

Key Points
The DLBCL microenvironment signature scoring system was established using nCounter-based profiling of GC-related microenvironmental genes.
DMS scores stratified DLBCL patients with different prognosis independently of existing prognostic models.

Abstract
Diffuse large B-cell lymphoma (DLBCL) is the most common B-cell malignancy, with varying prognosis after the gold standard rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). Several prognostic models have been established by focusing primarily on characteristics of lymphoma cells themselves, including cell-of-origin (COO), genomic alterations, and gene/protein expressions. However, the prognostic impact of the lymphoma microenvironment and its association with characteristics of lymphoma cells are not fully understood. Using the nCounter-based gene expression profiling of untreated DLBCL tissues, we assess the clinical impact of lymphoma microenvironment on the clinical outcomes and pathophysiological, molecular signatures in DLBCL. The presence of normal germinal center (GC)-microenvironmental cells, including follicular T cells, macrophage/dendritic cells, and stromal cells in lymphoma tissue indicates a positive therapeutic response. Our prognostic model, based on quantitation of transcripts from distinct GC-microenvironmental cell markers, clearly identified patients with graded prognosis independently of existing prognostic models. We observed increased incidences of genomic alterations and aberrant gene expression associated with poor prognosis in DLBCL tissues lacking GC-microenvironmental cells relative to those containing these cells. These data suggest that the loss of GC-associated microenvironmental signature dictates clinical outcomes of DLBCL patients reflecting the accumulation of “unfavorable” molecular signatures.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of B-cell non-Hodgkin lymphomas (NHL) with heterogeneous clinicopathologic features. By performing global gene expression profiling (GEP), Alizadeh et al have grouped DLBCL cases into 2 subtypes based on the cell-of-origin (COO) of lymphoma cells. The germinal center B-cell-like (GCB) type exhibits the signature of B cells in the germinal center (GC) of normal secondary lymphoid organs, while lymphoma cells of the activated B-cell (ABC)-like type resemble post-GC B cells that transit from the GC for plasmacytic differentiation.1 Among DLBCL patients treated with multiagent chemotherapy consisting of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), patients with ABC-type disease generally show significantly worse prognosis than do those with GCB-type disease.1 Other groups have proposed immunohistochemistry (IHC)-based algorithms for formalin-fixed paraffin-embedded (FFPE) tissues that recapitulate microarray-based COO classification (eg, Hans’ criteria).2,3 In clinical trials of patients undergoing R-CHOP (CHOP plus rituximab) regimens, IHC-based COO criteria have minimal prognostic power, presumably due to discordance between IHC-based classification and microarray-based “original” classification.4-8 Given that R-CHOP remains the gold standard after numerous attempts for improvement, such as intensification of rituximab or CHOP, the introduction of next-generation anti-CD20 antibodies, and addition of novel therapeutic agents (eg, proteasome inhibitor),9 the accurate stratification model is still in demand, which could predict clinical outcomes after R-CHOP.
A recent large-scale genomic study of >1,000 DLBCL patients revealed the mutational heterogeneity of DLBCL and concluded that the best performing predictive model could only be achieved by combining the DNA- and RNA-risk models.10 Currently, the International Prognostic Index (IPI) remains the most reliable predictive model, although it is based solely on clinical variables and patient status. Thus, a more accurate prognostic model, one that comprehensively reflects DLBCL pathophysiology and helps physicians in therapeutic decision-making, is still needed.
As naïve B cells develop into antibody-secreting plasma cells in lymph nodes (LNs), they undergo stage-specific genome editing activities to generate variable immunoglobulins. Although required for immune cell diversity, this process predisposes B cells to transformation if the editing machinery is hijacked.11 Dynamic B-cell development processes in the GC are controlled by cell-intrinsic activities and surrounding microenvironmental cells, including follicular T cells, stromal cells, dendritic cells (DCs), and macrophages. Microenvironmental cells are, in fact, necessary for GC formation and are recruited from the periphery in response to normal immune signals that govern GC formation.12,13 These observations suggest that the tumor microenvironment may play a role in DLBCL pathogenesis, originating from GC or post-GC B cells.
In the present study, we sought to identify prognostic factors through global GEP in clinical samples and demonstrated that microenvironment-related genes were tightly associated with clinical outcomes of DLBCL. We revealed characteristics of prognostic microenvironmental components, their prognostic impact, and, moreover, their correlation with molecular signatures of lymphoma cells.
Methods
Clinical data collection
We retrospectively collected 280 de novo DLBCL cases (30, 170, and 80 cases for pilot, training, and validation cohort, respectively), which were newly diagnosed by 2 or more experienced hematopathologists in the Kurume University Pathology Department from 2006 to 2013, based on the 2008 WHO classification of lymphoid neoplasms. Our cohorts include the high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (HGBL-DH/TH in the 2017 WHO classification14 ) with DLBCL morphology, while the high-grade B-cell lymphoma, not otherwise specified (HGBL-NOS14 ), which replaced the 2008 category of B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma (BCLU), were excluded. The use of materials and clinical information was approved by the Research Ethics Committee of Kurume University following the Helsinki Declaration. FFPE samples, which were collected with clinical data, were anonymized before shipping to Kyushu University. The present study was approved by the institutional ethics committee of the Kyushu University Graduate School of Medical Sciences.
GEP using nCounter system
To determine gene expression levels, 300 ng total RNA from DLBCL FFPE samples was analyzed using the nCounter system (NanoString Technologies, Seattle, WA). We used a nCounter Customer Assay Evaluation (CAE) kit to evaluate data reproducibility (supplemental Figure 1A). We used PanCancer Pathways, Immunology, and Kinase panels to extract candidate prognostic genes in the screening cohort. To validate the results of the screening, we used a custom gene set (NanoString Technologies) consisting of 447 genes. Detailed information about sample preparation and assay procedure is provided in the section with the same name in the supplemental Methods. The Gene Ontology (GO) enrichment analysis was performed using the Metascape web application (http://metascape.org).15
Multiplexed fluorescent immunostaining
Imaging analyses were performed using the Opal multiplex tissue staining system and the Mantra quantitative pathology workstation (PerkinElmer, Waltham, MA). Slides from biopsied tumor tissues were stained using an Opal multiplex tissue staining system (PerkinElmer, Waltham, MA). Antigen retrieval was performed by heating slides to 93 ± 2°C for 20 minutes in a high-pH antigen unmasking solution (H-3301, Vector Labs, Burlingame, CA), followed by blocking in 5% bovine serum albumin (BSA) (Jackson ImmunoResearch, Birmingham, AL) in phosphate-buffered saline (PBS). Cell phenotyping and counting were performed using the Mantra quantitative pathology workstation (PerkinElmer) within representative fields preselected by trained hematopathologists. Spatial distribution and marker intensity in target cells were analyzed using inForm® image analysis (PerkinElmer) and Spotfire (TIBCO Software, Palo Alto, CA) software.
Survival analysis
Data relevant to the observation period and survival and disease status at the last observation was available for all study patients. Overall survival (OS) was defined as the time from diagnosis to the last follow-up or death, and disease-free survival (DFS) was defined as the time from diagnosis to any recurrence or death. The cutoff values of gene expression levels were determined as the optimal value for predicting relapse or death events using the maximally selected rank statistics of the ‘maxstat’ R package.16
Survival probability was estimated using the Kaplan-Meier method, and P values were determined by log-rank test. Univariate or multivariate analysis using a Cox proportional hazards model was used to assess the predictive value of DLBCL Microenvironment Signature (DMS) scores, as defined by gene expression analysis.
Calculation of the DMS score
The DMS score was calculated based on the expression levels of 3 representative microenvironment genes (ICOS, CD11c, and FGFR1). To call positive (point = 1) or negative (point = 0) for each gene, cutoff values were defined using the ‘maxstat’ R package.16 The DMS score was determined as a sum of points from each gene (0 to 3 points).
Mutation analysis
Among 170 cases in the training cohort, we randomly selected patients for mutational analysis and evaluated 120 FFPE-derived DNA samples. Genomic DNA was extracted using a QIAamp DNA FFPE Tissue Kit (QIAGEN) based on the manufacturer’s instructions. Then, we undertook massively parallel sequencing using a solution-phase SureSelect hybrid capture kit (Agilent Technologies, Santa Clara, CA) and an HiSeq 2500 sequencer (Illumina). To detect genomic aberrations/rearrangements in FFPE samples, we used an OncoPanel assay (Brigham and Women’s Hospital) to evaluate exonic regions of 447 cancer genes and 191 regions across 60 genes.17 Among 120 cases, we analyzed data from 106 (14 samples were omitted due to insufficient data quality). Detailed information about the library preparation, sequencing, and the bioinformatic procedure is provided in the supplemental Methods.
Statistics
We used the Wilcoxon rank-sum test between 2 groups and the Steel-Dwass test for multiple comparisons for nonparametric testing to compare numerical data. Fisher’s exact test was used to compare the categorical data among subgroups. Univariate or multivariate analysis using a Cox proportional hazards model was used to assess the predictive value of prognostic models. Statistical analyses were performed using R (http://CRAN.R-project.org/) and JMP Pro software (SAS Institute Inc., Cary, NC). In the GO analysis, P values were adjusted using the Benjamini-Hochberg correction algorithm described in the original article.15
Results
Transcriptome profiling of untreated DLBCL tissues identifies prognostic genes
We collected 280 FFPE tissues from de novo DLBCL patients (30, 170, and 80 cases for pilot, training, and validation cohort, respectively) (Figure 1A). Cases of relapsed or transformed disease were excluded. All the patients had been treated with R-CHOP or R-THPCOP (tetrahydropyranyladriamycin, cyclophosphamide, vincristine, and prednisone).18,19 Median observation time for surviving patients was 3.67 and 5.60 years, and the 3-year DFS rate was 66.3% and 65.0% in the training and validation cohorts, respectively. Additional patient characteristics are provided in supplemental Table 1. To quantify fragmented mRNA in FFPE tissues, we employed the nCounter system, which enables highly sensitive and accurate RNA detection.20-22 First, we compared 2 replicated measurements of 48 representative genes (supplemental Table 2) with each other in an FFPE sample. The result showed exceptionally high measurement reproducibility, suggesting that this assay could detect RNAs expressed in specimens, whether they are from lymphoma cells or rare microenvironment components (supplemental Figure 1A).
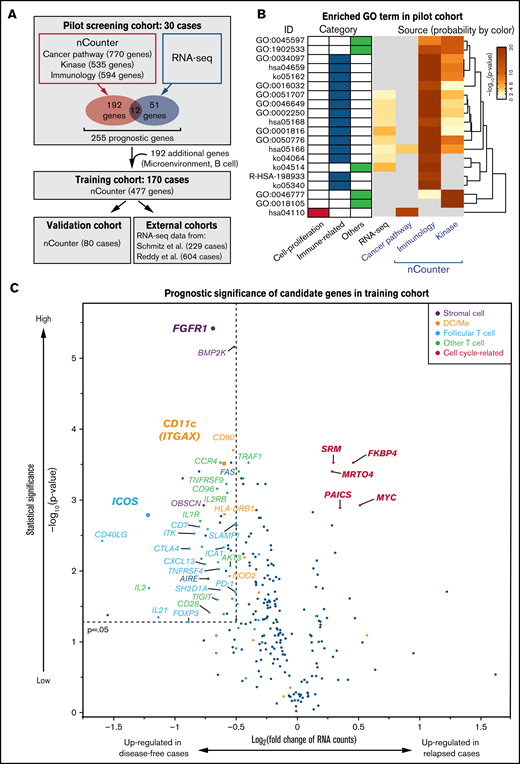
Identification of predicting factors for DLBCL outcomes. (A) Schematic representation of overall study design. Thirty cases of newly diagnosed DLBCL were recruited for a pilot screen using nCounter and RNA-seq methods, followed by analysis of 2 larger nCounter cohorts (training and validation cohort). (B) GO analysis of candidate prognostic genes from the pilot screen, which was performed using the Metascape web application (http://metascape.org). Enriched GO terms are shown by ID, category, and derived pilot cohort. Probabilities (adjusted P values) are depicted by color. Most prognostic genes were linked to immune-related terms, irrespective of source. (C) A volcano plot indicates differentially expressed genes as favorable (left) and poor (right) prognostic factors in the training cohort. Most unfavorable indicators were cell cycle-related (magenta). Note that many microenvironmental cell-related genes were associated with favorable prognoses. A Mann-Whitney U test (unpaired) with bootstrap was performed to calculate P values.
Identification of predicting factors for DLBCL outcomes. (A) Schematic representation of overall study design. Thirty cases of newly diagnosed DLBCL were recruited for a pilot screen using nCounter and RNA-seq methods, followed by analysis of 2 larger nCounter cohorts (training and validation cohort). (B) GO analysis of candidate prognostic genes from the pilot screen, which was performed using the Metascape web application (http://metascape.org). Enriched GO terms are shown by ID, category, and derived pilot cohort. Probabilities (adjusted P values) are depicted by color. Most prognostic genes were linked to immune-related terms, irrespective of source. (C) A volcano plot indicates differentially expressed genes as favorable (left) and poor (right) prognostic factors in the training cohort. Most unfavorable indicators were cell cycle-related (magenta). Note that many microenvironmental cell-related genes were associated with favorable prognoses. A Mann-Whitney U test (unpaired) with bootstrap was performed to calculate P values.
We performed a pilot screen to identify genes correlated with specific patient outcomes by analyzing FFPE samples obtained at initial diagnosis from 30 DLBCL patients: 15 showing favorable outcomes (>4 years in remission) and 15 showing poor prognosis (primary refractory or early relapse disease). We used commercially available probe sets that included 1899 genes related to immunology, cancer, or kinase pathway panels (Figure 1A). This screen identified 204 genes with a statistically significant association with either favorable or poor prognosis (q-value <0.05, supplemental Figure 1B, supplemental Tables 3-5). More than half of candidate prognostic genes were derived from the immunology panel, including many T-cell-related genes, especially related to follicular T cells (supplemental Figure 1B). To cover the limitation in the number of genes analyzed, we performed whole transcriptome analysis on the 30 samples of the pilot screen using RNA sequencing (RNA-seq) and extracted an additional 51 differentially-expressed genes as candidate prognostic markers (supplemental Table 6). GO analysis revealed that most prognostic genes were associated with immune responses, while only 1 cell proliferation-related term was enriched, suggesting that immune signature has a significant effect on DLBCL clinical outcomes (Figure 1B). Transcript levels of each of the 12 genes (ICOS, CD80, CTLA4, EGR2, CD58, TRAF1, C1QBP, IL21, CD40LG, FAS, MYC, and CD96) were identified as prognostic factors both in nCounter and RNA-seq analyses (Figure 1A). We added an additional 192 genes for reference and normalization: COO-defining genes,23 reported prognostic genes, B-cell-associated genes, and microenvironmental cells-specific genes,24,25 and designed a probe set containing a total of 447 genes for nCounter analysis (supplemental Table 7).
We next applied a similar analysis to 170 DLBCL samples from patients who had undergone R-CHOP-based regimens (Figure 1A). Among these, tissue specimens came from LNs of 100 patients and extranodal sites of 70 others (supplemental Figure 1C). The scatterplot shown in Figure 1C depicts genes upregulated in specimens representing poor (relapsed) vs favorable (disease-free) outcomes. Among genes upregulated in patients with poor prognosis (genes depicted in the right upper area in Figure 1C) were those related to cell proliferation, including MYC and its targets: FKBP4,26 SRM,27 and PAICS.28 All 4 of these genes were expressed at high levels in DLBCL cell lines relative to normal LN samples (supplemental Figure 2A). Kaplan-Meier curves of DFS confirmed that high expression of each proliferation-associated gene marks patients with poor prognosis with statistical significance (P < .005, supplemental Figures 2B,C).
Immune microenvironment-related genes define favorable prognosis in DLBCL
To our surprise, most genes in the analysis of 170 DLBCL samples described above were identified as favorable prognostic factors (Figure 1C, upper left box: P < .05 and log2[fold change] <−0.5). These factors were again linked to immune-related GO terms, especially those related to T-B interactions (supplemental Figure 3A). Of note, most favorable prognostic factors mark GC-related microenvironmental cells. Among them were ICOS, CXCL13, CD40LG, PD-1, SH2D1A (SAP), IL2RB, IL21, and SLAMF1, all of which are representative markers for CD4+ follicular T cells and regulate B-cell development in the normal GC.12,13,29,30 In addition, DC/macrophage- and interstitial stromal cells-associated genes were identified as favorable prognostic markers (orange and purple dots in Figure 1C). ITGAX, which encodes CD11c, and fibroblast growth factor 1 (FGFR1) were the most significant prognostic genes among DC/macrophage- and stromal cell-related genes as represented by the distance from the origin (Figure 1C). Of note, both ITGAX and FGFR1 were also identified as statistically significant genes in the pilot screen (supplemental Figure 1B, supplemental Tables 3-5). None of the above genes were expressed in DLBCL cell lines (Figure 2A), strongly suggesting they derived from nonlymphoma cells. Patients with DLBCL whose specimens show expression of these microenvironmental cell-related transcripts above the cutoff values (supplemental Figure 3B) exhibit a significantly (P < .005) more favorable prognosis in DFS (Figure 2B) and OS (supplemental Figure 3C).
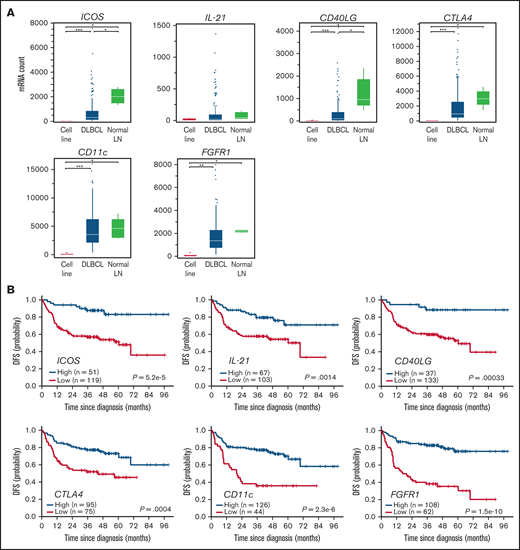
Clinical impact of extracted favorable prognostic genes in the training cohort. (A) Expression of 6 favorable prognostic genes in DLBCL cell lines (n = 6), primary DLBCL specimens (n = 170), and normal LNs (n = 5), as shown in a Box and Whisker plot (dots show outliers). Cell lines analyzed were RC-K8, SU-DHL-1, SU-DHL-4, SU-DHL-9, Karpas-422, and OCI-Ly-3. Lymphoma lines showed no or little candidate gene expression, suggesting genes are expressed in microenvironmental cells. Significance was determined using the Steel-Dwass test (*P < .05, **P < .005, and ***P < .0005). (B) Kaplan-Meier curves show the duration of DFS based on expression levels of favorable candidate genes. P values were calculated by log-rank test.
Clinical impact of extracted favorable prognostic genes in the training cohort. (A) Expression of 6 favorable prognostic genes in DLBCL cell lines (n = 6), primary DLBCL specimens (n = 170), and normal LNs (n = 5), as shown in a Box and Whisker plot (dots show outliers). Cell lines analyzed were RC-K8, SU-DHL-1, SU-DHL-4, SU-DHL-9, Karpas-422, and OCI-Ly-3. Lymphoma lines showed no or little candidate gene expression, suggesting genes are expressed in microenvironmental cells. Significance was determined using the Steel-Dwass test (*P < .05, **P < .005, and ***P < .0005). (B) Kaplan-Meier curves show the duration of DFS based on expression levels of favorable candidate genes. P values were calculated by log-rank test.
Favorable prognostic genes derive from specific lymphoma microenvironment cells
We next used multispectral fluorescence imaging to identify the origin of favorable prognostic genes in the DLBCL tissues. The infiltrating CD3+ ICOS+ follicular T cells (double-stained by cyan and green in Figure 3A, left) scattered around CD20+ lymphoma cells (red) in DLBCL tissues. Most CD3+ T cells (green) express ICOS protein (cyan) in disease-free cases, while not in relapsed cases. Note that the frequencies of CD20+ lymphoma B cells and overall CD3+ T cells were comparable between them. The number of follicular T cells in tissues from disease-free patients exceeded that in tissues from relapsed patients (P = .01, Figure 3A, right). Most CD11c (cyan) positive cells coexpressed pan-macrophage marker CD68 (yellow), but not CD3 or CD20, and exhibited a “dendritic” morphology (Figure 3B). CD11c+ CD68+ DC/macrophages were also enriched in samples from disease-free patients (P = .02), while CD68+ DC/macrophages did not express CD11c in relapsed case. In contrast, the expression of FGFR1, which is usually expressed by interstitial fibroblast or stromal cells,31 did not merge with any lineage-specific markers such as CD3, CD11c, CD20, or CD68, suggesting that they are tertiary microenvironment components (Figure 3C). We also performed multispectral imaging in normal second lymphoid tissues to assess ICOS, CD11c, and FGFR1 protein levels. Interestingly, all 3 proteins were expressed reactive LNs, preferentially in the GC region, but not in steady-state B-cell follicles (Figure 3D, supplemental Figure 3D). Overall, these observations support the idea that the presence in DLBCL specimens of specific microenvironmental cells seen in the normal GC is associated with favorable clinical outcome.12,32
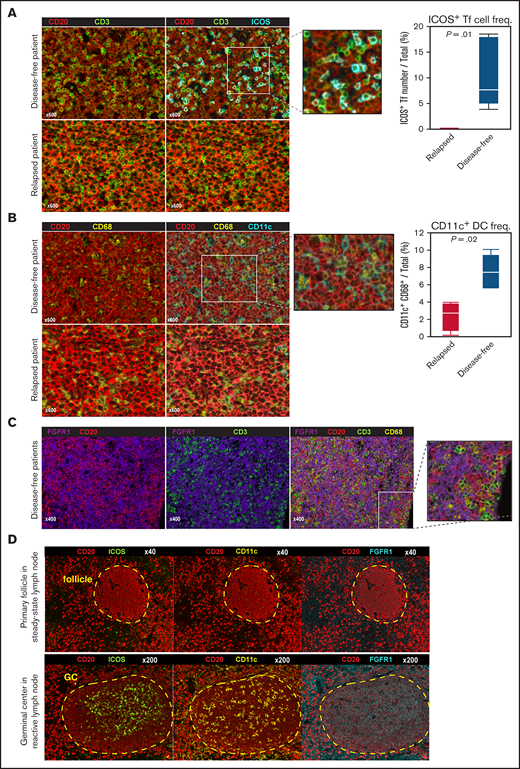
Identification of microenvironmental cell subtypes by single-cell expression profiling and imaging analysis. (A) Left, detection of follicular T (Tf) cells in DLBCL tissue based on indicated markers using multiplexed immunofluorescence imaging analysis and the Mantra system. Representative images taken from 10 patient samples analyzed are shown. We assigned each fluorescent signal to the preset pseudocolor (CD20: red; CD3: green; ICOS: cyan). Most CD3+ T cells (green) express ICOS protein (cyan) in disease-free cases, while not in relapsed cases. Note that the frequencies of CD20+ lymphoma B cells (red) and overall CD3+ T cells (green) were comparable between them. Right, Box and Whisker plot (dots indicate outliers) shows frequency of ICOS+ Tf cells, which were digitally counted in 5 fields. P values were calculated using the Wilcoxon rank-sum test. (B) Left, comparable imaging identified CD68+ DCs/macrophages (yellow) expressed CD11c (cyan) in favorable prognostic case. Note that CD68+ DC/macrophages (yellow) did not express CD11c in relapsed cases. Right, the proportion of CD11c+ CD68+ cells among total cells in 5 fields, shown as a Box and Whisker plot (dots indicate outliers), in relapsed vs disease-free cases, as calculated by the Wilcoxon rank-sum test. (C) Characterization of FGFR1+ cells (purple) based on staining with the lineage-specific markers CD3 (T cells), CD68 (DC/macrophages), and CD20 (B cells). (D) Multispectral imaging shows steady-state LN (upper) and reactive LN (lower) specimens stained with indicated markers. ICOS, CD11c, and FGFR1 are enriched during GC formation.
Identification of microenvironmental cell subtypes by single-cell expression profiling and imaging analysis. (A) Left, detection of follicular T (Tf) cells in DLBCL tissue based on indicated markers using multiplexed immunofluorescence imaging analysis and the Mantra system. Representative images taken from 10 patient samples analyzed are shown. We assigned each fluorescent signal to the preset pseudocolor (CD20: red; CD3: green; ICOS: cyan). Most CD3+ T cells (green) express ICOS protein (cyan) in disease-free cases, while not in relapsed cases. Note that the frequencies of CD20+ lymphoma B cells (red) and overall CD3+ T cells (green) were comparable between them. Right, Box and Whisker plot (dots indicate outliers) shows frequency of ICOS+ Tf cells, which were digitally counted in 5 fields. P values were calculated using the Wilcoxon rank-sum test. (B) Left, comparable imaging identified CD68+ DCs/macrophages (yellow) expressed CD11c (cyan) in favorable prognostic case. Note that CD68+ DC/macrophages (yellow) did not express CD11c in relapsed cases. Right, the proportion of CD11c+ CD68+ cells among total cells in 5 fields, shown as a Box and Whisker plot (dots indicate outliers), in relapsed vs disease-free cases, as calculated by the Wilcoxon rank-sum test. (C) Characterization of FGFR1+ cells (purple) based on staining with the lineage-specific markers CD3 (T cells), CD68 (DC/macrophages), and CD20 (B cells). (D) Multispectral imaging shows steady-state LN (upper) and reactive LN (lower) specimens stained with indicated markers. ICOS, CD11c, and FGFR1 are enriched during GC formation.
Immune microenvironmental signature dictates DLBCL prognosis independently of existing stratification models
ICOS, CD11c, or FGFR1 mRNAs were the top-ranked predictors of favorable prognosis with the significant statistical power and fold-changes (Figure 1C) and also with the smallest P values in the log-rank test (Figure 2B) and were representative of 3 different microenvironment components: follicular T cells, DC/macrophages, and stromal cells, respectively. Thus, we developed a simple scoring system based on the expression status of GC-associated microenvironmental signature that we call DMS. The DMS score was calculated based on ICOS, CD11c, and FGFR1 expression level of the tissue, as described in the supplemental Methods (Figure 4A). We then constructed Kaplan-Meier curves for DFS of patients segregated by DMS score (Figure 4B). Patients with DMS scores of 3, 2, 1, and 0 points exhibited statistically significant differences in probability of 3-year DFS survival of 0.903 (P = 1.2e-10, 95% confidence interval [CI], 0.818-0.998), 0.807 (95% CI, 0.710-0.917), 0.514 (95% CI, 0.375-0.703), and 0.246 (95% CI, 0.131-0.462), respectively. These significant differences were evident even among patients with extranodal tissues where microenvironmental cells must migrate (P = 2.1e-5, Figure 4C). The DMS score also predicted OS in nodal and extranodal cases (supplemental Figure 4A). Patients of GCB or non-GCB types based on the Hans COO classification were distributed equally among different DMS scores (P = .6627, Figure 4D, left). In contrast, based on Lymph2Cx analysis, which is a refined COO model based on the nCounter assay,23 GCB and ABC types more frequently corresponded to DMS-high and DMS-low cases, respectively (Figure 4E, left). Importantly, the DMS score could stratify patients with different prognoses within each COO type (Figure 4D,E), suggesting that the DMS score is a prognostic indicator independent of COO. Patients with high or low IPI were also distributed equally into groups with different DMS scores (P = .0548). In both IPI low and high groups, higher DMS scores marked patients with better outcomes (P = 6.5e-3 and 2.1e-4, Figure 4F). Importantly, the DMS score could extract patients with favorable prognoses even in the high IPI group. Furthermore, while patients who received R-THPCOP showed an inferior overall survival than those treated with R-CHOP (supplemental Figure 4B), the DMS score successfully stratified patients’ clinical outcomes regardless of the therapeutic regimen used (supplemental Figure 4C).
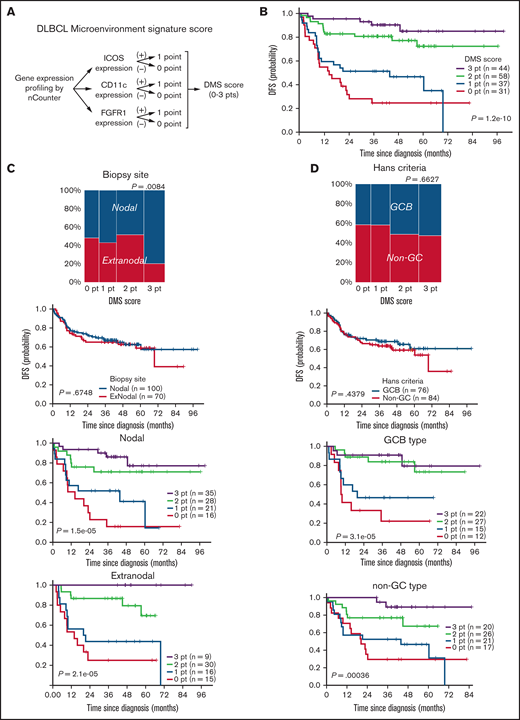
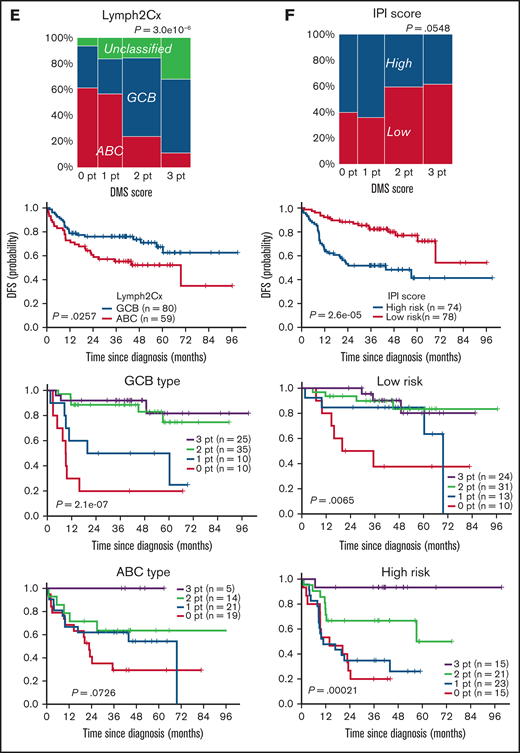
Clinical impact of DLBCL microenvironment-based stratification model. (A) Schematic representation showing the scoring system used to calculate DMS score. (B) Kaplan-Meier DFS curve based on DMS score. (C-F) Mosaic plots showing correlation of DMS score with disease site and canonical prognostic models: (C) disease site (n = 170), (D) Hans criteria (n = 160), (E) Lymph2Cx (n = 170), (F) IPI score (n = 152). The correlation was calculated using the Fisher's exact test. DMS-low cases were enriched in ABC-type DLBCL, based on Lymph2Cx, and vice versa, but we observed no correlation with Hans criteria or IPI score. Shown are Kaplan-Meier DFS curves based on disease site, Hans criteria, Lymph2Cx (31 unclassified cases were excluded), and IPI in all cases and by DMS score in each subgroup. The DMS score had prognostic value in all subtypes, based on these classifications. A log-rank test was used for survival analysis.
Clinical impact of DLBCL microenvironment-based stratification model. (A) Schematic representation showing the scoring system used to calculate DMS score. (B) Kaplan-Meier DFS curve based on DMS score. (C-F) Mosaic plots showing correlation of DMS score with disease site and canonical prognostic models: (C) disease site (n = 170), (D) Hans criteria (n = 160), (E) Lymph2Cx (n = 170), (F) IPI score (n = 152). The correlation was calculated using the Fisher's exact test. DMS-low cases were enriched in ABC-type DLBCL, based on Lymph2Cx, and vice versa, but we observed no correlation with Hans criteria or IPI score. Shown are Kaplan-Meier DFS curves based on disease site, Hans criteria, Lymph2Cx (31 unclassified cases were excluded), and IPI in all cases and by DMS score in each subgroup. The DMS score had prognostic value in all subtypes, based on these classifications. A log-rank test was used for survival analysis.
The independent cohorts validate the prognostic impact of DMS
We next validated the prognostic value of the DMS score using an independent cohort of 80 de novo DLBCL cases (Figure 1A, supplemental Table 1). As expected, the DMS scores defined by the same cutoff values as the training cohort successfully delineated clinical outcomes of patients receiving R-CHOP therapy: patients with different DMS scores exhibited statistically significant differences in the probability of DFS (P = 2.1e-5, Figure 5A) and OS (P = 2.1e-5, Figure 5B). Multivariate analysis revealed that the DMS score is a prognostic indicator independent of IPI criteria and Lymph2Cx (P = .0283, supplemental Table 8). We also validated the DMS score using publicly available RNA-seq datasets from 2 large studies that analyzed GEP and mutation profiling, composed of 229 and 604 DLBCL cases (Figure 1A).10,33 Patients with DLBCL whose specimens showed high expression of DMS-related transcripts exhibited a significantly better prognosis (supplemental Figure 5A,B). As expected, DMS scores, which were calculated using RNA-seq data values, stratified patients’ clinical outcomes in both cohorts (P = 8.9e-11 and 3.5e-7, Figure 5C,D). Of note, cutoff values were determined using maximally selected rank statistics (supplemental Figures 5C,D).16 The DMS score appeared to be independent of genetically defined prognostic factors identified by these original studies: low (0 pt) and high (3 pt) DMS scores were highly predictive in most subgroups (supplemental Figure 5E,F). Of note, the DMS score stratified the “genetically unclassifiable group,” which consists of more than 50% of reported cases33 into clinically distinct subgroups (P = .0026, supplemental Figure 5E). Furthermore, multivariate analysis revealed independence of the DMS score from these prognostic factors (P = 2.18e-4 and 1.26e-4, supplemental Tables 9 and 10). To assess potential overfitting in our model, we next applied a cruder method (median-based cutoff calculation) to define more optimized scores. Importantly, the optimized DMS scores also stratified patients’ clinical outcomes, attesting to the validity of our model (supplemental Figure 5G,H). We next assessed the impact of adding MYC, an anticorrelated gene (Figure 1C), to the DMS scoring system. While the overall trend did not significantly change, the addition of MYC further stratified the cases with low DMS scores (supplemental Figure 5I,J).
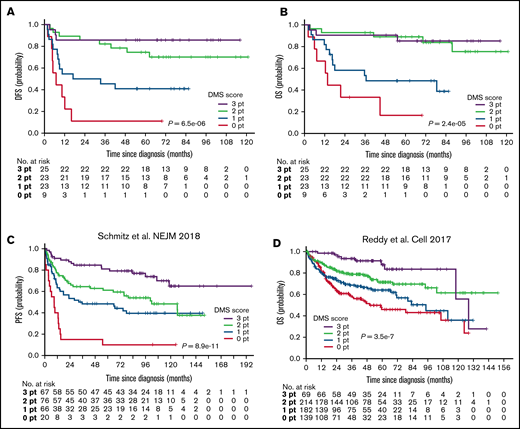
Validation of DMS score using independent cohort and 2 publicly available data sets. (A,B) Kaplan-Meier analyses of (A) DFS and (B) OS based on DMS score using a validation cohort. A log-rank test was used for survival analysis. (C,D) Kaplan-Meier analyses of (C) PFS in Schmitz cohort and (D) OS in Reddy cohort based on DMS score. A log-rank test was used for survival analysis.
Validation of DMS score using independent cohort and 2 publicly available data sets. (A,B) Kaplan-Meier analyses of (A) DFS and (B) OS based on DMS score using a validation cohort. A log-rank test was used for survival analysis. (C,D) Kaplan-Meier analyses of (C) PFS in Schmitz cohort and (D) OS in Reddy cohort based on DMS score. A log-rank test was used for survival analysis.
Multiomics analysis reveals molecular background for the prognostic impact of DMS
Finally, we tried to understand the molecular backgrounds for the predictive power of the DMS. For this purpose, we evaluated single nucleotide variants (SNVs), short insertion/deletions (indels), and copy number alterations (CNAs) in 106 DLBCL specimens from the training cohort and investigated the correlation between the DMS score and the genetic alterations serving as lymphoma cell hallmarks. The most frequently mutated gene was KMT2D (MLL2), followed by PIM1, BCL6, BCL2, CDKN2A, and TP53 (supplemental Figure 6A,B), in agreement with previous reports by us34 and others.35-39 Figure 6A summarizes the results of mutation analysis sorted by DMS score, plus GEP, COO classification, and “double-expressor” lymphoma (DEL) and “double-hit” lymphoma (DHL) status in 170 DLBCL patients of the training cohort.
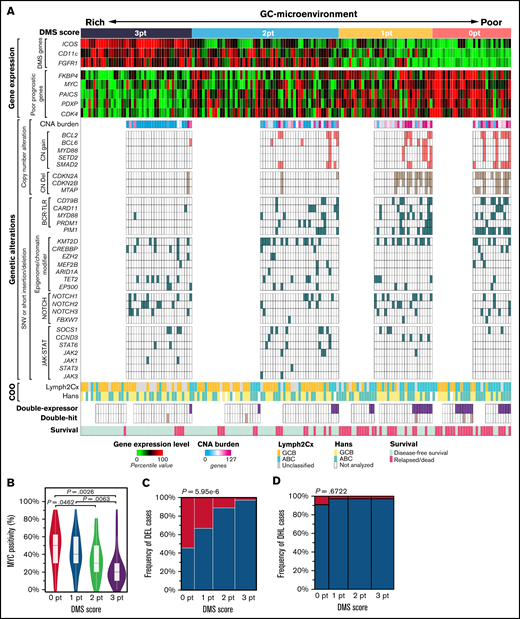
The relationship between the GC-microenvironmental signature and multiomics prognostic factors. (A) Integrated visualization of transcriptome data from nCounter analysis, genetic alterations, and COO classification combined with DMS score in 170 DLBCL cases. DMS scores were calculated based on enrichment of ICOS (follicular T cells), CD11c (DCs/macrophages), and FGFR1 (stromal cells). Gene expression levels were shown as percentile of the normalized expression values in each gene. CNA burden was represented by the number of genes involved in the CNA region. Tumor activity-related signatures, such as expression levels of proliferation-related genes or the number of genes showing CNA and BCR-TLR signaling mutations, were enriched as the DMS score decreased. (B) The difference of MYC protein expression by DMS score. MYC protein positivity was analyzed by IHC. P values were calculated using the Steel-Dwass test. (C) MYC and BCL2 protein levels were assessed by immunohistochemistry, and cases positive for both MYC and BCL2 were defined as DEL. DELs (magenta) exhibited low DMS scores. Fisher’s exact test was performed to calculate P value. (D) The frequency of DHL by each DMS score is shown. Fisher’s exact test was performed to calculate P value.
The relationship between the GC-microenvironmental signature and multiomics prognostic factors. (A) Integrated visualization of transcriptome data from nCounter analysis, genetic alterations, and COO classification combined with DMS score in 170 DLBCL cases. DMS scores were calculated based on enrichment of ICOS (follicular T cells), CD11c (DCs/macrophages), and FGFR1 (stromal cells). Gene expression levels were shown as percentile of the normalized expression values in each gene. CNA burden was represented by the number of genes involved in the CNA region. Tumor activity-related signatures, such as expression levels of proliferation-related genes or the number of genes showing CNA and BCR-TLR signaling mutations, were enriched as the DMS score decreased. (B) The difference of MYC protein expression by DMS score. MYC protein positivity was analyzed by IHC. P values were calculated using the Steel-Dwass test. (C) MYC and BCL2 protein levels were assessed by immunohistochemistry, and cases positive for both MYC and BCL2 were defined as DEL. DELs (magenta) exhibited low DMS scores. Fisher’s exact test was performed to calculate P value. (D) The frequency of DHL by each DMS score is shown. Fisher’s exact test was performed to calculate P value.
Expression of genes associated with poor prognosis, including cell cycle-related genes FKBP4, MYC, PAICS, PDXP, and CDK4 (see also supplemental Figure 2), became higher as DMS scores decreased (Figure 6A, upper). The anticorrelation between MYC protein levels and DMS score was also evident (Figure 6B). CNAs are dramatic structural changes in a genomic region (>1 kbp) and may involve multiple genes and alter global gene expression. Cases with a DMS score of 3 were almost devoid of CNAs, and the overall CNA burden (the number of genes with CNA) increased in low DMS cases (Figure 6A, middle). CNAs related to poor prognoses, such as CN gains of BCL240 and CN deletions in the tumor suppressor CDKN2A and the adjacent MTAP gene at the chromosome 9p21 locus41,42 were also more frequent in low relative to high DMS cases (Figure 6A, middle). Also, SNVs and short indels in BCR and Toll-like receptor (TLR) signaling genes and in the NF-kB regulator PIM143 were more frequent in DMS-low cases, with statistical significance (Figure 6A, middle). These mutations, as well as CN gains of MYD88 and adjacent SETD2, promote constitutive activation of antiapoptotic and proliferative NF-kB pathways and contribute to poor prognosis.10,34,43 -45 On the other hand, other major somatic mutations, including epigenome/chromatin modifiers, NOTCH- and JAK-STAT signaling pathways, were equally distributed among all DMS score groups.
We further assessed the DMS score in DEL and DHL, both of which reportedly exhibit poor prognoses.46 Among 123 DLBCL cases analyzed, 27 (22.0%) and 5 (4.1%, Figure 6A, lower) were identified as DEL and DHL, respectively. DEL were significantly enriched in DMS-low cases (Figure 6A,C). DHL cases appeared to be preferentially distributed in DMS-low cases, but without statistical significance, due to the small sample size (Figure 6A [lower], 6D). We also validated these results using the external cohort of 604 DLBCL cases.10 As expected, low DMS score was associated with high mRNA levels of genes related to poor prognosis (supplemental Figure 7A), presence of CNAs (supplemental Figure 7B), ABC phenotype (supplemental Figure 7C), DEL phenotype (supplemental Figure 7D) and MYD88 and PIM1 mutations (supplemental Figure 7E), as observed in our cohort (Figure 6). By contrast, no association was observed between DMS score and the DHL phenotype (supplemental Figure 7F). Of note, the genetic risk model defined by Reddy et al was highly correlated with the DMS score (supplemental Figure 7G).
Thus, lymphoma cell-intrinsic aggressiveness of DLBCL, which is characterized by proliferation-related gene expression, high frequency of specific genomic alterations, COO, and DEL status, is associated with loss of GC-microenvironment components as reflected by low DMS score. A GC-associated microenvironment comprehensively reflects multiomics’ unfavorable molecular signatures.
Discussion
GC formation is an essential step for normal B-cell development in LNs and requires the recruitment of microenvironmental cells. Here, by employing accurate GEP, we demonstrated that GC-associated microenvironmental components significantly impacted the clinical outcomes of DLBCL and established a DMS prognostic model. The DMS score successfully provided graded stratification of patients’ clinical responses to standard R-CHOP-based therapy. Moreover, DMS scoring had significant prognostic value even in patients’ groups stratified by either COO or the IPI score. These results suggest that the interaction of lymphoma cells with their microenvironment governs DLBCL malignancy.
RNA-seq is a robust transcriptome profiling method; however, data reproducibility of lowly-expressed genes is generally poor. Furthermore, constructing a high-quality library using FFPE-derived fragmented RNA is technically challenging. By contrast, the nCounter system is highly sensitive enough to quantify low abundance microenvironmental transcripts even from fragmented RNAs in FFPE samples.47,48 Therefore, observations through this system might highlight the subtle differences in microenvironmental immune cells among DLBCL tissues. In fact, we observed less differentially expressed genes (DEGs) in RNA-seq (51 genes) than in nCounter-based measurements (192 genes) (Figure 1A). A previous study showed that microenvironmental cells function in DLBCL pathogenesis: GEP of DLBCL tissues revealed positive associations between extracellular matrix (ECM) deposition by stromal cells and favorable prognosis (“stromal-1” signature) and tumor blood-vessel density and poor prognosis (“stromal-2” signature).49 These findings were validated by the recent study50 in which prognostic significance of the M2 macrophage signature was proposed. Ciavarella et al demonstrated a strong correlation between DLBCL prognosis and microenvironmental components such as myofibroblasts, DCs, and CD4+ T cells via GEP using a customized gene panel with the NanoString platform.51 Their results were validated in silico using the data from the Lenz et al study.49,51 More recently, Kotlov et al performed GEP using 4655 DLBCL samples and identified 4 types of lymphoma microenvironment signatures: “GC-like,” “mesenchymal,” “inflammatory,” and “depleted.”52 Tripodo et al proposed that GC-related aggressive B-cell lymphoma can be subdivided based on the dark- and light-zone microenvironment signatures using a digital spatial profiling method.53 Results from these studies strongly support our observations regarding the impact of DLBCL microenvironmental signature on clinical outcomes. In this study, we further uncover the characteristics of the prognostic microenvironment components and their correlation with multifaceted features that explained the malignant activity of DLBCL.
Our nCounter data suggest follicular T cells and DC/macrophages as critical microenvironment components in determining DLBCL prognosis. In the normal reactive GC, follicular T cells patrol the B-cell region to participate in B-cell selection via the BCR, and their activity determines whether B cells undergo apoptosis or proliferation.54,55 Recent studies indicate 2 subsets of follicular T cells: follicular helper T (Tfh) cells promoting survival, proliferation, and class switching of developing B cells,56 and FOXP3-positive follicular regulatory T (Tfr) cells, which function in suppression and elimination of inappropriate B-cell clones.30 The fact that specific markers for Tfh (IL-21) and Tfr (FOXP3 and CTLA4) were of the most significant prognostic genes (Figure 2B) suggests that the abundance of follicular T cells in DLBCL tissues is a strong indicator of a favorable prognosis. In addition to follicular T cells, CD11c+ DC/macrophages play a critical role in clearing apoptotic B cells32 after selection by follicular T cells and participate in antigen-presentation to developing B cells in normal GC. In our study, CD11c+ DC/macrophages were also enriched in primary DLBCL tissues from patients with favorable prognoses.
Given that the DMS score was calculated based on 3 independent GC-associated microenvironmental cells, these cells may cooperate to regulate DLBCL development or maintain DLBCL cells, comparable to their activity in the normal GC. Importantly, the DMS score stratified patients not only in cases of nodal DLBCL but also in those showing extranodal lesions (Figure 4C), suggesting that in the latter, the GC-associated microenvironment is reconstituted through recruitment of these cells into lymphoma tissue.
The nCounter probe set used in the pilot study broadly covers cancer and immune-related genes (1889 genes), enabling us to capture gene expression signatures associated with the DLBCL microenvironment with high precision. Given its high sensitivity and reproducibility in measuring fragmented RNAs from FFPE samples, a more unbiased analysis targeting all transcripts, including noncoding RNAs, may further elucidate DLBCL biology relevant to clinical practice. We noticed that the predictive power of DMS scores 1 and 2 is not as high as that seen in scores 0 and 3 in subgroup analyses (Figure 4C-F, supplemental Figure 5E,F). Although we don't know exactly what causes these results, a few plausible explanations come to mind. Since the DMS score recapitulates microenvironmental signatures of DLBCL tissues, the score may not necessarily reflect lymphoma cell-intrinsic signatures, such as mutational status (supplemental Figure 5E,F), and/or patients’ general characteristics, such as age (used for IPI calculation). It is also possible that the low number of cases in some subgroups could have caused inconsistent results (supplemental Figure 5E).
We believe that the simplicity of the DMS model is beneficial in terms of clinical utility; however, improvements in analytical methods and additional validation studies are necessary prior to clinical use. For example, adding more genes, such as those positively correlated with poor prognosis (such as FKBP4 and MYC) or those related to COO, may enhance the accuracy and reproducibility of the model. While an nCounter-based assay is used in the Prosigna Breast Cancer Gene Signature Assay, an FDA-approved clinical diagnostic test, because of its high sensitivity and reproducibility, our system, including analytical parameters, should be prospectively validated in a clinical trial setting.
Why does a high DMS score reflect a favorable prognosis in DLBCL? One assumption is that lymphoma cells in favorable cases are regulated by GC-associated microenvironmental cells comparably to normally developing B cells, and once they acquire cell-autonomous proliferation or survival properties through additional genetic events, they become microenvironment-independent. This hypothesis is supported by the fact that DMS-low cases tend to accumulate lymphoma cell-intrinsic abnormalities, including expression of c-Myc-related or cell cycle-related molecules, and genomic alterations, all related to unfavorable prognosis (Figure 6A).
In summary, our study shows that the GC-associated microenvironmental signature is tightly associated with clinical outcomes in DLBCL patients. These data strongly suggest that the evaluation of microenvironment components and their functions is the key to understanding DLBCL pathogenesis and malignancy. We believe that the DMS scoring system could help clinicians at multiple levels. First, it enables more accurate prognostic stratification when combined with the IPI Index (Figure 4F). Second, the DMS score could guide clinicians to choose better therapeutic options. For instance, while clinical trials are generally recommended for high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with DLBCL morphology (NCCN Guidelines v4.2021), those patients with high DMS scores may benefit from standard R-CHOP-based regimens. Thus, combining the DMS score system with current genomics- and/or COO-based systems enable precision medicine for DLBCL patients. Finally, the DMS score system is simple (based on the expression of 3 genes), fast, and easy to implement in a clinic.
Acknowledgments
The authors thank all laboratory members and especially Masao Seto (Kurume University) and Shinya Rai (Kindai University) for valuable discussions, Joji Shimono for collecting clinical information, Hiroshi Arima, Momoko Nishikori, and Akifumi Takaori-Kondo for A-seq data analysis, and Elise Lamar for proofreading of the manuscript.
This work was supported by Japan Agency for Medical Research and Development (AMED) under grant numbers (no.) JP17ck0106163, JP17cm0106507 (K.A.) and JP18ck0106196 (Y. Kikushige), and by Japan Society for the Promotion of Science (JSPS) under a Grant-in-Aid for Scientific Research (B) (T. Miyamoto, no. 16H05340), a Grant-in-Aid for Scientific Research (S) (K.A., no. 16H06391), a Grant-in-Aid for Challenging Exploratory Research (K.A., no. 18K19565), a Grant-in-Aid for Scientific Research (A) (17H01567, 20H00540), AMED under grant number 18063889 and a Grant-in-Aid for Scientific Research (S) (20H05699) (to T. Maeda), Grant-in-Aid for JSPS Research Fellow (K.M.), and a Grant-in-Aid for Young Scientists (K.M., no. 18K16120). This work was also supported in part by Takeda Science Foundation, The Shinnihon Foundation of Advanced Medical Treatment Research, and the Social Medical Corporation of the ChiyuKai Foundation.
Authorship
Contribution: K.M., T. Maeda, and K.A. designed experiments and wrote the manuscript; K.M., T.S., K.K., K.S., H.M., Y.S., Y. Kikushige, Y. Kunisaki, and F.K. performed experiments and analyzed data; and T. Maeda, T. Miyamoto, H.I., J.A., Y.M., and K.O. reviewed the data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Koichi Akashi, Department of Medicine and Biosystemic Science, Kyushu University Graduate School of Medical Sciences, 3-1-1, Maidashi, Higashi-Ku, Fukuoka 812-8582, Japan; e-mail: akashi@med.kyushu-u.ac.jp

