reverse primers were directed at exons 11 and 12, respectively, and the fluorogenic probe targeted a sequence across the junction of the exons 11 and 12. In the third set, a fluorogenic probe as well as forward and reverse primers were targeting sequences in exon 44, exons 43 and 44, and 44 and 45 junctions, respectively. The measurements were performed based on the comparative CT (ΔΔ CT) method. Measurements of VWF mRNA levels were normalized to endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or actin β (ACTB) mRNA. (C) Agarose gel electrophoresis of multiplex RT-PCR products amplified using primers designed across the junction of exons 2-3 (forward) and exons 51-52 (reverse) along with control internal primers (forward and reverse primers targeting sites in exon 12-13 junctions and exon 18, respectively). RT-PCR products of RNA obtained from the IP demonstrate a larger product (2820 bp) corresponding to the abnormal deleted VWF transcript derived from deleted VWF allele (del4-34) and a smaller fragment (979 bp) relevant to the normal transcript (lane 1). However, RT-PCR using RNA from healthy control as the template shows only the smaller normal fragment (lane 2). Molecular weight marker: GeneRuler 1kb ladder (Thermo Scientific, Germany).
ECFC characterization
The ECFCs isolated from the IP and healthy individuals exhibited typical endothelial cobblestone morphology and canonical endothelial cell surface markers. However, IP-ECFCs (all ECFC lines isolated on 3 different occasions) showed insufficiency in cell proliferation and expansion with continued culture in advanced passages. Furthermore, compared with healthy controls, PECAM-1 and VE-cadherin staining in many of the IP-ECFCs were altered. The IP-ECFCs showed a slight but significant reduction of PECAM-1 at cell junctions (proven by both immunostaining and the in-cell ELISA), most likely from the redistribution of molecules away from the junctions rather than downregulation of its expression, demonstrated by normal PECAM-1 mRNA levels of RNA-seq (Figure 3). Additionally, the IP-ECFCs exhibited a disorganized distribution of VE-cadherin at cell junctions.
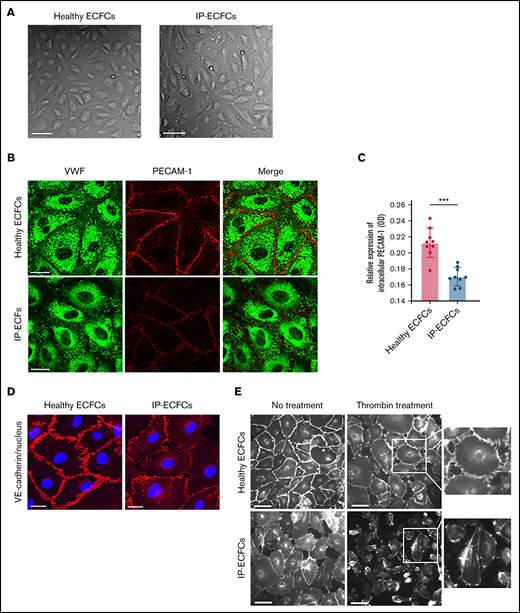
Endothelial cobblestone morphology and expression of endothelial cell adhesion proteins VE-cadherin and PECAM-1 at cell–cell junctions of ECFCs. (A) The typical endothelial cobblestone morphology of ECFCs isolated from a healthy individual and the IP. Scale bars, 100 µm. (B) VWF (green) and adherence junction protein PECAM-1 (red) are visualized with coimmunostaining of ECFCs derived from a healthy individual (upper) and patient (IP-ECFCs) (lower). Scale bars, 50 µm. (C) The intracellular level of PECAM-1 determined by in-cell ELISA assay (Abcam, UK). Cells of 3 healthy individuals and IP were seeded into collagen precoated 96-well microplate at a density of 2 × 104 cells/100 µL per well. Confluent ECFCs grown into 96-well plates were fixed, permeabilized, and stained with primary (CD31 antibody against PECAM-1) and horseradish peroxidase (HRP)-conjugated secondary antibodies. HRP-conjugated secondary antibodies were goat anti-mouse (Thermo Scientific, UK) and anti-rabbit (Abcam, UK) IgG. After the addition of the HRP-development solution, optical density at 650 (in a kinetic mode, within 60 minutes) was measured using a microplate reader (Synergy 2, BioTek). Last, The HRP signal of antibody-specific complexes was normalized to the Janus Green staining intensity to account for differences in cell density. Measurements were repeated for 3 independent experiments in triplicate (N = 9). Values are means ± SEM. ***P < .001 (unpaired Student t test). (D) ECFCs isolated from a healthy control (left) and the IP (right) were costained with antibodies recognizing VE-cadherin (red) and nucleus (DAPI staining, blue). The IP-ECFCs show defect in the expression of VE-cadherin at endothelial cell–endothelial cell junctions, associated with cytoplasmic red staining, indicating VE-cadherin internalization. Scale bars, 50 µm. (E) The response of the ECFCs to the thrombin stimulation by demonstrating gap formation between cells in the ECFCs monolayer. After thrombin treatment (10 nm, 5 minutes), cells were immunostained with an antibody targeted against VE-cadherin at endothelial cell junctions, and white-gray imaging was done by using inverted fluorescence microscopy. Scale bars, 100 µm. Boxes signify close-up views of VE-cadherin at cell-cell junctions.
Endothelial cobblestone morphology and expression of endothelial cell adhesion proteins VE-cadherin and PECAM-1 at cell–cell junctions of ECFCs. (A) The typical endothelial cobblestone morphology of ECFCs isolated from a healthy individual and the IP. Scale bars, 100 µm. (B) VWF (green) and adherence junction protein PECAM-1 (red) are visualized with coimmunostaining of ECFCs derived from a healthy individual (upper) and patient (IP-ECFCs) (lower). Scale bars, 50 µm. (C) The intracellular level of PECAM-1 determined by in-cell ELISA assay (Abcam, UK). Cells of 3 healthy individuals and IP were seeded into collagen precoated 96-well microplate at a density of 2 × 104 cells/100 µL per well. Confluent ECFCs grown into 96-well plates were fixed, permeabilized, and stained with primary (CD31 antibody against PECAM-1) and horseradish peroxidase (HRP)-conjugated secondary antibodies. HRP-conjugated secondary antibodies were goat anti-mouse (Thermo Scientific, UK) and anti-rabbit (Abcam, UK) IgG. After the addition of the HRP-development solution, optical density at 650 (in a kinetic mode, within 60 minutes) was measured using a microplate reader (Synergy 2, BioTek). Last, The HRP signal of antibody-specific complexes was normalized to the Janus Green staining intensity to account for differences in cell density. Measurements were repeated for 3 independent experiments in triplicate (N = 9). Values are means ± SEM. ***P < .001 (unpaired Student t test). (D) ECFCs isolated from a healthy control (left) and the IP (right) were costained with antibodies recognizing VE-cadherin (red) and nucleus (DAPI staining, blue). The IP-ECFCs show defect in the expression of VE-cadherin at endothelial cell–endothelial cell junctions, associated with cytoplasmic red staining, indicating VE-cadherin internalization. Scale bars, 50 µm. (E) The response of the ECFCs to the thrombin stimulation by demonstrating gap formation between cells in the ECFCs monolayer. After thrombin treatment (10 nm, 5 minutes), cells were immunostained with an antibody targeted against VE-cadherin at endothelial cell junctions, and white-gray imaging was done by using inverted fluorescence microscopy. Scale bars, 100 µm. Boxes signify close-up views of VE-cadherin at cell-cell junctions.
Expression and storage of endogenous and recombinant VWF
Immunostaining of the healthy ECFCs showed that the majority of VWF staining appeared as rod-like structures, representing WPBs. Further visual and quantitative assessment of the 3D immunofluorescence images demonstrated a significant reduction in the number and size of the WPBs within IP-ECFCs when compared with healthy ECFCs (Figure 4A).
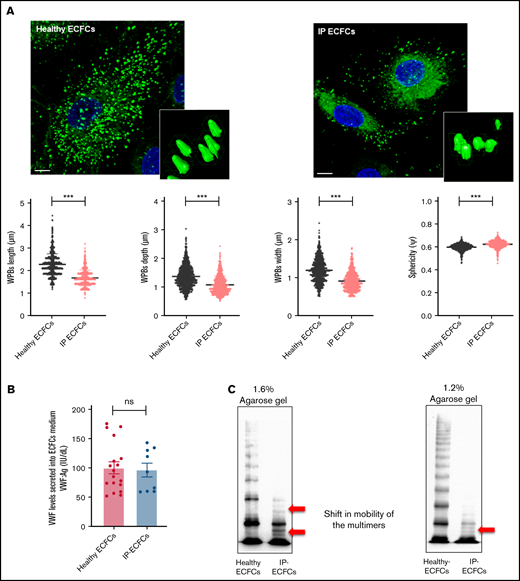
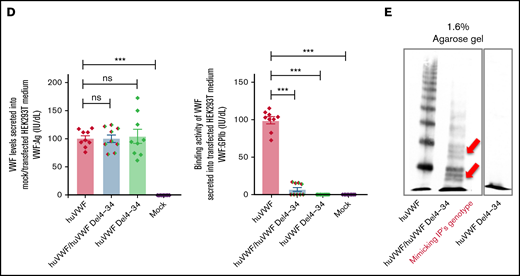
Intracellular storage and secretion of VWF in the ECFCs as well as expression of recombinant VWF in HEK293T cells. (A) Immunofluorescence images of ECFCs isolated from the IP and healthy individuals. In healthy ECFCs, VWF (green) is stored in rod-shaped organelles, resembling WPBs. However, VWF staining in the IP-ECFCs exhibited shorter WPBs than those observed in healthy ECFCs. The scale bar is 10 µm. Boxes signify close-up views of the processed 3D models of the WPBs granules surrounding with oriented bounds, generated by arivis Vision4D 3.2. The dot plot graphs demonstrate the quantitative morphological analysis of WPBs in healthy (black dot plot) and IP-ECFCs (pale red dot plot), including the average value of the longest (length), middle (depth), shortest sides (width), and sphericity factor (Ψ) of the selected 1000 WPBs. The sphericity factor, describing the roundness of the 3D granules, is represented as a value between 0 and 1, where 1 is an ideal sphere, and any particle that is not a sphere will have a sphericity < 1. IP-ECFCs demonstrated a decrease in length (1.69 ± 0.01 µm), depth (1.08 ± 0.01 µm), and width (0.91 ± 0.01 µm) of WPBs compared with wt (with an average length, depth, and width of 2.28 ± 0.01 µm, 1.37 ± 0.01 µm, and 1.2 ± 0.01 µm, respectively; P < .001). Furthermore, IP-ECFCs WPBs displayed a slightly increased sphericity value (0.61 ± 0.00 vs 0.59 ± 0.00 of wt WPBs; P < .001), favoring a less elongated shape. (B) The graph of the mean of VWF:Ag levels in the medium of ECFCs obtained from the IP (3 independent ECFCs isolation, each 3; N = 9) and 6 healthy donors (each 3 samples; N = 18). (C) The multimer profile of VWF in the supernatant of ECFCs analyzed by electrophoresis on 1.6% and 1.2% SDS-agarose gel. The multimer of VWF secreted from IP-ECFCs exhibited loss of large and intermediate multimers along with the shift in mobility of the small multimers (red arrows). (D) VWF:Ag levels (left) and VWF:GPIb (right) of secreted VWF into the medium of the transiently transfected HEK293T with huVWF (N = 9), huVWF/huVWF Del4-34 (coexpression at a ratio of 1:1; N = 9), and huVWF Del4-34 (N = 9) as well as untransfected cells (mock). (E) The multimer of recombinant VWF secreted into the medium of transfected huVWF, cotransfected huVWF/huVWF Del 4-34, and huVWF del 4-34 in HEK293T cell lines on 1.6% SDS-agarose gel. The multimer composition of secreted coexpressed huVWF/huVWF Del4-34 demonstrates the loss of large multimers along with a shift in mobility of the small multimers (red arrows) and homozygously expressed huVWF del 4-34 exhibited no multimer as expected. Error bars indicate the SEM. NS, not significant (P > .05); ***P < .001(unpaired Student t test).
Intracellular storage and secretion of VWF in the ECFCs as well as expression of recombinant VWF in HEK293T cells. (A) Immunofluorescence images of ECFCs isolated from the IP and healthy individuals. In healthy ECFCs, VWF (green) is stored in rod-shaped organelles, resembling WPBs. However, VWF staining in the IP-ECFCs exhibited shorter WPBs than those observed in healthy ECFCs. The scale bar is 10 µm. Boxes signify close-up views of the processed 3D models of the WPBs granules surrounding with oriented bounds, generated by arivis Vision4D 3.2. The dot plot graphs demonstrate the quantitative morphological analysis of WPBs in healthy (black dot plot) and IP-ECFCs (pale red dot plot), including the average value of the longest (length), middle (depth), shortest sides (width), and sphericity factor (Ψ) of the selected 1000 WPBs. The sphericity factor, describing the roundness of the 3D granules, is represented as a value between 0 and 1, where 1 is an ideal sphere, and any particle that is not a sphere will have a sphericity < 1. IP-ECFCs demonstrated a decrease in length (1.69 ± 0.01 µm), depth (1.08 ± 0.01 µm), and width (0.91 ± 0.01 µm) of WPBs compared with wt (with an average length, depth, and width of 2.28 ± 0.01 µm, 1.37 ± 0.01 µm, and 1.2 ± 0.01 µm, respectively; P < .001). Furthermore, IP-ECFCs WPBs displayed a slightly increased sphericity value (0.61 ± 0.00 vs 0.59 ± 0.00 of wt WPBs; P < .001), favoring a less elongated shape. (B) The graph of the mean of VWF:Ag levels in the medium of ECFCs obtained from the IP (3 independent ECFCs isolation, each 3; N = 9) and 6 healthy donors (each 3 samples; N = 18). (C) The multimer profile of VWF in the supernatant of ECFCs analyzed by electrophoresis on 1.6% and 1.2% SDS-agarose gel. The multimer of VWF secreted from IP-ECFCs exhibited loss of large and intermediate multimers along with the shift in mobility of the small multimers (red arrows). (D) VWF:Ag levels (left) and VWF:GPIb (right) of secreted VWF into the medium of the transiently transfected HEK293T with huVWF (N = 9), huVWF/huVWF Del4-34 (coexpression at a ratio of 1:1; N = 9), and huVWF Del4-34 (N = 9) as well as untransfected cells (mock). (E) The multimer of recombinant VWF secreted into the medium of transfected huVWF, cotransfected huVWF/huVWF Del 4-34, and huVWF del 4-34 in HEK293T cell lines on 1.6% SDS-agarose gel. The multimer composition of secreted coexpressed huVWF/huVWF Del4-34 demonstrates the loss of large multimers along with a shift in mobility of the small multimers (red arrows) and homozygously expressed huVWF del 4-34 exhibited no multimer as expected. Error bars indicate the SEM. NS, not significant (P > .05); ***P < .001(unpaired Student t test).
The quantification of the morphometric parameters of the 3D models of the WPBs within IP-ECFCs demonstrated a decrease in length of WPBs compared with wt (P < .001) (Figure 4A). Furthermore, IP-ECFCs WPBs displayed a slightly increased sphericity value (P < .001), favoring a less elongated shape (Figure 4A).
Further, the secreted VWF:Ag levels (IU/dL) into media of healthy and IP-ECFCs were quantified, and the IP-ECFCs VWF levels are presented here as a percentage of the mean value of VWF levels in healthy ECFC medium. Interestingly, the secreted VWF:Ag levels from the IP- ECFCs were not significantly different from that of the healthy donors (96.2% ± 11.9% of wt, P > .05) (Figure 4B). However, multimer analysis of the secreted VWF from IP-ECFCs showed loss of large- and intermediate-molecular-weight multimers along with a shift in mobility of small multimers (Figure 4C), demonstrating the dominant-negative impact of the deleted VWF on multimer assembly. In keeping with the ECFC ex vivo data, secretion of VWF from cotransfected HEK293T cells (huVWF/huVWF del4-34, mimicking the IP’s genotype) was normal relative to the secretion of huVWF (100.3% ± 6.7% of wt, P > .05), but with a defect in multimer composition (similar to the multimers of IP-ECFCs) and impaired VWF:GPIbM (reduced to 7.0% ± 2.8% of wt, P < .001), as expected (Figure 4D-E).
Alternative trafficking of pro-inflammatory Ang2 and P-selectin molecules
Immunostaining of healthy ECFCs revealed that Ang2, mainly demonstrating small punctate patterns, is mainly colocalized with VWF in WPBs (Figure 5A). In ∼10% of healthy ECFCs, partial localization of Ang2 in the nucleus was additionally observed. In contrast, in IP-ECFCS, Ang2 staining is not colocalized with VWF staining (correlation coefficient of 0.02 ± 0.01 vs 0.35 ± 0.02 for healthy controls, P < .001) (Figure 5A lower; Figure 5C), but we observed accumulation of Ang2 molecules in the nucleus of ∼70% of the cells (Figure 5B; supplemental Movies 1 and 2). The nuclear accumulation of Ang2 was detected in all 3 ECFCs isolated on 3 different episodes. The in-cell ELISA assay did not reveal any difference in intracellular Ang2 levels in IP-ECFCs (optical density: 0.22 ± 0.00 vs 0.23 ± 0.04 in healthy controls) compared with healthy individuals. We also observed a reduction in colocalization of VWF/P-selectin in IP-ECFCs (correlation coefficient of 0.27 ± 0.03 vs 0.60 ± 0.03, P < .001) (Figure 5D lower; Figure 5E).
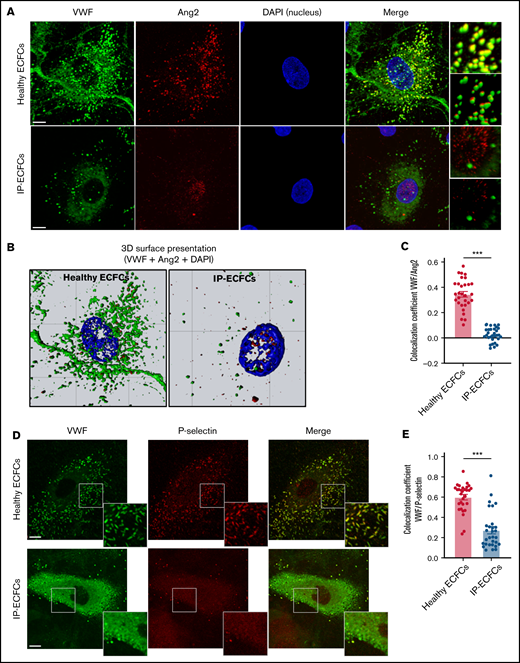
Impairment in the storage of the WPBs components, Ang2 and P-selectin, in patients’ endothelial cells. (A) Storage of VWF (green) and Ang2 (red) in WPBs was visualized by immunofluorescence staining of normal ECFCs (upper) and index patient ECFCs (IP-ECFCs; lower). The merge of green and red channels (with the focused region of interests) displays colocalization of VWF with Ang2 in healthy ECFCs, whereas strongly deficient VWF/Ang2 colocalization was perceived in IP-ECFCs. Scale bars, 10 µm. (B) The 3D topographic view of the merged channels of the ECFC images shown in panel A, generated by Zeiss Zen blue software. (C) The colocalization coefficients (of VWF and Ang2) were determined from at least 30 regions of interest from 2 independent experiments. (D) Fixed healthy ECFCs (upper) and IP-ECFCs (lower) were costained with antibodies detecting VWF (green) and P-selectin (red). The merge of green and red channels (with the focused regions of interest) demonstrates colocalization of VWF with P-selectin in healthy ECFCs, whereas IP-ECFCs show diminished colocalization of VWF/P-selectin. Scale bars, 20 µm. (E) Colocalization coefficients (of VWF and P-selectin) from at least 30 regions of interest from 2 independent experiments. Data are means ± SEM. ***P < .001 (unpaired Student t test).
Impairment in the storage of the WPBs components, Ang2 and P-selectin, in patients’ endothelial cells. (A) Storage of VWF (green) and Ang2 (red) in WPBs was visualized by immunofluorescence staining of normal ECFCs (upper) and index patient ECFCs (IP-ECFCs; lower). The merge of green and red channels (with the focused region of interests) displays colocalization of VWF with Ang2 in healthy ECFCs, whereas strongly deficient VWF/Ang2 colocalization was perceived in IP-ECFCs. Scale bars, 10 µm. (B) The 3D topographic view of the merged channels of the ECFC images shown in panel A, generated by Zeiss Zen blue software. (C) The colocalization coefficients (of VWF and Ang2) were determined from at least 30 regions of interest from 2 independent experiments. (D) Fixed healthy ECFCs (upper) and IP-ECFCs (lower) were costained with antibodies detecting VWF (green) and P-selectin (red). The merge of green and red channels (with the focused regions of interest) demonstrates colocalization of VWF with P-selectin in healthy ECFCs, whereas IP-ECFCs show diminished colocalization of VWF/P-selectin. Scale bars, 20 µm. (E) Colocalization coefficients (of VWF and P-selectin) from at least 30 regions of interest from 2 independent experiments. Data are means ± SEM. ***P < .001 (unpaired Student t test).
Differentially expressed genes
The whole transcriptome profiling was done to evaluate any alteration in gene expression profiling of IP-ECFCs. From a total of 18 912 identified mRNAs of RNA-seq analyses, there were 680 DEGs (428 upregulated and 252 downregulated) in IP-ECFCs, compared with healthy subjects (Figure 6A; supplemental Tables 4 and 5). The RNA-seq data demonstrated that expression of inflammatory molecules copacked with VWF inside WPBs, including IL-8 (CXCL8), IL-6, GROα (CXCL1), and P-selectin (SELP), is significantly upregulated in IP-ECFCs (mean difference>1, P < .05) (Figure 6B). The most enriched GO-biological process terms among the DEGs were regulation of cell spreading and differentiation (reflected by observed insufficiency in cell proliferation of the IP-ECFCs in ex vivo culture), leukocyte adhesion to vascular endothelial, blood vessel morphogenesis, and angiogenesis. (Figure 6C). Further, the upstream regulator analysis was done by IPA to predict potential regulators (cytokine, growth factor, or transcription regulators) that cause changes in expression in IP-ECFCs (P < .05 and a negative z-score). The network and list of the top 5 upstream regulators (thrombin as well as cytokines tumor necrosis factor [TNF], CSF1, IL-4, and IL-1α), which were predicted to be activated in IP-ECFCs, are illustrated in supplemental Figure 1.
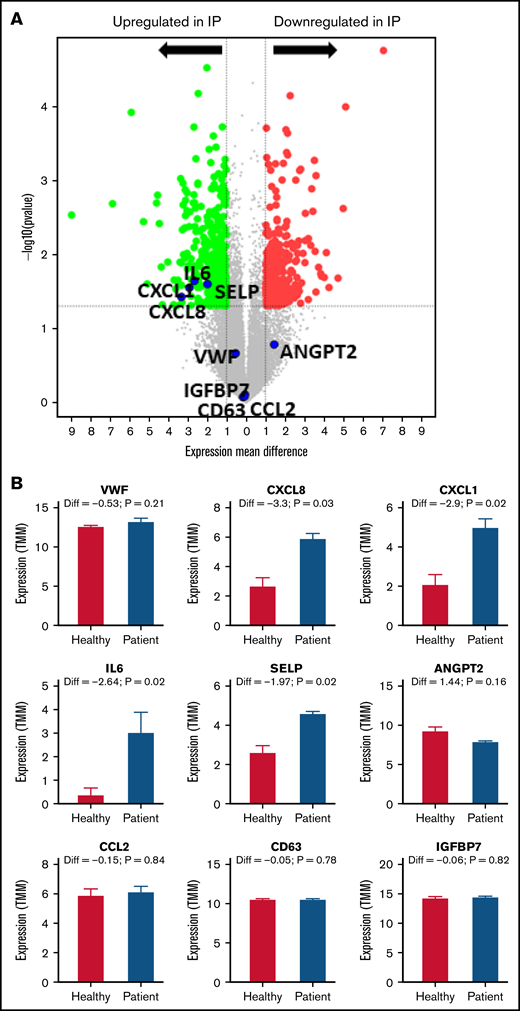
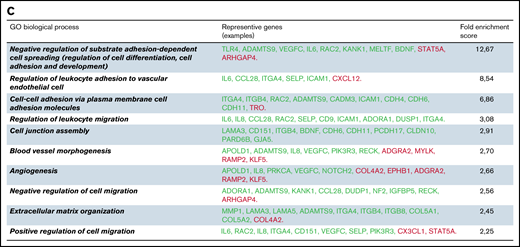
Differentially expressed genes in IP-ECFCs and the enriched GO-biological process terms. (A) Volcano plot illustrates significantly DEGs in IP-ECFCs in which –log10 (p) is plotted against the mean differences. Green dots represent the upregulated genes and red dots represent downregulated genes. Numbers of genes upregulated or downregulated are denoted. Dashed lines mark the threshold of statistical significance. DEGs were considered significant when P < .05 and fold-change was > 2 in respect of healthy control ECFC samples/or absolute log2FC (mean difference) was > 1. A negative mean difference value indicates lower expression in healthy controls and a positive mean difference indicates lower expression in IP-ECFCs. The blue dots illustrate the expression of WPB cargos, which are either upregulated (CXCL8, IL6, CXCL1, and SELP) or are expressed normally (VWF, Ang2, CCL2, CD63, and IGFBP7). (B) Graphs show the expression level (represented by TMM) of WPB constituents in healthy as well as index patient ECFCs. The mean difference values (Diff), calculating a difference between the expression of healthy controls and the IP, as well as P values are specified above the columns. There was no significant difference in expression (Diff) of the VWF, Ang2, CCL2, CD63, and IGFBP7 between healthy and IP-ECFCs (P > .05), though the expression of IL-8, CXCL1, IL-6, and P-selectin in IP was upregulated, demonstrating significant Diff (P < .05). (C) The table displays the list of top 10 enriched GO-biological process terms as well as the examples of the representative genes (green: upregulated genes; red: downregulated genes), and their Fold Enrichment Score estimated after carrying out GO enrichment analysis using the GO Consortium tool. TMM, trimmed mean of the M-values.
Differentially expressed genes in IP-ECFCs and the enriched GO-biological process terms. (A) Volcano plot illustrates significantly DEGs in IP-ECFCs in which –log10 (p) is plotted against the mean differences. Green dots represent the upregulated genes and red dots represent downregulated genes. Numbers of genes upregulated or downregulated are denoted. Dashed lines mark the threshold of statistical significance. DEGs were considered significant when P < .05 and fold-change was > 2 in respect of healthy control ECFC samples/or absolute log2FC (mean difference) was > 1. A negative mean difference value indicates lower expression in healthy controls and a positive mean difference indicates lower expression in IP-ECFCs. The blue dots illustrate the expression of WPB cargos, which are either upregulated (CXCL8, IL6, CXCL1, and SELP) or are expressed normally (VWF, Ang2, CCL2, CD63, and IGFBP7). (B) Graphs show the expression level (represented by TMM) of WPB constituents in healthy as well as index patient ECFCs. The mean difference values (Diff), calculating a difference between the expression of healthy controls and the IP, as well as P values are specified above the columns. There was no significant difference in expression (Diff) of the VWF, Ang2, CCL2, CD63, and IGFBP7 between healthy and IP-ECFCs (P > .05), though the expression of IL-8, CXCL1, IL-6, and P-selectin in IP was upregulated, demonstrating significant Diff (P < .05). (C) The table displays the list of top 10 enriched GO-biological process terms as well as the examples of the representative genes (green: upregulated genes; red: downregulated genes), and their Fold Enrichment Score estimated after carrying out GO enrichment analysis using the GO Consortium tool. TMM, trimmed mean of the M-values.
Assessing leukocyte adhesion on ECFCs
The adhesiveness of the IP-ECFCs (to leukocytes), which showed upregulation of the gene expression of the cell adhesion molecules P-selectin and ICAM, were assessed in comparison with healthy ECFCs. To enumerate the adhesive ECs, the number of adhered promyelocytic leukemia cell (HL60) clusters and the number of ECs in the field were counted, and the percentage of adhesive ECs was calculated. IP ECFCs exhibited an enhanced adhesiveness to the leukocytes (HL-60) compared with healthy individuals (supplemental Figure 2A-B). Furthermore, the adhered HL-60 (on IP-ECFCs) demonstrated lobed nucleus, indicating differentiation of the HL-60 to granulocyte-like cells (supplemental Figure 2C). Previously, it was recurrently reported that the HL-60 cells can be induced to differentiate to neutrophil-like cells.38 Besides, the adhered HL-60 cells revealed VWF staining (supplemental Figure 2D). Additionally, in the current study, the immunostaining of the neutrophils isolated from the IP showed VWF staining that was even stronger after the patient received VWF concentrates (after 3 hours) (supplemental Figure 3).
Clearance of recombinant huVWF in mice
Clearance studies, evaluating VWF plasma half-life, percentage recovery, and mean residence time, demonstrated that huVWF del4-34 is cleared at the same rate as full-length huVWF in VWF-deficient mice (supplemental Figure 4A-D).
Discussion
In this study, we showed pathomolecular mechanisms of a heterozygous in-frame large deletion, VWF del4-34, which was detected in a patient with type 3 VWD. The IP suffers from frequent spontaneous oral cavity bleeding despite prophylaxis treatment with a high dose of plasma-derived Voncento VWF/FVIII concentrates, associated with a short half-life of administered VWF in IP (5.2 hours vs on average 18.3 hours reported for Voncento VWF in adult patients with VWD).39 Initially, this single gene defect could not provide a plausible explanation for such low plasma VWF levels and severe clinical demonstration observed in the IP. Further transcript analysis assured the presence of a full-length normal VWF transcript, excluding any deficiency in the transcription of the second VWF allele. Interestingly, the RNA analysis revealed that the deleted VWF allele (del4-34) is still effectively transcribed, resulting in an aberrant in-frame mRNA that would encode a truncated protein (p.Asp75_Cys1948del). Because the VWF carboxyl-terminus domains are still preserved, it is therefore expected that the truncated protein participates in the dimerization and results in the formation of heterodimers (normal/del4-34). However, the heterodimers would terminate multimerization because of lacking the domains essential for the multimerization, verified by loss of large and intermediate multimers along with a shift in mobility of small multimers of secreted endogenous (IP-ECFCs) and recombinant coexpressed (huVWF/del4-34) VWF. Surprisingly secretion of the chimeric deleted VWF multimers was not diminished. This observation was an exception to previous observations reporting structurally abnormal VWF mutants.40-42 Casari et al reported a different in-frame VWF large deletion lacking exons 26 through 34 (p.P1127_C1948delinsR), which has a dominant-negative impact on both VWF multimer assembly and secretion, reporting a severe reduction in VWF secretion (∼25% of wt) in vitro.43 Furthermore, because the plasma VWF level in the IP was very low and the half-life of infused therapeutic VWF was markedly reduced (confirmed by PK evaluations), we concluded that the elimination of both the secreted anomalous VWF and infused VWF from the plasma is enhanced.
Immunostaining of the IP-ECFCs revealed that WPB biogenesis was severely impaired. Together with VWF, several inflammatory and angiogenic regulators are copackaged into WPBs, such as Ang2, P-selectin, IL-8, IL-6, GROα, CD63, IGFBP7, and MCP1 (CCL2), which can be released upon stimulation by inflammatory mediators (cytokines, such as TNF-α and IL-1, as well as thrombin).44-48 Here, we observed that abnormal WPBs in IP-ECFCs resulted in defective storage of other WPB constituents: inflammatory and angiogenesis regulators P-selectin and Ang2. Besides, the expression of 4 of 8 inflammatory molecules expressed in ECFCs, and known to be copackaged with VWF into WPBs, including P-selectin, chemokines IL-8, IL-6, and GROα were upregulated in IP-ECFCs. Although the expression of the Ang2 (ANGPT2) was not different in IP-ECFCs from healthy controls (proved by both RNA-seq and in-cell ELISA) (Figure 6B), its trafficking was altered and visualized by significant nuclear accumulation of Ang2, which is reported here for the first time in a patient with VWD. Ang2 has crucial roles in inflammation and angiogenesis; it promotes leukocyte adhesion as well as endothelial destabilization by triggering a network of cellular signaling pathways.49-51 The well-defined roles of Ang2 are mainly through Tie2 on the surface of endothelial, or by activation of β-1 integrin.52-54 However, previous studies, reporting a fractional localization of Ang2 in the nucleus of normal endothelial cells (similar to our observation) and also a prenuclear accumulation of Ang2 following stimulation of healthy endothelial cells with thrombin or histamine, indicate the potential roles of Ang2 in the nucleus.55,56 Although Ang2 primes endothelial responses to inflammatory stimuli such as TNF-α, thereby modulating the expression of genes promoting leukocyte adhesion and extravasation, previous studies have shown that the expression of Ang2 in ECs is sufficient to promote leukocyte recruitment even in the absence of preceding inflammatory stimuli.57,58 Remarkably, our RNA-seq indicated that enriched biological pathways in IP-ECFCs were related to inflammatory and immune responses, as well as cellular growth and angiogenesis (downstream signaling pathways induced by Ang2 acknowledged by IPA upstream regulator analysis). Further, our IPA upstream regulator analysis showed that the alternations in inflammatory and cellular growth pathways observed in the IP’s ECs are similar to perturbations induced by inflammatory mediators, such as TNF and thrombin, which stimulate WPB release. In the current RNA-seq exploration, the IP’s ECFCs were not treated with any of these inflammatory mediators, even though IPA predicted their activation with high confidence. We speculate that this is because the inflammatory factors such as Ang2 and P-selectin are not sequestered in the abnormal WPBs of IP-ECFCs because they are in healthy endothelial, mimicking circumstances that cells were induced with inflammatory mediators. In line with our RNA-seq data and pathway analysis, our cellular experiments demonstrated an increased adhesion of HL-60 leukocytes to IP-ECFCs (attributed to the observed upregulation of the leukocyte chemoattractants [eg, IL-6, IL-8, GROα, adhesion molecules P-selectin, ICAM]),49,59-65 as well as discontinuities in VE-cadherin presentation/low presentation of PECAM-1 at endothelial junctions. Therefore, we conclude that changes in gene expression and phenotype of the patient endothelial cells presumably contribute to the dislocalization of inflammatory cargos of WPBs, particularly transportation of Ang2 into nucleus.
Previous studies, exploring the effects of VWF deficiency on blood vessel formation (or inflammation) by using cellular and animal models, have demonstrated a range of abnormalities in angiogenesis and/or leukocyte adhesion, with no overall consistent feature. For instance, previous studies detected no (or very little) leukocyte rolling and leukocyte recruitment in VWF-deficient mice under inflammatory conditions.66,67 These studies concluded that absence of WPBs might lead to the deficiency in presentation of WPBs’ resident inflammatory proteins, such as P-selectin, which in turn affect leukocyte recruitment.68 Further, an increased leukocyte recruitment is observed in murine model of VWD-type 2B during an inflammatory response, which could be dependent on increased VWF platelet binding.69 Hence, altogether, these studies using mice models raised a question whether this deficient leukocyte recruitment is caused only by the absence of WPBs or whether it might be dependent of deficiency in plasma VWF and its platelet binding function. Our study, using ex vivo ECs, favor this notion that deficient WPBs and alternative trafficking of the WPBs’ inflammatory residents has led to the altered leukocyte recruitment. In sum, this heterogeneity of outcomes might be related to the extent and type of defect in VWF and its subsequent impact on WPBs biogenesis and intracellular trafficking of WPBs inflammatory cargos such as P-selectin, IL-8, and Ang2.69-76 Schillemans et al found relocation of Ang2 to the cell periphery, colocalized with Tie2, in CRISPR VWF knock-out ECFCs.77 This indicates that each mutation may uniquely affect the biogenesis of WPBs and trafficking of the other cargos, which might ultimately distinctively influence the downstream signaling pathways, consistent with the previously mentioned previous studies demonstrating heterogeneity in angiogenesis features and inflammatory responses between VWD patients.
In this study, we have also explored potential mechanisms leading to accelerated clearance of both abnormal endogenous and infused VWF from the plasma. Although our clearance studies showed that huVWF del4-34 is cleared at the same rate as huVWF in VWF knock-out mice, because of differences in clearance mechanisms between humans and mice, we cannot assuredly exclude faster elimination of deleted VWF from human circulation. Moreover, we excluded the possibility of any antibody development or increased plasma antigen levels and activity of ADAMTS-13. Remarkably, our RNA-seq data and cellular studies exhibited enhanced adherence of leukocytes (HL-60) to patient ECFCs. Furthermore, isolated neutrophils displayed VWF staining in both healthy controls and the IP, which was prominent after the IP received VWF concentrates. Hence, we propose that increased adhesiveness of endothelial cells to leukocytes, HL-60 cells (along with upregulated leukocyte chemoattractants IL-6 and IL-8) may cause activation of these immune cells, and might contribute to the rapid elimination of VWF from circulation. In keeping with our assumption, Pendu et al reported that polymorphonuclear leukocytes bind to VWF through leukocyte-specific β2 integrins (which can be increased 10-fold when activated with phorbol ester, PMA), suggesting it as an endocytic receptor candidate for VWF.18,78 Nonetheless, further investigations are required to confirm the contribution of the neutrophils to the internalization and clearance of the VWF, which is in progress in our laboratory.
To sum up, we show that the deleted VWF protein incorporates into VWF multimers and has a dominant-negative impact on the elongation of multimers and the biogenesis of WPBs. The malformed WPBs hinder the recruitment of cargos, such as Ang2 and P-selectin, thereby resulting in the alternative trafficking of these proteins in a unique way (nuclear accumulation) that mimics the stimulation of endothelial cells with inflammatory regulators (eg, thrombin), in turn causes distinctive perturbations in downstream signaling pathways, leading to endothelial cell dysfunction and increased endothelial cell adhesiveness.
Acknowledgments
Whole-genome expression profiling, including library preparation and A-sequencing analysis, was performed at the Life & Brain Genomics located in University Clinic Bonn. Quantitative evaluations of the 3D images were performed by the Vision4D 3.2 software, graciously supported by arivis AG, Imaging Science, Germany.
Authorship
Contribution: H.Y. designed the study; performed the experimental work, did bioinformatics gene ontology evaluations and the Ingenuity Pathway Analysis (IPA), interpreted the data, and wrote the paper; M.A.J. and O.E.-M. performed RNA-seq bioinformatics analysis and the IPA; J.M. contributed to the flow cytometry analysis of the endothelial colony-forming cells; N.M. contributed to the patient’s data and pharmacokinetic study; O.R. and D.L. designed, performed, and interpreted clearance studies in the mouse model and reviewed and edited the manuscript. U.B. evaluated the development of alloantibodies against von Willebrand factor; and J.O. supervised the study, reviewed, and edited the manuscript.
Conflict-of-interest disclosure: J.O. reports grants and personal fees from Bayer, Biotest, CSL Behring, Octapharma, Pfizer, SOBI, and Shire/Takeda, besides personal fees from Biogen Idec, Biomarin, Chugai, Freeline, Grifols, Novo Nordisk, Roche, Sanofi, and Sparks, outside the submitted work. N.M. reports grants and personal fees from Pfizer and personal fees from Bayer, Chugai, CSL Behring, Novo Nordisk, Octapharma, Roche, SOBI, and Shire/Takeda, outside the submitted work. J.O. and N.M. received personal fees for travel support, participation in Advisory Boards and participating in symposia as speaker. The remaining authors declare no competing financial interests.
Correspondence: Johannes Oldenburg, Institute of Experimental Haematology and Transfusion Medicine, University Clinic Bonn, Venusberg-Campus 1, Bonn 53105, Germany; e-mail: johannes.oldenburg@ukbonn.de; and Hamideh Yadegari, Institute of Experimental Haematology and Transfusion Medicine, University Clinic Bonn, Venusberg-Campus 1, Bonn 53105, Germany; e-mail: hamideh.yadegari@ukbonn.de.

