

Key Points
superFVa arrests severe bleeding and prevents the development of ATC after trauma.
superFVa therapy restores functional hemostasis when initiated after onset of ATC caused by traumatic bleeding.

Abstract
Acute traumatic coagulopathy (ATC) occurs in approximately 30% of patients with trauma and is associated with increased mortality. Excessive generation of activated protein C (APC) and hyperfibrinolysis are believed to be driving forces for ATC. Two mouse models were used to investigate whether an engineered activated FV variant (superFVa) that is resistant to inactivation by APC and contains a stabilizing A2-A3 domain disulfide bond can reduce traumatic bleeding and normalize hemostasis parameters in ATC. First, ATC was induced by the combination of trauma and shock. ATC was characterized by activated partial thromboplastin time (APTT) prolongation and reductions of factor V (FV), factor VIII (FVIII), and fibrinogen but not factor II and factor X. Administration of superFVa normalized the APTT, returned FV and FVIII clotting activity levels to their normal range, and reduced APC and thrombin-antithrombin (TAT) levels, indicating improved hemostasis. Next, a liver laceration model was used where ATC develops as a consequence of severe bleeding. superFVa prophylaxis before liver laceration reduced bleeding and prevented APTT prolongation, depletion of FV and FVIII, and excessive generation of APC. Thus, prophylactic administration of superFVa prevented the development of ATC. superFVa intervention started after the development of ATC stabilized bleeding, reversed prolonged APTT, returned FV and FVIII levels to their normal range, and reduced TAT levels that were increased by ATC. In summary, superFVa prevented ATC and traumatic bleeding when administered prophylactically, and superFVa stabilized bleeding and reversed abnormal hemostasis parameters when administered while ATC was in progress. Thus, superFVa may be an attractive strategy to intercept ATC and mitigate traumatic bleeding.
Introduction
Trauma is the leading cause of death and disability for young adults (age < 45 years).1-3 Trauma-induced coagulopathy (TIC) contributes significantly to mortality after traumatic injury despite improved protocols encompassing fluid resuscitation and transfusion strategies.4-12 TIC is a temporal syndrome that can present itself by multiple phenotypes of different etiology.13-17 An early endogenous component of TIC, and preceding iatrogenic effects, is acute traumatic coagulopathy (ATC).6,7,18,19 ATC is a major complication in the first few hours after trauma in approximately 30% of patients and contributes to uncontrollable bleeding with high mortality.5,20-23 ATC in association with resuscitation-induced coagulopathy, hypothermia, and acidosis (“lethal triad”) is thought to initiate complement activation and innate immune reactions resulting in a vicious cycle, fueling ATC/TIC and more bleeding.5,15,24 Notwithstanding that patients with trauma often have laboratory-defined coagulopathy without significant bleeding, the coagulopathy in these patients is associated with higher mortality, additional resuscitation needs, and significant morbidities in those who survive. It is therefore critical to intercept the coagulopathy, with the goal to stop bleeding, as early as possible. Advances in the management of ATC remain limited to optimization of goal-directed blood component transfusion strategies and tranexamic acid.3,10 Early use of tranexamic acid for catastrophic bleeding associated with trauma or postpartum hemorrhage has demonstrated consistently a relative mortality reduction of approximately 20% to 30%,24-26 leaving room for improvement. Thus, there is an unmet need for safe and effective antihemorrhagic/coagulopathy-mitigating therapies as there are currently no targeted treatments available to intervene before or after the development of ATC.
Although detailed molecular mechanisms of ATC remain to be elucidated, there is increasing evidence to support that ATC is generated by the exaggerated generation of the natural anticoagulant, activated protein C (APC), resulting in reduced availability of factor Va (FVa) and factor VIIIA (FVIIIa), as well as unchecked fibrinolysis. ATC is characterized by rapid depletion of FV and FVIII, as well as fibrinogen consumption because of hyperfibrinolysis.27-34 ATC appears qualitatively distinct from other coagulopathies, such as diffuse intravascular coagulation (DIC), because levels of FV, FVIII, and fibrinogen are selectively diminished, whereas levels of factor X (FX) and factor II (FII) remain relatively normal.27 Moreover, normal or moderately increased thrombin generation in vitro in plasma from patients with ATC supports the notion that the capacity to generate thrombin is preserved and may not be representative of the in vivo hemostatic potential, highlighting that bleeding in ATC is complex and invokes pathways that are not based on procoagulant defects alone.27,35,36
APC is a plasma serine protease generated on the endothelial cell surface by the thrombin–thrombomodulin complex and the endothelial protein C receptor.37-39 As a potent natural anticoagulant, APC proteolytically and irreversibly inactivates the activated cofactors, FVa and FVIIIa, facilitated by protein S as a nonenzymatic cofactor and various lipids.37,38,40,41 Growing evidence in humans and animal models suggests that excessive APC anticoagulation can exacerbate bleeding.27,28,42,43 As a cofactor in the prothrombinase complex, FVa enhances the rate of thrombin generation approximately 10 000-fold.44-46 APC rapidly inactivates FVa via proteolytic cleavage at Arg506, followed by a slower cleavage at Arg306.44,47,48 Mutations of these cleavage sites, such as Arg506Gln (FVLeiden), augment its ability to enhance thrombin generation.49,50 The first clinical evidence that APC-resistant FV mutations may attenuate bleeding stems from observations in patients with hemophilia with the FVLeiden mutation.51-53 Data in a murine model of ATC support the concept that APC is a major instigator of ATC and that targeting the APC anticoagulant pathway in early stages of trauma improves survival.27,28,54 Thus, a targeted strategy to intercept the APC anticoagulant pathway appears attractive.
superFVa is an engineered activated FV (FVa) resistant to APC-mediated inactivation because of mutation of the 3 APC cleavage sites (Arg306Gln, Arg506Gln, and Arg679Gln), whereas a disulfide-linkage introduced between the A2 and A3 domain conveys a three- to fourfold increased specific activity.47,53 superFVa demonstrated efficient normalization of hemostasis in hemophilic mice and plasma from patients with hemophilia with and without inhibitors.53,55 superFVa also ablated APC-exacerbated traumatic bleeding and bleeding associated with direct oral anticoagulants in wild-type mice.43,56 Mechanistically, superFVa contributes uniquely to hemostasis because it not only reduces bleeding because of resistance to APC but also by circumventing the FV activation threshold and prothrombinase complex enhancement.43 Here we evaluated the extent to which superFVa can intercept ATC and control hemorrhage in 2 murine trauma models, involving trauma/shock and liver laceration-induced bleeding, because the liver is the most commonly injured organ in adult trauma with high mortality because of hemorrhage.57,58
Methods
Materials
Human prothrombin–, FVIII-, and FX-deficient plasma was purchased from George King Bio-Medical (Overland Park, KS). Human FV-deficient plasma and thrombin were purchased from Enzyme Research Laboratories (South Bend, IN). Hirudin was from Calbiochem (La Jolla, CA).
Recombinant superFV
Recombinant superFV was made on a B-domain–deleted S2183A platform and purified from conditioned media of stably transfected BHK cells by a combination of affinity chromatography using anti-FV 3B1 and HV5101 monoclonal antibodies as described.47,53,59 superFV was activated with 2 nM thrombin for 20 minutes at 37°C in prothrombinase buffer consisting of 50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 150 mM NaCl, 0.5% bovine serum albumin (BSA), 5 mM CaCl2 and 0.1 mM MnCl2, and activation was terminated by adding 1.1 molar equivalent of hirudin. The concentration of superFVa was determined by absorbance at 280 nm using ɛ1%, 1 cm = 15.460 and enzyme-linked immunosorbent assay (Enzyme Research Laboratories, South Bend, LA). Characterization of protein purity, disulfide linkage, APC resistance, and specific activity were performed as previously described.53
Animal models
All animal protocols were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute, La Jolla, CA. C57BL/6J mice were obtained from The Scripps Research Institute internal breeding facility. Male and female mice 10 to 20 weeks old were used for experiments.
Mouse model of ATC induced by trauma and shock
ATC was induced using the validated model described by Chesebro et al28 with minor modifications (supplemental Figure 1). In brief, mice were anesthetized with 1.5% isoflurane and 2 L/min O2 and secured in supine position on a metal board over a heating pad connected to a circulating water bath to maintain the body temperature at 37°C throughout the experiment. The left jugular vein was cannulated with PE-10 tubing prefilled with saline. A high-fidelity 1.2-F rodent Pressure Catheter (Transonic) was inserted into the right carotid artery for monitoring of the mean arterial pressure (MAP). The pressure catheter was connected to a SP200 pressure control unit (Transonic) and to a LabTrax 4-Channel Data Acquisition module (World Precision Instruments). The arterial waveform was recorded continuously using LabScribe software with the blood pressure module (iWorx Systems) to calculate the MAP in real time. Soft tissue trauma was induced 20 minutes before start of the experiment by a sterile 2-cm midline laparotomy, whereupon the wound was immediately closed with wound clips (AutoClip kit; Fine Science Tools). Shock was induced by slowly withdrawing blood (450-550 µL) from the jugular vein catheter until the MAP reached 35 ± 5 mm Hg. This MAP was maintained throughout the 1-hour experimental period. Mice were divided into different experimental groups: trauma (T) mice underwent laparotomy; shock (S) mice underwent blood withdrawal; trauma and shock (TS) mice underwent laparotomy and blood withdrawal; and control baseline (BL) mice underwent neither. Irrespective of grouping, all mice were subjected to the same anesthetic procedures and catheter and pressure probe placement and were maintained for 60 minutes. At the end of the 60-minute experimental period, blood was collected by cardiac puncture, and the mice were killed.
Mouse model of ATC induced by liver laceration and hemorrhage
ATC was induced using a liver laceration model that causes severe internal hemorrhage as described previously.61 Mice were anesthetized with 1.5% isoflurane and 2 L/min O2, secured in supine position on a metal board over a heating pad to maintain the body temperature at 37°C throughout the experiment. Mice were given 0.1 mg/kg buprenorphine (Buprenox) before surgery. The abdominal cavity was exposed by midline laparotomy. Two triangle-cut preweighed pieces of filter paper (approximately 0.05 g each) were inserted into the abdominal cavity, and 75% of the left liver lobe was removed with sharp scissors. The abdominal skin was securely closed using wound clips (AutoClip kit) to avoid leakage of blood. Mice were returned to their cages and monitored for 60 minutes. At the end of the 60-minute experimental period, mice were anesthetized, wound clips were removed, and filter papers were collected. A third piece of preweighed filter paper was used to absorb any remaining blood in the abdominal cavity. Bleeding was determined by weighing the blood-soaked filter papers using a standard curve of defined amounts of blood added to the filter papers. Control mice underwent the same procedures except liver laceration. In separate experiments, the jugular vein was cannulated and/or a pressure probe was inserted into the carotid artery to gain information about the MAP as described for the trauma/shock model.
superFVa administration
superFVa was diluted to a concentration of 0.4 or 0.8 mg/kg in 0.9% sterile saline for injection (Hosperia) and administered either as a single bolus by retro-orbital injection 5 minutes before liver laceration or by continuous infusion 30 minutes after induction of trauma/shock or liver laceration. Continuous infusion was administered via the jugular vein catheter by a GenieTouch syringe pump (Kent Scientific) at a rate of 5 µL/min for 20 minutes. Control mice received an equal volume of sterile saline. No resuscitation was administered in these animals.
Blood collection
At the end of the experimental period, blood was collected by cardiac puncture into 3.8% sodium citrate (1:9 volume dilution). For APC measurements, 100 mM benzamidine (Sigma) was added to the blood collection tubes. Samples were centrifuged twice (2000g for 10 minutes and 13 500g for 5 minutes) before plasma was stored at −80°C until analysis.
Determination of plasma clotting factor activity levels
Clotting times were recorded using an Amelung KC 4a micro coagulometer (Sigma Diagnostics, St Louis, MO). Activated partial thromboplastin time (APTT) was determined by mixing 25 µL mouse plasma with 25 µL APTT-XL reagent (Pacific Hemostasis; ThermoFisher Scientific), followed after 3 minutes at 37°C by the addition of 25 µL CaCl2 (25 mM) in HEPES-buffered saline (HBS; 20 mM HEPES, 147 mM NaCl, 3 mM KCl, pH 7.4).62 To determine the FII, FV, or FX activity levels, murine plasma (5 µL) was mixed with FII-, FV-, or FX-deficient plasma (20 µL) and 25 µL of HBS 0.5% BSA and incubated at 37°C for 1 minute. The clotting time was recorded following the addition of Innovin (DADE Behring, Marburg, Germany). To determine FVIII activity levels, murine plasma (5 µL) was mixed with FVIII-deficient plasma (20 µL) and APTT-XL reagent and incubated at 37°C for 3 minutes following the addition of 25 µL CaCl2 (25 mM) in HBS 0.5% BSA. The fibrinogen concentration was measured using the Clauss method.63 APC levels in mouse plasma were measured as described using recombinant mouse APC as standard.64 Thrombin-antithrombin (TAT) levels were determined using the TAT Enzygnost kit (Siemens Healthcare, Erlangen, Germany) per the manufacturer’s instructions.
Statistical analysis
All data are expressed as mean ± standard deviation (SD). Coagulation factors were analyzed using an unpaired t test or analysis of variance (ANOVA) with Dunnett’s multiple comparisons test. Bleeding volume and clotting times were compared using the nonparametric Mann-Whitney test or Kruskal-Wallis with Dunn’s multiple comparisons test. P < .05 was considered statistically significant.
Results
Validation of the trauma and shock model
In 2009, Chesebro et al28 reported that the combination of soft tissue trauma and shock resulted in elevated levels of circulating APC associated with the development of ATC. We emulated this method with minor adaptations because of equipment differences. Using this model, the induction of shock (Figure 1A) in combination with trauma caused by laparotomy resulted in a significant prolongation of the APTT clotting time (Figure 1B) and an approximately 10-fold increase in the circulating APC levels (Figure 1C) in agreement with previous reported findings.28 Neither shock nor trauma alone induced the same changes in APTT clotting times or APC levels.
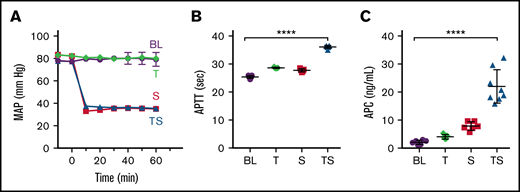
Induction of ATC by trauma and shock. Mice were subjected to a midline laparotomy denoted as trauma (T), acute blood withdrawal (approximately 500 µL) to induce shock (S), or both trauma and shock (TS). Control baseline (BL) mice were catheterized but were not subjected to trauma or shock (supplemental Figure 1). (A) MAP, monitored with a pressure probe inserted into the carotid artery (n = 3). (B) APTT at 60 minutes (n = 5-7). (C) APC plasma levels measured at 60 minutes (n = 5-8). Results are shown as mean ± SD. Statistical significance was determined by (B) Kruskal-Wallis 1-way ANOVA with Dunn’s multiple comparisons test and (C) 1-way ANOVA with Dunnett’s multiple comparisons test. ****P < .0001.
Induction of ATC by trauma and shock. Mice were subjected to a midline laparotomy denoted as trauma (T), acute blood withdrawal (approximately 500 µL) to induce shock (S), or both trauma and shock (TS). Control baseline (BL) mice were catheterized but were not subjected to trauma or shock (supplemental Figure 1). (A) MAP, monitored with a pressure probe inserted into the carotid artery (n = 3). (B) APTT at 60 minutes (n = 5-7). (C) APC plasma levels measured at 60 minutes (n = 5-8). Results are shown as mean ± SD. Statistical significance was determined by (B) Kruskal-Wallis 1-way ANOVA with Dunn’s multiple comparisons test and (C) 1-way ANOVA with Dunnett’s multiple comparisons test. ****P < .0001.
Development of ATC after trauma and shock
The combination of trauma and shock induced severe reductions of FV (Figure 2A) and FVIII (Figure 2B) activity levels in plasma at 60 minutes, as well as a partial reduction of fibrinogen (Figure 2C), whereas the prothrombin (Figure 2D) and factor X (Figure 2E) activity levels remained within the normal range. The selective reductions of FV, FVIII, and fibrinogen are consistent with the presumed major role for APC and hyperactive fibrinolysis in the development of ATC and distinguishes ATC from other more broadly consumptive coagulopathies, such as DIC.
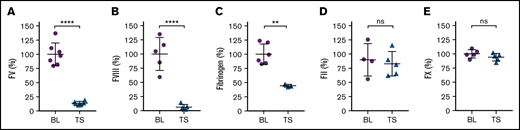
Trauma and shock are associated with changes in coagulation parameters. Mice were subjected to both trauma and shock (TS). Control baseline (BL) mice were catheterized but were not subjected to trauma or shock (supplemental Figure 1). Plasma samples were collected 60 minutes after trauma and shock. Shown are the plasma activity levels of (A) FV (n = 6-7), (B) FVIII (n = 5), (C) fibrinogen (n = 3-6), (D) FII (n = 4-5), and (E) FX (n = 5). Results are shown as mean ± SD. Statistical significance was determined by 1-way ANOVA with Dunnett’s multiple comparisons test. **P < .01; ****P < .0001; ns, not significant.
Trauma and shock are associated with changes in coagulation parameters. Mice were subjected to both trauma and shock (TS). Control baseline (BL) mice were catheterized but were not subjected to trauma or shock (supplemental Figure 1). Plasma samples were collected 60 minutes after trauma and shock. Shown are the plasma activity levels of (A) FV (n = 6-7), (B) FVIII (n = 5), (C) fibrinogen (n = 3-6), (D) FII (n = 4-5), and (E) FX (n = 5). Results are shown as mean ± SD. Statistical significance was determined by 1-way ANOVA with Dunnett’s multiple comparisons test. **P < .01; ****P < .0001; ns, not significant.
Normalization of clotting parameters and ATC by superFVa
To determine the effect of superFVa on the development of ATC, superFVa or saline control was infused continuously starting 30 minutes after induction of laparotomy and shock and continued for 20 minutes at a rate of 5 μL/min (supplemental Figure 1). Blood pressure profiles demonstrated that the infusion did not result in correction of shock because the MAP remained in the 35 mm Hg range (supplemental Figure 2). Despite the persistence of shock, superFVa corrected the APTT prolongation measured at 60 minutes at a dose of 0.8 mg/kg, whereas the APTT at 0.4 mg/kg superFVa remained unaffected (Figure 3A). superFVa increased the FV (Figure 3B) and FVIII (Figure 3C) activity levels in a dose-dependent manner. However, superFVa did not restore FV and FVIII protein levels (supplemental Figure 3), indicating that superFVa reconstituted FV activity and bypassed FVIII as was previously shown in FVIII-deficient plasma.53,65 In contrast, superFVa had little effect on fibrinogen levels that remained reduced by 50% (Figure 3D). The normalization of coagulation parameters by superFVa (0.8 mg/kg) coincided with a significant reduction of TAT complexes (Figure 3E) and of circulating APC levels (Figure 3F). These observations indicate that superFVa contributed substantially to the correction of APC-mediated induction of ATC, dampening excessive protein C activation during trauma and shock, and resulting in normalization of FV and FVIII activity levels, as well as thrombin formation.

Correction of coagulation parameters with superFVa after trauma and shock–induced ATC. Mice were subjected to both trauma and shock (TS). Control baseline (BL) mice were catheterized but were not subjected to trauma or shock (supplemental Figure 1). superFVa (SFVa) therapy (0.4 or 0.8 mg/kg) or saline control (0 mg/kg SFVa) was initiated 30 minutes after the induction of trauma and shock and administered as a continuous infusion at a rate of 5 µL/min for 20 minutes. Plasma samples were collected 60 minutes after trauma and shock. Shown are (A) APTT (n = 4-8), the plasma activity levels of (B) FV (n = 4-6), (C) FVIII (n = 5), and (D) fibrinogen (n = 3-6), (E) plasma TAT levels (n = 5-7), and (F) APC plasma levels (n = 5-10). Results are shown as mean ± SD. Statistical significance was determined by (A) Mann-Whitney test and (B-F) 1-way ANOVA with Dunnett’s multiple comparisons test. ***P < .001; ****P < .0001; ns, not significant.
Correction of coagulation parameters with superFVa after trauma and shock–induced ATC. Mice were subjected to both trauma and shock (TS). Control baseline (BL) mice were catheterized but were not subjected to trauma or shock (supplemental Figure 1). superFVa (SFVa) therapy (0.4 or 0.8 mg/kg) or saline control (0 mg/kg SFVa) was initiated 30 minutes after the induction of trauma and shock and administered as a continuous infusion at a rate of 5 µL/min for 20 minutes. Plasma samples were collected 60 minutes after trauma and shock. Shown are (A) APTT (n = 4-8), the plasma activity levels of (B) FV (n = 4-6), (C) FVIII (n = 5), and (D) fibrinogen (n = 3-6), (E) plasma TAT levels (n = 5-7), and (F) APC plasma levels (n = 5-10). Results are shown as mean ± SD. Statistical significance was determined by (A) Mann-Whitney test and (B-F) 1-way ANOVA with Dunnett’s multiple comparisons test. ***P < .001; ****P < .0001; ns, not significant.
Bleeding-induced ATC caused by liver laceration
Next, we used a model where ATC is induced by severe bleeding caused by traumatic organ injury (supplemental Figure 4). Severe bleeding was induced by laparotomy and liver laceration involving the removal of 75% of the left lobe of the liver (Figure 4A). Liver laceration caused blood loss of approximately 0.5 mL at 60 minutes (Figure 4B), which was associated with a noticeable drop of the MAP to about 35 mm Hg (Figure 4C). This drop in blood pressure was similar to the target value for the MAP used to induce shock in the trauma/shock model. The acute blood loss and shock resulted in a significant prolongation of the APTT at 60 minutes (Figure 4D), with changes in coagulation parameters very similar to those observed in the trauma/shock model. Changes included markedly elevated circulating APC levels (Figure 5A), nearly undetectable levels of FV (Figure 5B) and FVIII (Figure 5C) activity (>90% reduction), and a reduction of fibrinogen by 42% (Figure 5D). Also similar to the laparotomy/shock model, FII (supplemental Figure 5A) and FX (supplemental Figure 5B) activity levels were only minimally affected (reduced by 17% and 11%, respectively). Taken together, these observations are consistent with the development of ATC in association with severe bleeding and shock caused by liver laceration.
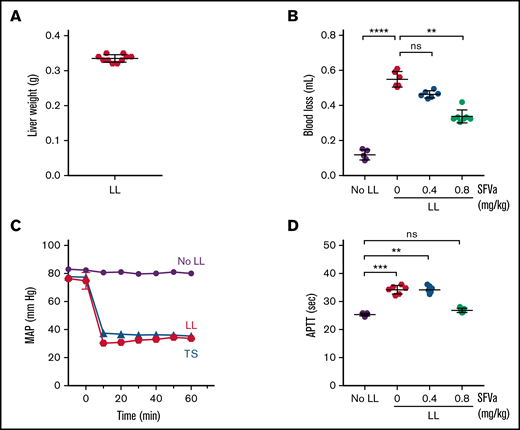
Development of ATC after severe bleeding and prevention by superFVa prophylaxis. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 4). Control mice (No LL) underwent the same procedures except liver laceration. In some experiments, a pressure probe was inserted into the carotid artery to gain information about the MAP. Mice received superFVa (SFVa) prophylaxis (0.4 or 0.8 mg/kg) or vehicle control (0 mg/kg SFVa) as a bolus IV administration (100 µL) shortly before liver laceration. Plasma samples were collected 60 minutes after liver laceration. (A) Weight of the excised liver (n = 10). (B) Blood loss after 60 minutes (n = 5-7). (C) MAP, monitored using a pressure probe inserted into the carotid artery (n = 3). MAP after trauma and shock (TS, supplemental Figure 1) is shown for comparison. (D) APTT at 60 minutes (n = 5-7). Results are shown as mean ± SD. Statistical significance was determined by Kruskal-Wallis 1-way ANOVA with Dunn’s multiple comparisons test. **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Development of ATC after severe bleeding and prevention by superFVa prophylaxis. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 4). Control mice (No LL) underwent the same procedures except liver laceration. In some experiments, a pressure probe was inserted into the carotid artery to gain information about the MAP. Mice received superFVa (SFVa) prophylaxis (0.4 or 0.8 mg/kg) or vehicle control (0 mg/kg SFVa) as a bolus IV administration (100 µL) shortly before liver laceration. Plasma samples were collected 60 minutes after liver laceration. (A) Weight of the excised liver (n = 10). (B) Blood loss after 60 minutes (n = 5-7). (C) MAP, monitored using a pressure probe inserted into the carotid artery (n = 3). MAP after trauma and shock (TS, supplemental Figure 1) is shown for comparison. (D) APTT at 60 minutes (n = 5-7). Results are shown as mean ± SD. Statistical significance was determined by Kruskal-Wallis 1-way ANOVA with Dunn’s multiple comparisons test. **P < .01; ***P < .001; ****P < .0001; ns, not significant.
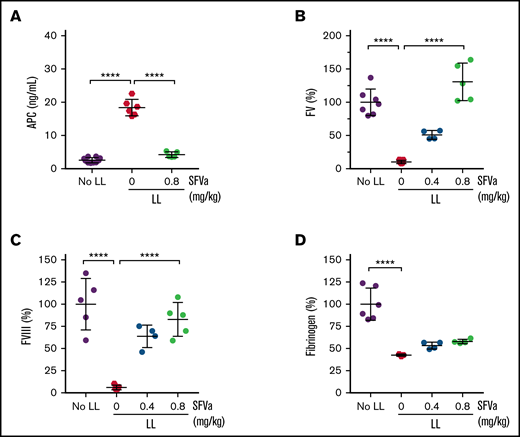
Changes in ATC-associated coagulation parameters by superFVa prophylaxis. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 4). Control mice (No LL) underwent the same procedures except liver laceration. Mice received superFVa (SFVa) prophylaxis (0.4 or 0.8 mg/kg) or vehicle control (0 mg/kg SFVa) as a bolus IV administration (100 µL) shortly before liver laceration. Plasma samples were collected 60 minutes after liver laceration. Shown are (A) APC plasma levels (n = 5-10) and plasma activity levels of (B) FV (n = 4-7), (C) FVIII (n = 4-5), and (D) fibrinogen (n = 3-6). Results are shown as mean ± SD. Statistical significance was determined by 1-way ANOVA with Dunnett’s multiple comparisons test. ****P < .0001.
Changes in ATC-associated coagulation parameters by superFVa prophylaxis. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 4). Control mice (No LL) underwent the same procedures except liver laceration. Mice received superFVa (SFVa) prophylaxis (0.4 or 0.8 mg/kg) or vehicle control (0 mg/kg SFVa) as a bolus IV administration (100 µL) shortly before liver laceration. Plasma samples were collected 60 minutes after liver laceration. Shown are (A) APC plasma levels (n = 5-10) and plasma activity levels of (B) FV (n = 4-7), (C) FVIII (n = 4-5), and (D) fibrinogen (n = 3-6). Results are shown as mean ± SD. Statistical significance was determined by 1-way ANOVA with Dunnett’s multiple comparisons test. ****P < .0001.
Prevention of blood loss and development of ATC by superFVa after liver laceration
To determine whether superFVa can prevent the development of ATC, superFVa was administered as an IV bolus shortly before the procedure at 2 different doses (0.4 or 0.8 mg/kg; supplemental Figure 4). The prophylactic bolus injection of superFVa reduced blood loss in a dose-dependent fashion, as assessed 60 minutes after liver laceration (Figure 4B). The higher dose of superFVa resulted in 50% bleed reduction and was associated with near correction of the APTT (Figure 4D). superFVa also prevented the severe depletion of FV (Figure 5B) and FVIII activity levels (Figure 5C) in a dose-dependent fashion, whereas it had little effect on FV or FVIII protein levels (supplemental Figure 3) or fibrinogen levels (Figure 5D). In addition, with the reduction of bleeding and normalization of coagulation parameters, superFVa also prevented the abnormal increase in circulating APC levels (Figure 5A), indicating that prophylactic administration of superFVa can prevent the development of bleeding-induced ATC.
Reversal of ongoing ATC by superFVa after liver laceration
Next, we asked the question whether superFVa can reverse the effects of ongoing ATC. To determine the timing of onset of ATC after liver laceration, a time course of bleeding with corresponding APTT clotting times after liver laceration was performed. Most of the bleeding was immediate and occurred within the first 15 minutes (approximately 0.4 mL; Figure 6A), with additional gradual blood loss of an additional approximate 0.2 mL during the following 45 minutes. Blood loss was associated with a corresponding prolongation of the APTT (Figure 6B) until termination of the experiment at 60 minutes, indicating continued and gradual development of ATC. The 30-minute time point (ATC in progress) was chosen to initiate treatment with superFVa by continuous infusion (0.8 mg/kg for 20 minutes at 5 µL/min; supplemental Figure 6). superFVa stabilized bleeding, with no additional blood loss after 30 minutes compared with saline-treated mice (Figure 6A). A corresponding pattern of APTT correction was noted, where superFVa reversed APTT prolongation to normal, but treatment with saline did not (Figure 6B). FV (Figure 7A) and FVIII (Figure 7B) activities dropped continuously over the 60-minute period in saline-treated mice but were restored to their normal range by reconstitution of FV and bypassing of FVIII by superFVa (supplemental Figure 3). Furthermore, superFVa reduced elevated TAT levels (Figure 7C), indicating that late administration of superFVa was able to reverse the perpetuation of ATC and improve clinical hemostasis.
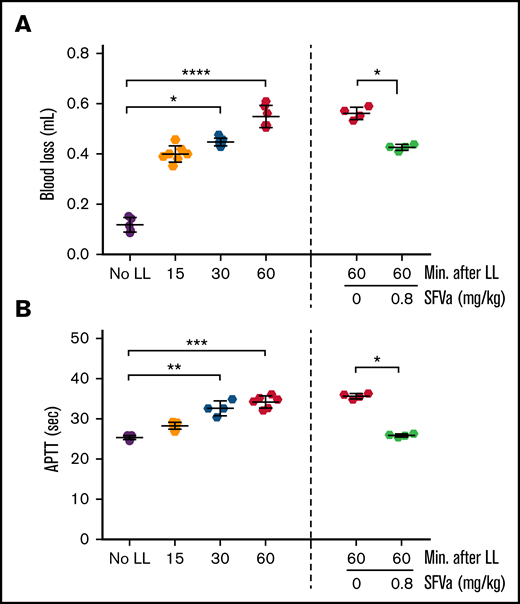
Time course of bleeding and reversal of ongoing ATC by superFVa after liver laceration. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 6). Control mice (No LL) underwent the same procedures except liver laceration. superFVa (SFVa) therapy (0.8 mg/kg) or saline control (0 mg/kg SFVa) was initiated 30 minutes after the induction of liver laceration and administered as a continuous infusion at a rate of 5 µL/min for 20 minutes. Plasma samples were collected 15 to 60 minutes after liver laceration. (A) Blood loss at 15 to 60 minutes after liver laceration (left, n = 5-7) and at 60 minutes after liver laceration with superFVa or vehicle administration (right, n = 4). (B) APTT at 15 to 60 minutes after liver laceration (left, n = 4-7) and at 60 minutes after liver laceration with superFVa or vehicle administration (right, n = 4). Results are shown as mean ± SD. Statistical significance was determined by Kruskal-Wallis 1-way ANOVA with Dunn’s multiple comparisons test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Time course of bleeding and reversal of ongoing ATC by superFVa after liver laceration. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 6). Control mice (No LL) underwent the same procedures except liver laceration. superFVa (SFVa) therapy (0.8 mg/kg) or saline control (0 mg/kg SFVa) was initiated 30 minutes after the induction of liver laceration and administered as a continuous infusion at a rate of 5 µL/min for 20 minutes. Plasma samples were collected 15 to 60 minutes after liver laceration. (A) Blood loss at 15 to 60 minutes after liver laceration (left, n = 5-7) and at 60 minutes after liver laceration with superFVa or vehicle administration (right, n = 4). (B) APTT at 15 to 60 minutes after liver laceration (left, n = 4-7) and at 60 minutes after liver laceration with superFVa or vehicle administration (right, n = 4). Results are shown as mean ± SD. Statistical significance was determined by Kruskal-Wallis 1-way ANOVA with Dunn’s multiple comparisons test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
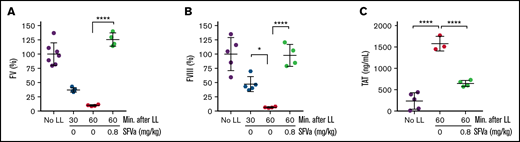
Correction of ATC-associated changes in coagulation parameters by superFVa treatment. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 6). Control mice (No LL) underwent the same procedures except liver laceration. superFVa (SFVa) therapy (0.8 mg/kg) or saline control (0 mg/kg SFVa) was initiated 30 minutes after the induction of liver laceration and administered as a continuous infusion at a rate of 5 µL/min for 20 minutes. Plasma samples were collected 30 and 60 minutes after liver laceration (representing the before and after superFVa therapy time points). Shown are the plasma activity levels of (A) FV (n = 4-7), (B) FVIII (n = 4-5), and (C) plasma TAT levels (n = 3-5). Results are shown as mean ± SD. Statistical significance was determined by 1-way ANOVA with Dunnett’s multiple comparisons test. ****P < .0001.
Correction of ATC-associated changes in coagulation parameters by superFVa treatment. Severe bleeding was induced by a midline laparotomy followed by liver laceration (LL) involving the removal of 75% of the left lobe of the liver (supplemental Figure 6). Control mice (No LL) underwent the same procedures except liver laceration. superFVa (SFVa) therapy (0.8 mg/kg) or saline control (0 mg/kg SFVa) was initiated 30 minutes after the induction of liver laceration and administered as a continuous infusion at a rate of 5 µL/min for 20 minutes. Plasma samples were collected 30 and 60 minutes after liver laceration (representing the before and after superFVa therapy time points). Shown are the plasma activity levels of (A) FV (n = 4-7), (B) FVIII (n = 4-5), and (C) plasma TAT levels (n = 3-5). Results are shown as mean ± SD. Statistical significance was determined by 1-way ANOVA with Dunnett’s multiple comparisons test. ****P < .0001.
Discussion
For decades, transfusion management has been the mainstay for the treatment of traumatic hemorrhage, which continues to carry an unacceptably high mortality rate, exposing an unmet clinical need for more effective, safe, and targeted interventions. Mortality is particularly high in trauma victims who develop TIC, including ATC. ATC usually develops within the first few hours after injury, resulting in acceleration of uncontrollable bleeding.5 Therefore, early intervention of ATC appears critical to limit its appearance, limit hemorrhage, and hopefully, decrease mortality.2 The coagulopathy of ATC involves a selective depletion of FV, FVIII, and fibrinogen levels because of the exaggerated activation of the protein C and fibrinolytic pathways after vascular disruption, perpetuating hemorrhage and shock.27,30 Previous reports indicated that, in mouse models of ATC, targeting the exaggerated APC generation effectively prevented ATC, either by genetic manipulation of thrombomodulin to render it defective in supporting protein C activation (TMPro/Pro mouse) or with activity-selective anti-APC antibodies.27,28,54 However, mice homozygous for FVLeiden that conveys partial APC resistance were only minimally protected against ATC.27 Toward this end, the characteristics of APC-resistant superFVa provided a unique opportunity to further elucidate the mechanism of ATC and to study the potential of superFVa as a targeted therapy to prevent and correct ATC and its attendant hemorrhage.
Using 2 mouse models in which trauma was combined with either artificially induced shock or severe hemorrhage, ATC was characterized by a pronounced increase in APC as previously observed by others.27-29,31 This was accompanied by selective depletion of FV, FVIII, and fibrinogen, whereas other clotting factors (FII and FX) remained within the normal range. A time course of bleeding after liver laceration demonstrated that ATC, assessed by APTT as a surrogate parameter for FV- and/or FVIII-dependent coagulopathy, developed gradually within the first 30 minutes and matured over the next 30 minutes. These observations support the concept that excessive APC generation resulted in targeted depletion of FV and FVIII as previously suggested by others.29,30,66 We therefore stipulate that low fibrinogen may be explained more by hyperfibrinolysis than consumption (as would be the case in DIC) given that FII and FX activity remained largely unaffected. APC-mediated hyperfibrinolysis and fibinogenolysis have been shown previously to occur with trauma and/or ATC,27,67 whereby patients with elevated APC levels, hyperfibrinolysis, and early depletion of fibrinogen lacked a systemic consumptive process (ie, DIC).27 These observations suggest that the coagulopathy initially encountered in ATC differs from DIC, although temporal and iatrogenic effects may change the coagulopathy qualitatively as traumatic bleeding progresses.
superFVa corrected ATC, restored hemostasis, and reduced hemorrhage in 2 mouse models of ATC. These effects were observed when superFVa was administered prophylactically by bolus a few minutes before injury, as well as therapeutically as a continuous infusion started after the onset of coagulopathy. The optimal total dose, regardless of being administered as bolus or by continuous infusion, was 0.8 mg/kg, corresponding to doses found to be effective in other bleeding models.43,53,55,56 Interestingly, rather than increasing TAT complex formation as might be expected from a prohemostatic agent, superFVa decreased TAT complex formation, consistent with normalization of thrombin formation, while still achieving clinically effective hemostasis and the half-life of TAT complexes in the circulation (2-10 minutes68,69 ). In turn, the normalization of thrombin generation appeared to deescalate the excessive activation of PC, with recovery of FV and FVIII plasma activity levels by superFVa, although endogenous FV and FVIII protein levels remained low. Of note, superFVa did not correct fibrinogen levels, which differs from the results obtained in TMPro/Pro mice that showed a significant attenuation of fibrinolysis after trauma and hemorrhage.27 Whether this suggests that APC activities other than those involving FVa contribute to hyperfibrinolysis and fibrinogenolysis or that there may be some overlap between ATC and DIC and/or a dynamic transition phase as previously suggested remains to be determined.38,70 Importantly, the targeted improvement of APC-induced derangement of hemostatic mediators by superFVa in our study translated into hemorrhage control despite the absence of fibrinogen elevation. Of note, superFVa not only prevented excessive bleeding from severe trauma when given prophylactically but also prevented bleed progression and stabilized hemorrhaging when administered 30 minutes after trauma, when ATC was florid. These observations support that ATC is at least in part facilitated by an excessive increase of APC in the setting of shock and traumatic injury, fueling hemorrhage, which can be interrupted and reversed by targeted intervention with superFVa.
These findings may translate into a number of practical clinical applications. It is conceivable that superFVa could be used not only therapeutically but also prophylactically to prevent fatal traumatic hemorrhage (eg, military combat). In this context, it is important to mention that superFVa has high stability in solution and presumably a short circulating half-life in humans based on murine half-life studies (approximately 20-30 minutes).65 A short half-life is desirable in trauma, where rapid “on-off” effects are advantageous to minimize the risk of undesirable thrombogenic complications while controlling hemorrhage. This feature distinguishes superFVa from approaches using anti-APC antibodies with long half-lives to curb bleeding.42 In contrast to enzymatic prohemostatics such as rhFVIIa (Novo7, NovoNordisk),71,72 superFVa is a nonenzymatic cofactor with restricted function at sites of active injury and dependent on the endogenous generation of FXa, thereby exhibiting a presumably low thrombogenic risk. Our current observations would support this concept because superFVa normalized, rather than enhanced, coagulation. The thrombogenic potential of therapeutic prohemostatic agents in trauma is of concern because trauma victims are at higher risk of thromboembolic complications despite excessive bleeding.73 Preliminary studies demonstrated that superFVa was nonthrombogenic in a murine lung fibrin deposition model,65 but more studies in larger animals are warranted to better characterize thrombogenicity. Another potential risk is the development of inhibitory antibodies that may cross-react with endogenous FV, but, given the anticipated one-time short-term administration and the conservative mutations used in the engineering of superFVa, this risk is considered low.65
Although thrombogenicity and inhibitor formation are important considerations for the development of recombinant proteins such as superFVa, the clinical possibility of rescuing rapidly fatal bleeding, which has no good alternative therapeutic treatments, should bring perspective to any risk/benefit analysis. It appears that superFVa possesses unique characteristics that differentiate superFVa from other prohemostatic agents.
In conclusion, intervention with superFVa mitigated ATC and traumatic bleeding by targeted interception of APC-induced derangement of coagulation. These observations pave the way for further development of superFVa for traumatic bleeding.
Acknowledgments
The authors thank Tine Wyseure, Hiroshi Deguchi, and Srila Karthik-Gopal for technical advice.
This work was supported by National Institutes of Health (NHLBI) grants HL104165, HL142975, and HL148096 (L.O.M.) and University of California San Diego discretionary funds (A.v.D.).
Authorship
Contribution: B.C.J. performed and designed experiments, interpreted results, and wrote the manuscript; B.Y.M. and M.J.C. provided critical expertise and generated preliminary data demonstrating the effect of superFVa in ATC; C.T.E. provided critical reagents; A.v.D. and L.O.M. designed the study, provided study oversight, interpreted results, and wrote the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: University of California San Diego and The Scripps Research Institute hold intellectual property rights related to superFVa on which A.v.D. and L.O.M. are listed as inventors. A.v.D. and L.O.M. are founders of Hematherix LLC, a biotech company that is developing superFVa therapy for bleeding complications, and are members of the board of directors of Hematherix LLC. A.v.D. has received honoraria for participating in scientific advisory board panels, consulting, and speaking engagements for Sanofi, Biomarin, Takeda, and Uniqure and has received research funding from Sanofi and Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Laurent O. Mosnier, Department of Molecular Medicine (IMM-315), The Scripps Research Institute, 10550 North Torrey Pines Rd, La Jolla, CA 92037; e-mail: lmosnier@scripps.edu; and Annette von Drygalski, Division of Hematology/Oncology, Department of Medicine, Hemophilia and Thrombosis Treatment Center, University of California San Diego, 9333 Genesee Ave, Suite 310, La Jolla, CA 92121; e-mail: avondrygalski@ucsd.edu.

