

Key Points
Disruption of cell-cycle control and DNA damage response pathways are synthetic lethal with Srsf2P95H/+.
Palbociclib preferentially targets Srsf2P95H/+ cells.

Abstract
Current strategies to target RNA splicing mutant myeloid cancers proposes targeting the remaining splicing apparatus. This approach has only been modestly sensitizing and is also toxic to non-mutant-bearing wild-type cells. To explore potentially exploitable genetic interactions with spliceosome mutations, we combined data mining and functional screening for synthetic lethal interactions with an Srsf2P95H/+ mutation. Analysis of missplicing events in a series of both human and murine SRSF2P95H mutant samples across multiple myeloid diseases (acute myeloid leukemia, myelodysplastic syndromes, chronic myelomonocytic leukemia) was performed to identify conserved missplicing events. From this analysis, we identified that the cell-cycle and DNA repair pathways were overrepresented within the conserved misspliced transcript sets. In parallel, to functionally define pathways essential for survival and proliferation of Srsf2P95H/+ cells, we performed a genome-wide Clustered regularly interspaced short palindromic repeat loss-of-function screen using Hoxb8 immortalized R26-CreERki/+Srsf2P95H/+ and R26-CreERki/+Srsf2+/+ cell lines. We assessed loss of single guide RNA representation at 3 timepoints: immediately after Srsf2P95H/+ activation, and at 1 week and 2 weeks after Srsf2P95H/+ mutation. Pathway analysis demonstrated that the cell-cycle and DNA damage response pathways were among the top synthetic lethal pathways with Srsf2P95H/+ mutation. Based on the loss of guide RNAs targeting Cdk6, we identified that palbociclib, a CDK6 inhibitor, showed preferential sensitivity in Srsf2P95H/+ cell lines and in primary nonimmortalized lin−cKIT+Sca-1+ cells compared with wild-type controls. Our data strongly suggest that the cell-cycle and DNA damage response pathways are required for Srsf2P95H/+ cell survival, and that palbociclib could be an alternative therapeutic option for targeting SRSF2 mutant cancers.
Introduction
Mutations in the core RNA splicing machinery (termed the spliceosome) have been identified as 1 of the most frequently mutated pathways in myeloid neoplasms.1 There is increasing evidence linking specific mutations in the spliceosome with distinct phenotypic and clinical outcomes. The RNA splicing factor SRSF2 is mutated in 11% to 15% of myelodysplastic syndromes (MDS) and 28% to 47% of chronic myelomonocytic leukemia.2 Mutations in SRSF2 cluster at or around proline 95, with proline to histidine (P95H) being the most common mutation. SRSF2P95H has been shown to lead to altered recognition of binding motifs on the exonic splicing enhancer, resulting in aberrantly spliced transcripts.3,4 So far, various groups have attempted to decipher the link between SRSF2P95H-specific missplicing events and MDS pathogenesis. Despite identification of recurrently misspliced transcripts, such as HNRNPA2B1 and FYN, it remains unclear how the spliceosome mutations and the resultant misspliced transcripts contribute to the initiation and development of MDS and related myeloid neoplasms.3-5
In parallel to understanding the effect of splicing factor mutations on hematopoiesis, therapies targeting splicing mutations have been proposed. Spliceosome mutations are nearly always heterozygous and mutually exclusive, which suggests that the remaining wild-type (WT) allele in the mutant cells is crucial for maintaining a level of splicing necessary for cell survival.6-8 Small molecule spliceosome inhibitors, such as Pladienolide B, E7107, Spliceostatin A, and H3B-8800, interfere with the binding of SF3B1 and cause cell-cycle arrest in splicing mutant cells.9-11 However, 2 phase 1 clinical trials (NCT00459823, NCT00499499) observed unexpected ophthalmic adverse effects with spliceosome inhibitors E7107.12 A preliminary report of a recent phase 1 clinical trial of H3B-8800 (NCT02841540) indicates that the compound failed to induce complete or partial clinical response in patients with acute myeloid leukemia, MDS, and chronic myelomonocytic leukemia.13 The lack of effective treatments for MDS and related myeloid cancers and the high prevalence of spliceosomal mutations highlights the need to explore the therapeutic targeting of splicing mutant cells without disruption of splicing catalysis.
An attractive therapeutic strategy for cancer is the identification of synthetic lethal interactions, exemplified by the use of PARP inhibitors in BRCA-mutated cancers.14 A synthetic lethal interaction involves 2 genes in which mutation of either one is viable, but perturbation of both genes simultaneously is lethal to the cell.15 Thus, it provides opportunities to advance specific anticancer therapies, especially when the driver mutation is not readily druggable.16 From RNA interference screens in yeast to Clustered regularly interspaced short palindromic repeats (CRISPR) screens in mammalian in vivo systems, larger scale and more clinically relevant models have been developed and have identified new targets in their respective cancers settings.17 These approaches are now widely applicable with the availability of genetic perturbation tools and the development of inducible, physiologically expressed mutations found in human cancers in both human cell lines and in murine models.
Here, we report a pooled in vitro CRISPR loss of function screen to decipher the genetic vulnerabilities of Srsf2P95H/+ cells. We have made use of cell lines derived from our genetically engineered conditional knock-in model of Srsf2P95H/+ mutation.18,19 This model recapitulates the core characteristics of human disease associated with SRSF2 mutation and develops an MDS/myeloproliferative neoplasm phenotype in vivo. Using this unbiased whole genome approach, we identified that cell-cycle control and DNA damage response pathways play an important role in the survival of Srsf2P95H/+ cells. By overlaying the genetic interactors with the drug–gene interaction database, we identified that the CDK6 inhibitor palbociclib could preferentially target the Srsf2P95H/+ mutant cells. Collectively, these results demonstrate that there are potentially exploitable genetic vulnerabilities in Srsf2P95H/+ mutant cells outside of the RNA splicing machinery.
Methods
Mice
All mouse experiments were approved by the Animal Ethics Committee, St. Vincent’s Hospital, Melbourne, Australia (AEC#001/16 and 007/19). Mice used in the experiments were housed at the BioResource Centre located at St. Vincent’s Hospital. Rosa26-CreERT2Srsf2P95H/+ mice have been previously described.18
DNA isolation and genotyping
Genomic DNA was extracted from cell pellets using a Bioline Genomic DNA kit as per manufacturer’s instructions. Genotyping was performed by polymerase chain reaction (PCR) in the Mastercycler Pro PCR machine (Eppendorf). The oligonucleotides (IDT, Singapore) used for Srsf2 P95H genotyping PCR are:
SRSF2 5'loxP forward primer: 5'-GTTATGATCCACACCTCTCACC-3';
SRSF2 5'loxP reverse primer: 5'-ATAAACGTTTATGTCCGCTACC-3'.
Product size: WT = 327 bp; recombined = 400 bp; 5'loxP (floxed) intact = 446 bp.
Generation of Hoxb8 immortalized and Cas9 expressing cell lines
Hoxb8 immortalized cell lines. Briefly, Ficoll separated bone marrow cells from 3 R26-CreERT2Srsf2P95H/+ and 3 R26-CreERT2Srsf2+/+ mice, were stimulated for 48 hours in complete Iscove modified Dulbecco medium (IMDM [Sigma Aldrich] containing 20% fetal bovine serum [FBS, Assay Matrix], 1% penicillin/streptomycin [Gibco], 1% glutamine [Gibco]) supplemented with recombinant mouse stem cell factor (50 ng/mL, PeproTech), recombinant mouse interleukin-3 (10 ng/mL, PeproTech), and recombinant human interleukin-6 (10 ng/mL, Amgen). After stimulation, 1 × 106 cells were spin-infected with Hoxb8 retrovirus20 (Hoxb8 plasmids were generously provided by Mark Kamps, University of California San Diego) and polybrene at 1100g for 90 minutes. After 48 hours, cells were then passaged into complete IMDM containing 1% granulocyte-macrophage colony-stimulating factor (GM-CSF) conditioned medium (from BHK-HM5 cell conditioned medium).
Cas9 expressing cell lines. Hoxb8 immortalized cell lines were infected with Cas9-blasticidin lentivirus (lentiCas9-Blast was a gift from Feng Zhang; Addgene plasmid #52962)21 by spin infection at 1100g for 90 minutes. The infected cells were then cultured in IMDM-Cas9 medium (IMDM, 10% FBS, 1% GM-CSF, and 3 µg/mL blasticidin) for 2 weeks to select for a Cas9-expressing population.
Srsf2P95H/+ CRISPR knockout pooled library screen
See supplemental Methods for full method description.
CRISPR screen sample preparation and sgRNA library sequencing
Genomic DNA was extracted from the cell pellets using the Gentra Puregene kit (Qiagen). The extracted DNA was quantified on a Nanodrop spectrophotometer. DNA libraries were generated by PCR amplification of the integrated single guide RNA (sgRNA) constructs (as described at https://portals.broadinstitute.org/gpp/public/resources/protocols). In total, 24 libraries were generated and sequenced on the Illumina platform using 150-bp paired end reads by Novogene (China). CRISPR screen analysis was performed using MaGeCK, MaGeCK-VISPR, and the CRISPRBetaBinomial package (v1.3.0).22-24 The heatmap for negatively selected pathway analysis was generated with Metascape.25
RNA-sequencing
RNA was isolated from Cas9 R26-CreERT2Srsf2P95H/+ and R26-CreERT2Srsf2+/+ cells (n = 3 per genotype) at 7 days after tamoxifen treatment. RNA was ribosome depleted, subjected to library preparation (Kapa Stranded RNA-Seq Library Kit; Kapa Biosystems),18,26,27 and sequenced at 150-bp paired-end reads on the Illumina platform by Novogene (China). Differential expression was assessed using DESeq2.28 Splicing analysis was performed using rMATs as described previously.18 Alternative splicing analysis was performed using PSI-Sigma (v1.9) using nreads = 5.29 The Sashimi plots were generated using ggSashimi.30
Palbociclib sensitivity assay
Hoxb8 R26-CreERT2Srsf2P95H/+ and R26-CreERT2Srsf2+/+ cell lines were treated with 400 nM 4-hydroxy tamoxifen for 4 days. At 48 hours after tamoxifen cessation, 1000 cells were plated in 50 µL of IMDM with 10% FBS and 1% GM-CSF in a 96-well tissue culture plate and 50 µL of a 2× concentration of drugs was added in triplicate per dose. For the lin-c-Kit+Sca-1+ (LKS+) in vitro drug assay, sorted LKS+ cells were cultured for 24 hours in cytokine-supplemented complete IMDM (20% FBS, 50 ng/mL recombinant mouse stem cell factor, 10 ng/mL recombinant mouse interleukin-3, 10 ng/mL recombinant human interleukin-6), and then treated with 400 nM 4-hydroxy tamoxifen for 4 days. The treated LKS+ cells were then plated for drug assays as described previously. A dimethyl sulfoxide or water control was included in all assays. Cell viability was assessed after 4 days of drug culture using the ATP-Lite Luminescence Assay System (Perkin Elmer) and an Enspire plate reader (Perkin Elmer). The 50% inhibitory concentration (IC50) was calculated in Prism 8 (GraphPad) using log(inhibitor) vs normalized response-variable slope. Statistical analysis was performed with extra sum-of-squares F test. P < .05 is considered as significant.
Results
Cas9 Srsf2P95H/+ cell lines have similar splicing changes as primary cells from the in vivo models
To obtain a cell line model suitable for genome-wide CRISPR knock-out screening, we generated Hoxb8 immortalized myeloid cell lines from the bone marrow of 3 R26-CreERT2Srsf2P95H/+ and 3 R26-CreERT2Srsf2+/+ mice18 (supplemental Figure 1). These were then engineered to express Cas9. To determine if the Cas9 expressing Hoxb8 Rosa26-CreERT2ki/+Srsf2P95H/+ cells (referred to as Cas9 Srsf2P95H/+ cells) had splicing changes similar to primary Srsf2P95H/+ cells,18 we tamoxifen-treated the Cas9 Srsf2P95H/+ and Cas9 Srsf2+/+ cells (n = 3 per genotype) to activate the mutation and performed RNA-sequencing (RNA-seq) on cells at 7 days after tamoxifen treatment (Figure 1A). At this time point, there were a limited number of differentially expressed transcripts detected between the Cas9 Srsf2P95H/+ and Srsf2+/+ cells (Figure 1B; supplemental Figure 2). Using rMATS31 to identify differential alternative splicing events, we found that in the Srsf2P95H/+ cells skipped exons (n = 910) were the most frequent type of altered splicing, followed by mutually exclusive exons (n = 360), alternative 5' splice sites (n = 73), alternative 3' splice sites (n = 85), and retained introns (n = 83) (Figure 1C). We used an inclusion cutoff of 0.05 to identify changes in splicing, meaning that to be included in the analysis, exons were required to be at least 5% differentially spliced. At a false discovery rate (FDR) of 5%, less than 2% of the total events were significantly different for skipped exons and 3% of total events were significantly different for mutually exclusive exons (Figure 1D). The magnitude of splicing changes observed in Cas9 Srsf2P95H/+ cells was therefore similar to other SRSF2P95H mutant cell lines, as well as in vivo models.3,5,18 We then assessed specific genes that were reported previously, including from our and other studies. Indeed, previously identified misspliced targets of Srsf2P95H/+ such as Dap3 and Hnrnpa2b1, were aberrantly spliced in the Cas9 Srsf2P95H/+ cells (Figure 1E).4,5,18 These data demonstrate that the Cas9 Srsf2P95H/+ cells are recapitulating changes in splicing observed in primary cells and represent a tractable system to further study the effect of Srsf2 mutation in vitro, without the interference of other cooccurring mutations in myeloid leukemia.
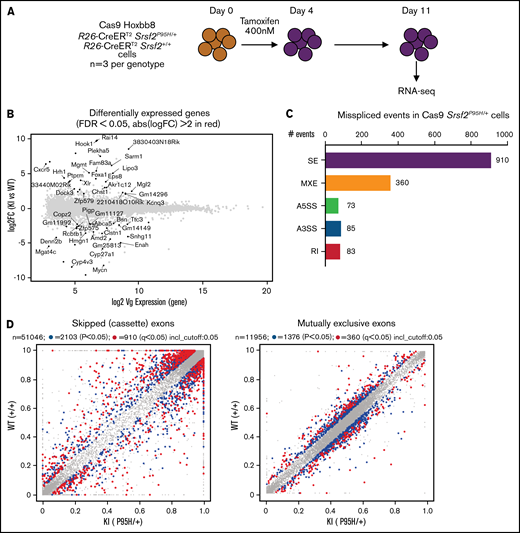
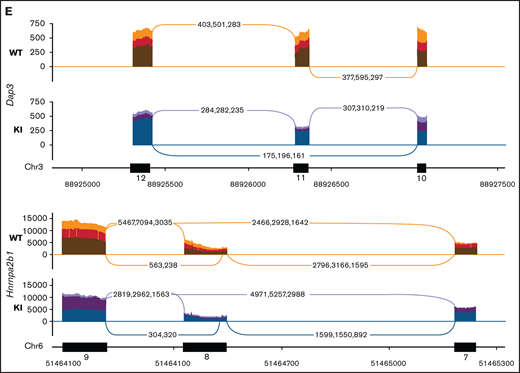
Cas9 Srsf2P95H/+ cell lines show similar missplicing changes as primary cells. (A) A schematic diagram depicting the experimental design for RNA sequencing. (B) An MA plot showing differentially expressed genes in the Srsf2P95H/+ cells compared with Srsf2+/+; red dots = FDR < .05, absolute fold change >2, genes without symbols are not labeled. (C) The number of misspliced events in GM-CSF Srsf2P95H/+ cells. A3SS alternative 3' splicing site; A5SS, alternative 5' splicing site; MXE, mutually exclusive exon; RI, retained intron; SE, skipped exon. (D) Scatter plots of the GM-CSF Srsf2P95H/+ cells showing the skipped (cassette) exons and mutually exclusive exon events. Gray dots, <5% difference between genotypes; blue dots, P < .05 and >5% difference between genotypes; red dots, q < .05 and >5% difference between genotypes. (E) Sashimi plot of selected missplicing events in the myeloid cell lines.
Cas9 Srsf2P95H/+ cell lines show similar missplicing changes as primary cells. (A) A schematic diagram depicting the experimental design for RNA sequencing. (B) An MA plot showing differentially expressed genes in the Srsf2P95H/+ cells compared with Srsf2+/+; red dots = FDR < .05, absolute fold change >2, genes without symbols are not labeled. (C) The number of misspliced events in GM-CSF Srsf2P95H/+ cells. A3SS alternative 3' splicing site; A5SS, alternative 5' splicing site; MXE, mutually exclusive exon; RI, retained intron; SE, skipped exon. (D) Scatter plots of the GM-CSF Srsf2P95H/+ cells showing the skipped (cassette) exons and mutually exclusive exon events. Gray dots, <5% difference between genotypes; blue dots, P < .05 and >5% difference between genotypes; red dots, q < .05 and >5% difference between genotypes. (E) Sashimi plot of selected missplicing events in the myeloid cell lines.
Cell-cycle regulation and DNA repair pathways are overrepresented within the misspliced transcript set of SRSF2 mutant cells
To understand the landscape of missplicing in SRSF2 mutated cells, we literature-curated misspliced genes from 16 different human and murine datasets of SRSF2 mutation, including the Cas9 Srsf2 cells described previously (supplemental Table 1).3-5,18,32-36 Metascape pathway enrichment analysis was performed on genes that were identified in more than 4 of the 16 datasets, regardless of the type of misspliced event.25 This analysis revealed that in SRSF2 mutant cells, messenger RNA (mRNA) processing was the most frequently overrepresented pathway of the top 20 pathways that were most significantly enriched for misspliced transcripts (Figure 2A). In the network map of enriched pathways, pathways associated with regulation of the cell cycle, chromatin and cellular organization, mitochondrion, complex assembly, DNA repair, and protein modification were also identified (Figure 2B). Individual genes identified as having altered splicing in 10 of 16 datasets are listed in Table 1. This includes known targets of SRSF2P95H, such as HNRNPA2B1 and FYN, as well as genes such as ATF2, which has been shown to be involved in cell-cycle and DNA repair regulation.37,38
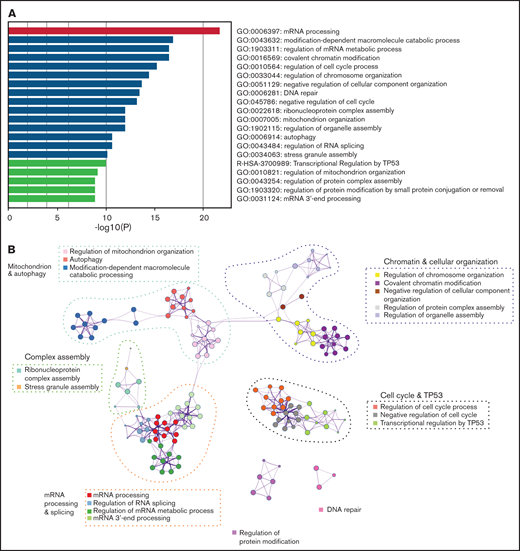
Common misspliced pathways in 16 Srsf2/SRSF2 mutant cells. (A) The pathways most frequently represented within the misspliced transcript sets in the Srsf2/SRSF2 mutant cells. (B) Protein–protein interaction networks between the common transcripts misspliced in Srsf2/SRSF2 mutant cells.
Common misspliced pathways in 16 Srsf2/SRSF2 mutant cells. (A) The pathways most frequently represented within the misspliced transcript sets in the Srsf2/SRSF2 mutant cells. (B) Protein–protein interaction networks between the common transcripts misspliced in Srsf2/SRSF2 mutant cells.
Genes identified as misspliced in Srsf2/SRSF2 mutant cells in at least 10 of 16 datasets
| Gene | Description | No. of datasets (human/murine) |
|---|---|---|
| ATF2 | Activating transcription factor 2 | 13 (7H/6M) |
| MTMR14 | Myotubularin related protein 14 | 13 (9H/4M) |
| HNRNPA2B1 | Heterogenous nuclear ribonucleoprotein A2/B1 | 11 (5H/6M) |
| SEC24B | SEC24 family member B | 11 (6H/5M) |
| AGTPBP1 | ATP/GTP binding protein 1 | 10 (5H/5M) |
| PRPSAP2 | Phosphoribosyl pyrophosphate synthetase-associated protein 2 | 10 (6H/4M) |
| EHMT2 | Euchromatic histone-lysine N-methyltransferase 2 | 10 (5H/5M) |
| STRADA | STE20-related kinase adaptor α | 10 (5H/5M) |
| TIA1 | TIA1 cytotoxic granule-associated RNA binding protein | 10 (5H/5M) |
| MTF2 | Metal response element binding transcription factor 2 | 10 (5H/5M) |
| RSRC2 | Arginine/serine-rich coiled-coil 2 | 10 (7H/3M) |
| CEP57 | Centrosomal protein 57kDa | 10 (8H/2M) |
| FIP1L1 | Factor interacting with PAPOLA and CPSF1 | 10 (7H/3M) |
| FYN | FYN proto-oncogene, Src family tyrosine kinase | 10 (6H/4M) |
| ABI1 | Abl-interactor 1 | 10 (6H/4M) |
| HUWE1 | HECT, UBA, and WWE domain containing 1, E3 ubiquitin protein ligase | 10 (5H/5M) |
| TPD52L2 | Tumor protein D52-like 2 | 10 (8H/2M) |
| ZMYND8 | Zinc finger, MYND-type containing 8 | 10 (7H/3M) |
| NSD1 | Nuclear receptor binding SET domain protein 1 | 10 (5H/5M) |
| Gene | Description | No. of datasets (human/murine) |
|---|---|---|
| ATF2 | Activating transcription factor 2 | 13 (7H/6M) |
| MTMR14 | Myotubularin related protein 14 | 13 (9H/4M) |
| HNRNPA2B1 | Heterogenous nuclear ribonucleoprotein A2/B1 | 11 (5H/6M) |
| SEC24B | SEC24 family member B | 11 (6H/5M) |
| AGTPBP1 | ATP/GTP binding protein 1 | 10 (5H/5M) |
| PRPSAP2 | Phosphoribosyl pyrophosphate synthetase-associated protein 2 | 10 (6H/4M) |
| EHMT2 | Euchromatic histone-lysine N-methyltransferase 2 | 10 (5H/5M) |
| STRADA | STE20-related kinase adaptor α | 10 (5H/5M) |
| TIA1 | TIA1 cytotoxic granule-associated RNA binding protein | 10 (5H/5M) |
| MTF2 | Metal response element binding transcription factor 2 | 10 (5H/5M) |
| RSRC2 | Arginine/serine-rich coiled-coil 2 | 10 (7H/3M) |
| CEP57 | Centrosomal protein 57kDa | 10 (8H/2M) |
| FIP1L1 | Factor interacting with PAPOLA and CPSF1 | 10 (7H/3M) |
| FYN | FYN proto-oncogene, Src family tyrosine kinase | 10 (6H/4M) |
| ABI1 | Abl-interactor 1 | 10 (6H/4M) |
| HUWE1 | HECT, UBA, and WWE domain containing 1, E3 ubiquitin protein ligase | 10 (5H/5M) |
| TPD52L2 | Tumor protein D52-like 2 | 10 (8H/2M) |
| ZMYND8 | Zinc finger, MYND-type containing 8 | 10 (7H/3M) |
| NSD1 | Nuclear receptor binding SET domain protein 1 | 10 (5H/5M) |
Genes are ranked by the number of datasets the transcript was identified in. The number of human (H) or murine (M) datasets is indicated in parentheses.
Selection of MDS related genes in Srsf2 mutant cells mirrors human genetic interactions
In parallel to the literature analysis, we performed a genome-wide pooled CRISPR knock-out screen using the Cas9 expressing cells to explore the genetic vulnerabilities induced by Srsf2P95H mutation (Figure 3A). Cas9 Srsf2P95H/+ or Cas9 Srsf2+/+ cells (n = 3 biological replicates per genotype) were transduced with the Brie sgRNA library, which targets 19c674 genes with 4 sgRNAs per gene.39 After selection for cells infected with the sgRNA library with puromycin, the cells were used for screening. At day 0 of the screen, cells expressing sgRNAs were treated with tamoxifen (400 nM) for 4 days to activate expression of the Srsf2P95H/+ mutation. Cells were then cultured for up to 14 days after tamoxifen treatment. Genomic DNA from each cell line was collected at days 0, 4, 11, and 18 and sequenced to determine the sgRNA representation compared with the baseline (day 0) sgRNA counts (outlined in Figure 3A).
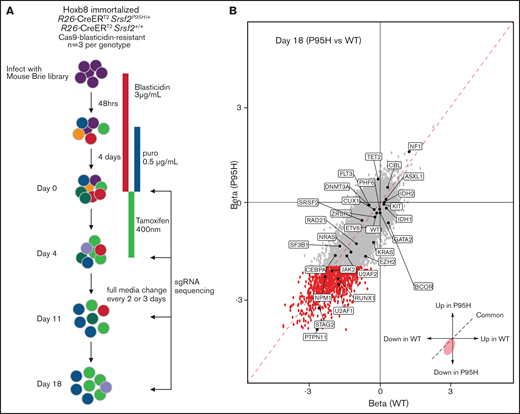
Genome-wide CRISPR knock-out Srsf2P95H/+ screen. (A) Schematic outline of the pooled CRISPR knock-out Srsf2 screen. (B) Visualization of performance of sgRNA against genes mutated in MDS and MDS/myeloproliferative neoplasm at day 18 of the screen. Gray dots, all the sgRNA identified; red dots, sgRNA with FDR < .01 and β score <−2.
Genome-wide CRISPR knock-out Srsf2P95H/+ screen. (A) Schematic outline of the pooled CRISPR knock-out Srsf2 screen. (B) Visualization of performance of sgRNA against genes mutated in MDS and MDS/myeloproliferative neoplasm at day 18 of the screen. Gray dots, all the sgRNA identified; red dots, sgRNA with FDR < .01 and β score <−2.
Analysis of sgRNA counts was performed with both CRISPRBetaBinomial and MaGeCK VISPR.22,23 The number of mapped reads per sample ranged from 24 to 33 million, with an average mappability of 85%. The average copy number of the sgRNAs was 355 (supplemental Table 2) and was equally distributed across the cell lines, suggesting equivalent sgRNA representation across samples (supplemental Table 3A). As expected, the diversity of the sgRNA pool decreased over time (supplemental Table 3B). The correlation between Cas9 Srsf2P95H/+ and Cas9 Srsf2+/+ cells also decreased, indicating mutant and WT cells responded differently to the pooled genome knockout (supplemental Table 4). We next assessed the depletion of essential genes from the samples in the screen. Using the Cluster of the Essential Gene (CEG2) database, we annotated and visualized the negative selection of essential genes using MaGeCK maximum likelihood estimation (supplemental Table 5).23,40 At day 4, essential genes were equally depleted from Srsf2P95H/+ and Srsf2+/+ cells. Genes from the essential ribosome pathways, such as Rps15A, Rps7, Rpl23, Rplp2, Rps8, and Rpl8, were among the most depleted genes in the screen. In addition, cell line-specific essential genes such as Hoxb8 and Csf2ra (indicated in green), were depleted in both genotypes. Both Hoxb8 and Csf2ra (GM-CSF receptor) would be expected to be lost because the cell lines are immortalized with Hoxb8 and dependent on GM-CSF for survival and proliferation.
We then assessed the performance of sgRNA against genes that are recurrently mutated in human MDS and related myeloid neoplasms (Figure 3B). At day 18, multiple spliceosome genes, such as Srsf2, Sf3b1, U2af1, U2af2, and Zrsr2 were all negatively selected in both Srsf2P95H/+ and Srsf2+/+ cells. This is expected because complete loss of key spliceosome genes resulted in cell death or embryonic lethality in murine models.34,41 Looking at genes of which loss of function is known to be cooperative with SRSF2P95H mutation, sgRNA targeting Tet2 (FDR = 0.05), or Cbl (FDR = 0.02) showed a positive enrichment in Srsf2P95H/+ cells. This is consistent with the human genetics where TET2 loss-of-function mutations frequently cooccur with SRSF2 mutation.42 A caveat with this analysis is that genes with recurrent truncating or point mutations are unlikely to be captured with this approach because Cas9 cleavage results in indels and therefore most likely gene disruption near the target sites.
Pooled CRISPR knock-out screen reveals genetic vulnerabilities of Srsf2 mutant cells in cell-cycle and DNA repair pathways
To determine candidate genes that are negatively selected in Cas9 Srsf2P95H/+ compared with the Cas9 Srsf2+/+ cells, we set inclusion criteria as FDR < 0.01, β score <−1, no CEG2 essential genes, and the number of sgRNA recovered ≥3. The β score represents the degree and direction a gene is selected. Overall, the number of negatively selected sgRNA was higher in the Srsf2P95H/+ cells than in the Srsf2+/+ cells (Figure 4A). At each time point, the majority of genes depleted in the WT cells overlapped with the genes depleted in the P95H cells. This suggested that WT cells share most of their genetic sensitivities with Srsf2P95H/+ cells, whereas mutant cells are sensitive to an additional set of genetic perturbations. We then performed analysis of the genes and pathways negatively selected in Srsf2P95H/+ and Srsf2+/+ cells using Metascape.25 The top 20 most significant pathways are listed in Figure 4B. From the analysis, sgRNA within the RNA metabolism, ribosome, and translation pathways were significantly depleted in both P95H and WT cells, although they tended to be more significantly depleted in P95H cells. Interestingly, there were a number of pathways that were specific to the P95H cells or also present in the WT cells but at a much lower level of significance. These pathways include histone modification/chromosome organization, DNA damage response/DNA repair, and cell-cycle pathways. The results indicate that potential genetic vulnerabilities of Srsf2P95H/+ cells are contained within these pathways.
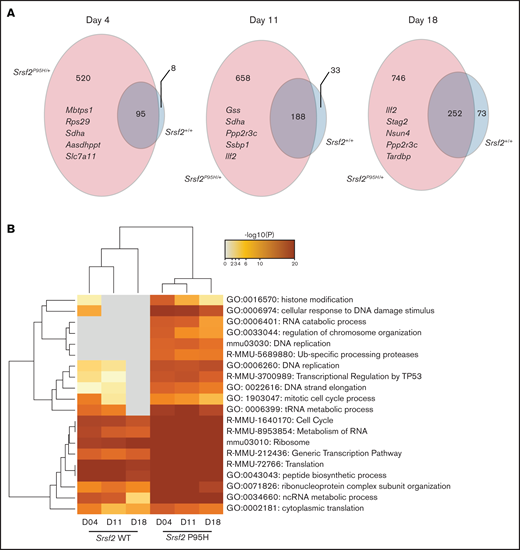
DNA replication, cell-cycle, and DNA damage response are among the top negatively selected pathways in screen. (A) The number of genes negatively selected at days 4, 11, and 18 in Srsf2P95H/+ (pink) and Srsf2+/+ (blue) cells. The top 5 negatively selected genes for Srsf2P95H/+ cells ranked by β score of all 3 cell lines at each timepoint are listed. FDR < 0.01, β score <−1, and any CEG2 essential genes are excluded. (B) Pathway analysis of the negatively selected genes for Srsf2+/+ and Srsf2P95H/+ at 3 timepoints showing DNA replication, cell-cycle, and DNA damage response pathways have a more negative effect on Srsf2P95H/+ cell survival when perturbed.
DNA replication, cell-cycle, and DNA damage response are among the top negatively selected pathways in screen. (A) The number of genes negatively selected at days 4, 11, and 18 in Srsf2P95H/+ (pink) and Srsf2+/+ (blue) cells. The top 5 negatively selected genes for Srsf2P95H/+ cells ranked by β score of all 3 cell lines at each timepoint are listed. FDR < 0.01, β score <−1, and any CEG2 essential genes are excluded. (B) Pathway analysis of the negatively selected genes for Srsf2+/+ and Srsf2P95H/+ at 3 timepoints showing DNA replication, cell-cycle, and DNA damage response pathways have a more negative effect on Srsf2P95H/+ cell survival when perturbed.
Misspliced genes in cell-cycle pathways are essential for Srsf2P95H/+ cells survival
After determining the negatively selected sgRNAs in Cas9 Srsf2P95H/+ cells, we tested if any of the genes were contained within the differentially spliced transcripts of the Srsf2P95H/+ cells. Aberrant transcripts generated by missplicing could potentially lead to changes in protein function and promote cell survival and proliferation of the mutant cells. Knock-out of the genes, as undertaken in the screen, could prevent these aberrant transcripts arising and therefore cause cell death. We searched for overlap between the misspliced transcripts generated from Cas9 Srsf2P95H/+ cells at day 11 (Figure 1) and the negatively selected genes during the whole course of the screen. Upon comparison, there was little overlap (less than 10%) between the 2 datasets (Figure 5A). Among the overlapping genes, Npm1, Dap3, and Runx1 were identified at all the timepoints, whereas Hnrnpa2b1 was only identified at day 18. Interestingly, genes belonging to the cell-cycle pathway were identified as both misspliced and negatively selected in Srsf2P95H/+ cells (Figure 5B). Other pathways found in the negatively selected list, such as translation, RNA metabolism, and chromosome organization were also pathways enriched in misspliced transcripts in Srsf2P95H/+ cells. Together, the data suggest that these pathways have a close genetic interaction with Srsf2P95H/+, especially the cell-cycle pathways.
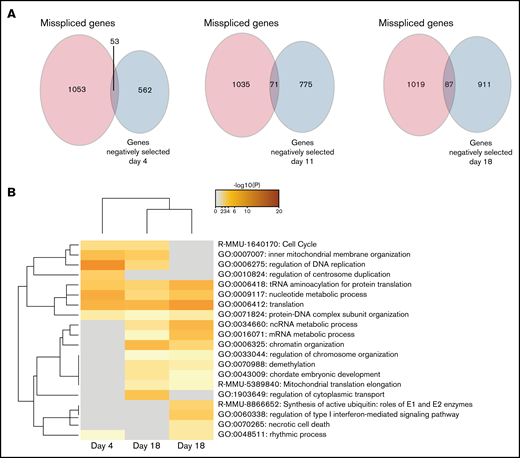
Genes from the cell-cycle and regulation of DNA replication pathways are both misspliced and negatively selected in Srsf2P95H/+ cells. (A) Venn diagrams showing the overlap between the genes that are misspliced in the GM-CSF Srsf2P95H/+ cells and the genes that are negatively selected at days 4, 11, and 18 of the Srsf2 CRISPR screen. (B) Pathway analysis showing that genes associated with cell-cycle and DNA replication regulation are misspliced and cause cell death when deleted in Srsf2P95H/+ cells.
Genes from the cell-cycle and regulation of DNA replication pathways are both misspliced and negatively selected in Srsf2P95H/+ cells. (A) Venn diagrams showing the overlap between the genes that are misspliced in the GM-CSF Srsf2P95H/+ cells and the genes that are negatively selected at days 4, 11, and 18 of the Srsf2 CRISPR screen. (B) Pathway analysis showing that genes associated with cell-cycle and DNA replication regulation are misspliced and cause cell death when deleted in Srsf2P95H/+ cells.
Palbociclib, a CDK6 inhibitor, preferentially inhibits Srsf2P95H/+ cell growth
After analyzing the functional relationships of the negatively selected genes, we searched for druggable targets by comparing the negatively selected genes at day 18 with the Drug-Gene Interaction database.43-45 Selected candidates were further tested using in vitro dose response assays to validate the synthetic lethality. The cell cycle–related gene Cdk6, among the top 30 negatively selected genes at day 18 using MaGeCK24 analysis (supplemental Figure 6A), was prioritized as a druggable target. Evaluation of the Cdk6 sgRNA from the screen indicated that there was a substantial drop in the number of guides in all 3 Cas9 Srsf2P95H/+ cell lines, whereas significantly less depletion was observed in 2 of the 3 Cas9 Srsf2+/+ cell lines (Figure 6A). Subsequent analysis of the cell lines showed no inherent differences between the cell-cycle profiles of Srsf2P95H/+ and WT cells (Figure 6B). We then tested the sensitivity of the cell lines (n = 3 per genotype) to inhibition of CDK6 using palbociclib. Roscovitine which is a CDK2/5 inhibitor was used as a negative control because we did not find depletion of Cdk2 sgRNA from the P95H cells (supplemental Figure 6B). We also tested Pladienolide B, a spliceosome inhibitor, as a positive control (Figure 6C, top). The results showed that Srsf2P95H/+ cells had a near threefold reduction in the IC50 value of palbociclib (P < .0001, extra sum-of-squares F test), compared with Srsf2+/+ cells. Consistent with the screen results, roscovitine did not show any specific inhibition of Srsf2P95H/+ cells, whereas Pladienolide B was approximately twofold more toxic to Srsf2P95H/+ cells than WT cells, which is in agreement with previous reports.10,18 In addition to Cdk6, we performed drug assay using tigecycline46 and murbritinib47 because several mitochondrial respiratory genes including Uqcrc1, Cox6b1, Atp5a1, and Uqcc2 were among the top 30 negatively selected genes in Srsf2 mutant cells (supplemental Figure 6A). However, both drugs showed no effect in targeting Srsf2 mutant cells (supplemental Figure 6D).
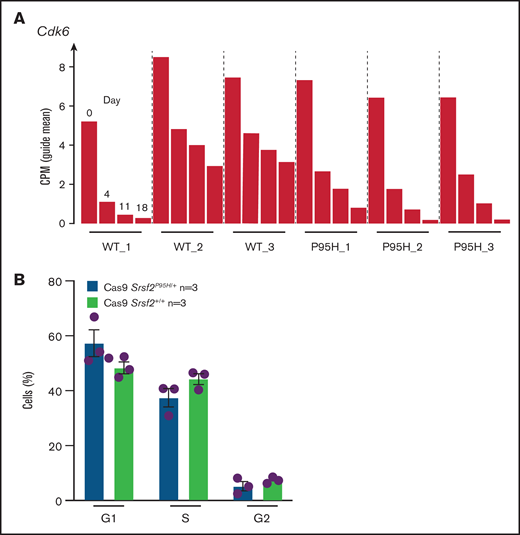
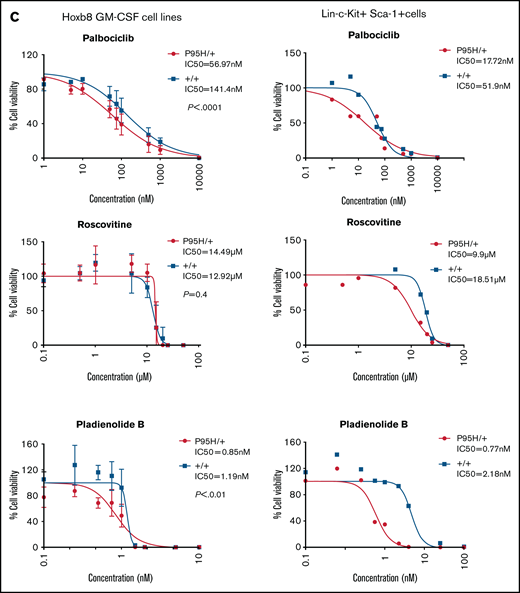
Srsf2P95H/+ cells are sensitive to the CDK6 inhibitor palbociclib and the spliceosome inhibitor Pladienolide B. (A) The guide count for CDK6 sgRNA in Srsf2+/+ and Srsf2P95H/+ cells over the course of the screen. (B) The cell-cycle stage distribution of Srsf2P95H/+ and Srsf2+/+ cells at day 18 after tamoxifen cessation. No statistical difference detected between P95H and WT cells. (C) Hoxb8 GM-CSF Srsf2P95H/+ cell lines and sorted primary Srsf2P95H/+ LKS+ cells are sensitive to palbociclib and Pladienolide B after 4 days of drug culture. Hoxb8 GM-CSF cell lines: n = 3 per genotype for roscovitine and Pladienolide B, n = 2 for palbociclib. Extra sum-of-squares F test. LKS+ cells, n = 1 per genotype. LKS+, lin−ckit+Sca-1+.
Srsf2P95H/+ cells are sensitive to the CDK6 inhibitor palbociclib and the spliceosome inhibitor Pladienolide B. (A) The guide count for CDK6 sgRNA in Srsf2+/+ and Srsf2P95H/+ cells over the course of the screen. (B) The cell-cycle stage distribution of Srsf2P95H/+ and Srsf2+/+ cells at day 18 after tamoxifen cessation. No statistical difference detected between P95H and WT cells. (C) Hoxb8 GM-CSF Srsf2P95H/+ cell lines and sorted primary Srsf2P95H/+ LKS+ cells are sensitive to palbociclib and Pladienolide B after 4 days of drug culture. Hoxb8 GM-CSF cell lines: n = 3 per genotype for roscovitine and Pladienolide B, n = 2 for palbociclib. Extra sum-of-squares F test. LKS+ cells, n = 1 per genotype. LKS+, lin−ckit+Sca-1+.
We then tested palbociclib on primary Srsf2P95H/+ cells. LKS+ cells were sorted from R26-CreERT2ki/+Srsf2+/+ and R26-CreERT2ki/+Srsf2P95H/+ mice, tamoxifen treated in vitro before coculturing with palbociclib, roscovitine, and Pladienolide B for 4 days (Figure 6C, bottom). The results showed that primary Srsf2P95H/+ cells had a threefold reduction in the IC50 value of palbociclib as well as Pladienolide B. Roscovitine showed a twofold difference between Srsf2P95H/+ and Srsf2+/+ cells. Taken together, the results demonstrate that palbociclib displays a preferential inhibition of Srsf2P95H/+ cells in both Hoxb8 immortalized cell lines and in primary LKS+ cells.
Discussion
Despite recent efforts to identify means of targeting splicing-mutant myeloid neoplasms and leukemia, targeting of the remaining WT splicing apparatus has proven toxic and with a limited therapeutic window.7,12,18,48 By comparing multiple human and murine datasets, we identified cell cycle, DNA repair, and chromatin organization and modification as overrepresented pathways within the misspliced transcripts of both human and mouse Srsf2P95H/+ cells. RNA-seq analysis of Cas9 Srsf2P95H/+ cells, a cell model generated to exclude confounding mutations often detected in existing cell lines, showed similar missplicing changes compared with the in vivo model. Using these Cas9 cells, we performed a functional screen to experimentally determine genetic sensitivities of Srsf2P95H/+ mutation. With 3 biological replicates per group, we discovered that cell-cycle and DNA damage response pathways are coessential with Srsf2P95H/+ mutation.
It is unknown whether the genetic interaction we observed is an SRSF2 mutant-specific phenomena or if it would apply to other recurrent splicing mutations such as SF3B1K700E and U2AF1S34. Previously, it has been demonstrated that RNA splicing and cell cycle are interconnected. Adequate splicing of cell-cycle genes, such as CDC25 and CDK2, are required for a proper transition between different cell-cycle phases49,50 and disruption of core spliceosome components, such as deletion of SRSF2, leads to G2-phase cell-cycle arrest and apoptosis.51-53 It is possible that mutant splicing factors diminish the capacity of the spliceosome, which is critical for normal cell-cycle progression, and that mutant cells heavily rely on the remaining WT allele for survival. This is supported by the observation that no homozygous (Srsf2P95H/P95H) or hemizygous (Srsf2P95H/−) mutations have been reported in human cancers. Our analysis indicates that splicing mutant cells are unable to cope with further disruption of cell-cycle pathways that indirectly results in cell death. It is also possible that disruption of the cell cycle has a more direct impact on RNA splicing. A recent study by Rimel et al reports that inhibition of cell-cycle regulator CDK7 could lead to widespread splicing defects.54 This is similar to the effect of spliceosome inhibition and may at least partly be responsible for the synergistic interaction that we observed.
Through our analysis of negatively selected genes and the direct testing of genetic interactions, we were able to identify CDK6, a cell-cycle regulator, as a potential new therapeutic target for Srsf2P95H/+ mutation. Pharmacological inhibition of CDK6 with palbociclib resulted in a threefold reduction in IC50 in both Srsf2P95H/+ cell lines and primary Srsf2P95H/+ cells, compared with the respective WT cells. Although palbociclib is also a potent inhibitor of CDK4, unlike Cdk6 sgRNA, Cdk4 sgRNA was not preferentially depleted in the P95H cells (supplemental Figure 6C). The inhibitory effect of palbociclib on P95H cell growth is thus likely because of CDK6 inhibition. This is the first report that a cell-cycle inhibitor could be used to target splicing mutant cells. In normal HSCs, CDK6 is associated with the regulation of hematopoietic stem cell quiescence.55,56 In MLL-AF9 leukemic cell lines, inhibition of CDK6 has been shown to attenuate the growth and to overcome the block to myeloid differentiation.57,58 In breast cancer patients,59 however, neutropenia is a known side effect of palbociclib treatment. It is possible that in vivo palbociclib inhibits excessive proliferation of myeloid population. Because this is a key phenotypic feature of SRSF2P95H/+ mutation,4,5,18 this could be an advantage in the treatment of SRSF2 mutant MDS. Exactly how inhibition of CDK6 induces lethality in our Srsf2P95H/+ cells is unclear. Nevertheless, the functional and pharmacological investigation demonstrates a close genetic interaction between cell-cycle regulation pathways and Srsf2P95H/+ mutation.
Besides cell cycle, the DNA damage response pathway was also identified as a genetic interactor with Srsf2P95H/+ mutation. Aberrant RNA splicing has been reported to lead to genome instability via increased R-loop formation and possible suboptimal splicing of key transcripts for genome maintenance.60 On 1 hand, impairment of the splicing machinery, such as depletion of SRSF2 and SRSF2P95H mutation, may lead to accumulation of R-loops, which is linked with replication folk stalling and DNA replication stress.61,62 On the other hand, key response elements of DNA repair, such as ATR, are misspliced in Srsf2P95H/+ mutant cells. It is possible that 1 or both of these factors may contribute to the synthetic lethality observed with the inhibition of the DNA damage response pathway. Additional investigations may expose more intrinsic vulnerabilities of splicing factor mutant cells and decipher how splicing mutation induced DNA damage may contribute to cancer development.
Overall, we have identified that the cell-cycle and DNA damage response are overrepresented within the recurrently misspliced transcripts of SRSF2P95H/+ across both human and mouse samples. Using agnostic genome-wide screening, we demonstrate that targeting of these pathways in combination with Srsf2P95H/+ mutation reveals a previously unrecognized vulnerability to CDK6 inhibition by palbociclib. Therefore, our study identifies a close genetic relationship between the cell-cycle and DNA damage response/DNA repair pathway with Srsf2P95H/+ mutation and a new therapeutic opportunity outside of spliceosome inhibition.
Acknowledgments
The authors thank J. Heierhorst, L. Purton, J. Heraud-Farlow, J. Pimanda, A. Unnikrishnan, K.P. Truong, and M. Pillai for comments and discussion; S. Taylor for technical assistance; St. Vincent’s Hospital BioResources Centre for care of experimental animals; St. Vincent’s Institute Flow Cytometry Core Facility; M Kamps for HoxB8 plasmids; Addgene for plasmid distribution; and Novogene for DNA and A sequencing.
This work was supported by the Cancer Council of Victoria (C.R.W., A.M.C., APP1126010), Victorian Cancer Agency Research Fellowship (C.R.W., MCRF15015), and in part by the Victorian State Government OIS to St Vincent’s Institute. The Victorian Centre for Functional Genomics (K.J.S.) is funded by the Australian Cancer Research Foundation, Phenomics Australia through funding from the Australian Government’s National Collaborative Research Infrastructure Strategy program, and the Peter MacCallum Cancer Centre Foundation.
Authorship
Contribution: J.J.X., M.F.S., and C.R.W. conceptualized the study; J.J.X., M.F.S., I.N., K.J.S., and C.R.W. designed the experiments; J.J.X., M.F.S., and A.M.C. performed the experiments; J.J.X. and A.M.C. produced the figures; J.J.X. wrote the original manuscript; J.J.X., M.F.S., A.M.C., and C.R.W. reviewed and edited the manuscript; A.M.C. and C.R.W. were responsible for funding acquisition; I.N. and K.J.S. provided resources; and M.F.S., K.J.S., and C.R.W. provided supervision.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carl Walkley, St Vincent’s Institute, 9 Princes St, Fitzroy 3065 VIC, Australia; e-mail: cwalkley@svi.edu.au; and Monique Smeets, St Vincent’s Institute, 9 Princes St, Fitzroy 3065 VIC, Australia; e-mail: msmeets@svi.edu.au.

