

Abstract
Activating mutations of MPL exon 10 have been described in a minority of patients with idiopathic myelofibrosis (IMF) or essential thrombocythemia (ET), but their prevalence and clinical significance are unclear. Here we demonstrate that MPL mutations outside exon 10 are uncommon in platelet cDNA and identify 4 different exon 10 mutations in granulocyte DNA from a retrospective cohort of 200 patients with ET or IMF. Allele-specific polymerase chain reaction was then used to genotype 776 samples from patients with ET entered into the PT-1 studies. MPL mutations were identified in 8.5% of JAK2 V617F− patients and a single V617F+ patient. Patients carrying the W515K allele had a significantly higher allele burden than did those with the W515L allele, suggesting a functional difference between the 2 variants. Compared with V617F+ ET patients, those with MPL mutations displayed lower hemoglobin and higher platelet levels at diagnosis, higher serum erythropoietin levels, endogenous megakaryocytic but not erythroid colony growth, and reduced bone marrow erythroid and overall cellularity. Compared with V617F− patients, those with MPL mutations were older with reduced bone marrow cellularity but could not be identified as a discrete clinicopathologic subgroup. MPL mutations lacked prognostic significance with respect to thrombosis, major hemorrhage, myelofibrotic transformation or survival.
Introduction
The myeloproliferative disorders, comprising essential thrombocythemia (ET), polycythemia vera (PV) and idiopathic myelofibrosis (IMF), are hematopoietic stem cell disorders characterized by the overproduction of one or more mature myeloid lineages. Although the clonal nature of these disorders was recognized some 3 decades ago,1 their molecular basis remained obscure until the identification of the activating JAK2 V617F mutation.2-5 The vast majority of patients with PV harbor a mutation in JAK2, with JAK2 V617F seen in around 97%3 and mutations in JAK2 exon 12 found in many of the remainder.6-8 PV patients with JAK2 exon 12 mutations differ from those with JAK2 V617F, presenting at a younger age with higher hemoglobin levels, lower platelet counts, and lower white cell counts at diagnosis.6 The JAK2 V617F mutation is present in around half of patients with ET and is associated with features resembling a “forme-fruste” of PV, including increased erythropoiesis, increased granulopoiesis, and increased rates of venous thrombosis compared with the JAK2 V617F− group.9-12 The JAK2 V617F mutation is also present in around half of patients with IMF, and is associated with higher neutrophil and platelet counts at diagnosis, reduced likelihood of transfusion dependence, and probably a poorer survival compared with the JAK2 V617F− group.13-15
Mutations in the juxtamembrane region of the thrombopoietin receptor MPL have recently been described in IMF and as a rare occurrence in ET.16,17 Biochemical and cell line studies indicate an autoinhibitory role for this region, with disruption leading to receptor activation in the absence of thrombopoietin binding.18 Expression of the MPL W515L allele resulted in cytokine-independent growth of 32D, UT7, and BaF3 cell lines, together with constitutive phosphorylation of JAK2, STAT3, STAT5, AKT and ERK.16 Transplantation of mice with bone marrow expressing the MPL W515L allele resulted in a myeloproliferative disorder (MPD)–like disease characterized by marked thrombocytosis, splenomegaly, splenic infarction, and reduced life expectancy.16 MPL mutations in IMF patients have been associated with lower hemoglobin levels at diagnosis and increased risk of transfusion dependence compared with both JAK2 V617F+ and JAK2 V617F− patients.19 Mutations in MPL have been reported in a small minority of patients with ET17 but their clinical significance remains unclear. Here we describe the prevalence of MPL mutations in a retrospective cohort of unselected patients with ET or IMF, and also report the clinical and laboratory features associated with MPL mutations in the large prospective Primary Thrombocythaemia 1 (PT-1) cohort.
Methods
Retrospective cohort: patients and samples
Patients aged 18 years or over, who met the Polycythaemia Vera Study Group (PVSG) criteria for either ET or IMF20 were recruited from MPD clinics in Cambridge, London, Sheffield, Birmingham, and Odense. Institutional and multiregion Ethics Committee approval was obtained and the study was carried out in accordance with the principals of the Declaration of Helsinki.
Granulocytes were prepared by centrifugation of whole blood through a Ficoll density gradient, and T cells were isolated from the mononuclear cell layer by anti-CD2 magnetic beads (DynaBeads; Invitrogen, Paisley, United Kingdom). Mean purity was greater than 95% for granulocytes and 91% for T cells. Platelets were isolated from whole blood by 3 rounds of centrifugation at 150g for 20 minutes at room temperature, followed by negative selection of CD45+ cells using magnetic beads (DynaBeads, Invitrogen). RNA was prepared from TRI reagent (Sigma-Aldrich, Gillingham, United Kingdom) as per the manufacturer's instructions. cDNA was synthesised using M-MLV (Invitrogen) as per the manufacturer's instructions.
PT-1 study: patients and samples
Newly diagnosed and previously treated patients, aged 18 years or over, who met the Polycythaemia Vera Study Group (PVSG) criteria for ET,20 were recruited into one of 3 multicenter PT-1 studies: the Medical Research Council high-risk trial, in which high-risk patients were randomly assigned to either hydroxyurea plus aspirin or to anagrelide plus aspirin21 ; the National Cancer Research Institute intermediate-risk study, a randomization between aspirin alone or hydroxyurea plus aspirin; or the National Cancer Research Institute low-risk study, a prospective observational study of low-risk patients given aspirin alone. Patients entered a higher risk study if they developed appropriate features. The study protocol was approved by institutional ethics committees in all participating centers, and written informed consent was obtained in accordance with the Declaration of Helsinki from all patients. This study is registered at http://isrctn.org as #72251782 and at http://eudract.emea.europa.eu/ as #2004-000245-38. Further information regarding the trial can be accessed at http://www.ctsu.ox.ac.uk/projects/leuk/pt1.
Details obtained at trial entry included diagnostic features such as blood counts, cytogenetics, and clinical complications at or preceding diagnosis. Follow-up forms were completed every year by the patient's clinician, documenting medications, blood counts, and clinical events for which standard definitions were used.21 All data were collected prospectively with more than 99% of patients having complete follow-up. The median follow-up in this study was 36.5 months. Bone marrow trephines were independently reviewed by 3 hematopathologists who were aware of the patient's age and sex but unaware of JAK2 and MPL status, and scored for reticulin grade on a 0 to 4 scale, cellularity, megakaryocyte clustering, and atypical megakaryocyte nuclear morphology.22 Where there was disagreement, the mean reticulin score and the mode of other scores were used.
Samples of peripheral blood were requested at trial entry from all patients, and 776 samples were received from the 1022 patients entered. Whole-blood genomic DNA was extracted commercially (Whatman International, Ely, United Kingdom) and used for genotyping and mutant allele quantitation.
Mutation detection
JAK2 V617F mutation status was determined by allele-specific polymerase chain reaction (PCR) as previously described.3 MPL exon 10 screening was performed by direct sequencing of PCR products from peripheral blood granulocytes. Allele-specific PCR assays were developed for the MPL W515L, MPL W515K and MPL S505N alleles, and the sensitivity of each assay was established by mixing experiments using normal and mutant genomic DNA.
Mutant allele quantitation
Pyrosequencing assays were established for each MPL-mutant allele to quantitate mutant allele burden. Mixing experiments using cloned PCR products were performed for each assay, and the results used to plot a dilution curve from which the mutant allele burden was read (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). For the MPL S505N and MPL W515L alleles, the dilution curve was plotted against the allele proportion generated by the pyrosequencing software. As the software is unable to quantitate allele proportion where 2 or more adjacent bases are changed, MPL W515Ki and MPL W515Kii alleles were quantitated using the following formulas:
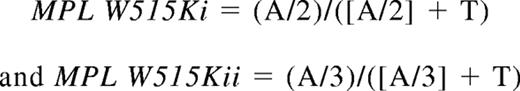
where A and T are the pyrosequencing peak heights for the mutant and wild-type bases, respectively (Figure S1).
Statistical analysis
Due to the large number of hypothesis tests performed in the paper, we elected to use a threshold of P less than .01 to define statistical significance, thus reducing the risk of false positive results. Pairwise univariate analyses comparing diagnostic variables between the MPL-mutant and either JAK2 V617F+ or JAK2 V617F− groups were performed using the t test for continuous variables, Fisher exact test for 2 × 2 tables, and Cochran-Armitage test for trend with exact P values for ordinal variables. Multivariate analyses for the associations of ordinal variables with mutation status, accounting for the effects of age, were performed using proportional odds logistic regression. Complication rates during follow-up were assessed using Kaplan-Meier life tables and log-rank analyses. Confidence intervals for odds ratios (OR) of complications in the year prior to diagnosis were calculated using the asymptotic formula 1/a + 1/b + 1/c + 1/d to estimate the variance of the log(OR). Mulitvariate survival analyses were performed using Cox proportional hazards models. S-plus v 7.0 (Insightful, Seattle, WA) was used for all statistical analyses, apart from the exact methods, for which SAS v9.0 (SAS, Cary, NC) was used.
Colony analysis
For erythroid colony analysis, mononuclear cells were plated at 105 cells/mL in Methocult (H4531; StemCell Technologies, Vancouver, BC) in the presence or absence of erythropoietin (2 U/mL). For megakaryocyte colony analysis, mononuclear cells were plated at 3 × 105/mL in Megacult (StemCell Technologies) in the presence or absence of cytokines (thrombopoietin 50 ng/mL, IL-3 10 ng/mL and IL-11 50 ng/mL). Cultures were incubated for 14 days at 37°C, high humidity, and 5% CO2. Individual erythroid colonies were placed in water and heated to 95°C for 8 minutes for DNA preparation. Single colonies were genotyped for the MPL W515L mutation by either pyrosequencing or direct sequencing. Colonies were classified as heterozygous if equal amounts of wild-type and mutant alleles were seen, and as wild-type or homozygous if only the wild-type or mutant allele was present, respectively. Whole megakaryocyte cultures were dehydrated and stained for GpIIb/IIIa expression following the manufacturer's instructions (StemCell Technologies).
Results
MPL mutations outside exon 10 are uncommon in ET patients negative for the JAK2 V617F mutation
To investigate the possibility that JAK2 V617F− patients might harbor MPL mutations other than those previously described,16,17 we sequenced the entire MPL coding region in 18 patients with ET and 2 patients with IMF. As previous studies have indicated that JAK2 V617F− ET patients have a more isolated megakaryocyte proliferation,9 platelet-derived cDNA was used for mutation screening. One patient with ET carried a mutation in MPL exon 10 (MPL W515L); all other base changes seen were previously reported single nucleotide polymorphisms (data not shown). These results indicate that mutations outside MPL exon 10 are not common in V617F− ET patients, although our data do not exclude the existence of such mutations in a small minority of patients. To identify different types of mutations within exon 10, we proceeded to sequence MPL exon 10 in granulocyte DNA from 200 patients. Of these, 88 had ET (47 V617F+ and 41 V617F−) and 112 had IMF (57 V617F+ and 55 V617F−). Mutations were identified in 11 patients. Two carried MPL S505N mutations (1 ET and 1 IMF), 7 carried MPL W515L mutations (2 ET and 5 IMF) and 2 carried MPL W515K mutations (both IMF; 1 W515Ki and 1 W515Kii). In this cohort, the prevalence of MPL mutations was 3.4% in ET and 7.1% in IMF. None of the MPL-mutant patients in this cohort had a coexisting JAK2 V617F mutation. Two different mutations resulting in the same MPL W515K amino acid substitution were observed (MPL W515Ki and MPL W515Kii, respectively, Figure 1A). The MPL S505N mutation has been described as an inherited mutation in a Japanese pedigree with familial thrombocythemia.23 However, in our ET patient, the majority of T cells (Figure 1A) and buccal cells (data not shown) did not carry the mutation, strongly suggesting that it was acquired in this individual.

Detection of MPL exon 10 mutations. (A) Sequence traces of the 4 MPL exon 10 mutant alleles, showing 2 different mutations leading to the same MPL W515K substitution (MPL W515Ki and MPL W515Kii), and MPL S505N as an acquired mutation. (B) Allele-specific PCR strategy showing the common forward and reverse intronic primers and the allele-specific primer within MPL exon 10. (C) Mixing experiments with normal and mutant DNA demonstrating the sensitivity of the allele-specific PCR assays used for mutation screening. The images were captured on a Gel Doc 200 imager (Bio-Rad, Hercules, CA) using Quantity One software (Bio-Rad). NA indicates T cell sample not available from this patient; Pt, patient sample; WT, wild-type; NTC, no template control.
Detection of MPL exon 10 mutations. (A) Sequence traces of the 4 MPL exon 10 mutant alleles, showing 2 different mutations leading to the same MPL W515K substitution (MPL W515Ki and MPL W515Kii), and MPL S505N as an acquired mutation. (B) Allele-specific PCR strategy showing the common forward and reverse intronic primers and the allele-specific primer within MPL exon 10. (C) Mixing experiments with normal and mutant DNA demonstrating the sensitivity of the allele-specific PCR assays used for mutation screening. The images were captured on a Gel Doc 200 imager (Bio-Rad, Hercules, CA) using Quantity One software (Bio-Rad). NA indicates T cell sample not available from this patient; Pt, patient sample; WT, wild-type; NTC, no template control.
Development of assays for the detection and quantitation of MPL exon 10 mutations
To investigate the clinical significance of MPL exon 10 mutations in patients with ET, we wished to assess the prevalence of these mutations in samples from patients entered into the PT-1 studies, for whom comprehensive diagnostic and prospectively acquired follow-up data are available. However, these samples were from unfractionated whole blood. Because JAK2 or MPL mutations are absent from or found at a low level in lymphocytes, and because a variable proportion of the granulocytes were also likely to be normal, it was important to develop sensitive assays for each mutant MPL allele. We therefore developed 3 separate allele-specific PCR assays to detect the MPL S505N, MPL W515L and MPL W515K alleles, with the W515K assay being able to detect both the W515Ki and W515Kii mutations (Figure 1B). To assess the sensitivity of the allele-specific PCR assays, we quantitated mutant allele burden in patient samples by pyrosequencing (see “Mutant allele quantitation” and Figure S1). Allele-specific PCR was then performed on dilutions corresponding to 1% to 9% mutant allele burden. As shown in Figure 1C, the assays for MPL S505N and MPL W515L could detect a mutant allele burden of approximately 1%, whereas the assay for MPL W515Ki and MPL W515Kii could detect a mutant allele burden of approximately 3% to 5%.
Clinical and laboratory features of MPL mutations in ET
Patients in the PT-1 cohort (n = 776) were genotyped for the MPL S505N, MPL W515L and MPL W515K mutations using allele-specific PCR. MPL mutations were detected in 32 patients, accounting for 4.1% of all ET patients (95% confidence interval [CI] 2.9%-5.8%, Table 1) and 8.5% of JAK2 V617F− ET patients. MPL W515L was the most common mutation, seen in 24 patients; 5 patients had MPL W515K (all MPL W515Ki allele) and 3 patients had MPL S505N mutations. In all patients with MPL W515K or MPL S505N alleles, the presence of the mutation was confirmed by both direct sequencing and pyrosequencing. Of the 24 MPL W515L mutations, 19 were detected by both direct sequencing and pyrosequencing, 3 were detected by pyrosequencing but not direct sequencing, and 2 were detected by allele-specific PCR alone. In these 2 patients, the presence of the mutation was confirmed by repeating the allele-specific PCR using an independent blood sample. Of the 32 MPL-mutant patients, one also carried the JAK2 V617F mutation, and is included in the MPL-mutant group in the statistical analysis. No patient was positive for more than one MPL-mutant allele.
Laboratory and clinical features of the MPL-mutant group were compared with both the JAK2 V617F+ and JAK2 V617F− groups, both of which lack MPL mutations. As shown in Table 1, MPL-mutant patients were significantly older at diagnosis than JAK2 V617F− patients (mean vs mean; P < .001). Compared with JAK2 V617F+ patients, MPL-mutant patients had lower hemoglobin and higher platelet levels at diagnosis (mean vs mean; P < .001 and mean vs mean; P = .006, respectively). There were no differences, however, in diagnostic blood counts between the MPL-mutant and JAK2 V617F− groups. There were also no significant differences in the presence of splenomegaly or bone marrow cytogenetic abnormalities between the groups.
Bone-marrow trephine biopsies at diagnosis were available from 311 patients, including 13 patients with MPL mutations, comprising 2 S505N, 2 W515K, and 9 W515L patients. These were assessed independently by 3 hematopathologists who were aware of the patients' age and sex but unaware of JAK2 or MPL mutation status (Table 2). Given the known association between age and bone marrow cellularity, we included patient age as a variable in the statistical analyses for cellularity. There were no differences in the mean reticulin grade, megakaryocyte cellularity, or the presence of megakaryocyte clusters and atypia between the MPL-mutant, JAK2 V617F+ and JAK2 V617F− groups. However, trephine biopsies from the MPL-mutant group were less cellular than both the JAK2 V617F+ and JAK2 V617F− groups (P < .001 [< .001 with age] and P = .005 [.003 with age], respectively). Compared with the JAK2 V617F+ group, both erythroid and granulocytic cellularity were reduced in the MPL-mutant group (P < .001 [< .001with age] and P = .009 [.02 with age], respectively). Compared with the JAK2 V617F− group, the MPL mutant group showed reduced erythroid cellularity (P = .005 [.004 with age]). Thus MPL-mutant patients exhibited a more isolated megakaryocytic proliferation at diagnosis, with a reduction in overall cellularity compared with both the JAK2 V617F+ and JAK2 V617F− groups. There was, however, considerable overlap between the histologic appearances observed in the 3 groups of patients, and our data indicate that MPL mutations as a whole do not define a distinct histologic subtype of ET. It remains formally possible that specific MPL mutations are associated with particular histologic features, but the number of patients with each individual MPL mutation was too small to address this issue.
Serum erythropoietin levels at trial entry in MPL mutation patients were significantly higher than JAK2 V617F+ but not JAK2 V617F− patients (mean vs mean; P = < .001 and P = .6, respectively). There were no significant differences in iron status between MPL-mutant patients and both comparator groups, as assessed by serum ferritin and erythrocyte mean cellular volume (Table 2).
Cytokine-independent colony formation is one of the hallmark features of the myeloproliferative disorders, with the presence of both endogenous megakaryocyte and erythroid colonies reported in patients carrying the JAK2 V617F mutation.3,24 To investigate the association of MPL mutations and cytokine-independent colony formation, colony assays were performed using peripheral blood from 2 hydroxyurea-treated ET patients, and an IMF patient receiving transfusion support only, all of whom carried the MPL W515L mutation, and all of whom had only heterozygous and wild-type erythroid colonies. These investigations demonstrated the presence of thrombopoietin-independent megakaryocyte colony growth in all 3 patients studied. In contrast to the JAK2 V617F mutation, however, we did not observe endogenous erythroid colonies in any of 5 patients with the MPL W515L mutation (4 ET, 1 IMF). These data confirm previous studies in IMF and extend these findings to patients with ET.25,26
Clinical outcome of patients with MPL mutations
As part of the PT-1 studies, comprehensive clinical and outcome data were collected prospectively. Clinical events were independently adjudicated according to predefined criteria by a panel of experts blinded to treatment allocation.21 The median follow-up was 36.5 months, although the relatively small numbers of patients in the MPL-mutant cohort meant that only a few end-point events were recorded in this group. There were no significant differences between the 3 comparator groups in rates of arterial thrombosis either before or after trial entry (Table 3); compared with JAK2 V617F− patients, MPL-mutant patients had a higher rate of venous thrombosis after trial entry (odds ratio 34.3, 95% CI: 1.6-725, P = .02). In view of the large number of hypothesis tests performed, we elected to use a threshold for statistical significance of P less than or equal to .01. Moreover, the association appeared weaker in multivariate analysis including patient age, sex, and past history of venous thrombosis (hazard ratio [HR], 4.7; 95% CI, 0.7-29.5; P = .09), and there was no increased rate of venous thrombosis in the year before diagnosis. MPL-mutant patients had an increased rate of death compared with the JAK2 V617F− group on univariate analysis, but this was due to the significantly older age of the MPL-mutant patients (HR, 1.2; 95% CI, 0.5-3.5 after correction for patient age; P = .7).
Biologic aspects of MPL mutations
To investigate possible differences between the MPL W515L and MPL W515K alleles, mutant allele burden was quantitated by pyrosequencing using whole blood obtained at PT-1 trial entry. Patients with MPL W515K had a significantly higher mutant allele burden than those with MPL W515L; the number of patients with a mutant allele burden more than 50% was 0 of 24 for W515L and 4 of 5 for W515K (P < .001, Figure 2A). There were no significant differences in age at diagnosis (P = .6), disease duration (P = .5), use of cytoreductive therapy (P = .2) or proportion of peripheral blood neutrophils at time of sampling (W515L, 0.67 ± 0.11; W515K, 0.75 ± 0.05; P = .2) to account for this difference. To examine the phenotypic correlates of the higher MPL W515K mutant allele burden, blood counts at diagnosis were compared between MPL W515L and MPL W515K patients in the PT-1 cohort. Despite the higher mutant allele burden in the MPL W515K group, there were no significant differences in diagnostic hemoglobin level (W515L, 132 ± 11g/L; W515K, 135 ± 16g/L; P = .7), neutrophil count (W515L, 6.5 ± 2.0 × 109/L; W515K, 8.7 ± 2.7 × 109/L; P = .07) or platelet count (W515L, 1010 ± 242 × 109/L; W515K, 1124 ± 397 × 109/L; P = .4). Moreover, there were no significant differences in blood counts at trial entry (data not shown).

Biologic aspects of MPL W515L and MPL W515K mutations. (A) Whole blood mutant allele quantitation at trial entry showing significant difference in mean mutant allele burden for MPL W515L (mean mutant allele burden 17% ± 11%,) and MPL W515K (mean mutant allele burden 66% ± 26%, P < .001). (B) Genotyping of single erythroid colonies from 5 ET patients, 4 with MPL W515L and 1 with MPL S505N; a single homozygous colony was identified in patient 3.
Biologic aspects of MPL W515L and MPL W515K mutations. (A) Whole blood mutant allele quantitation at trial entry showing significant difference in mean mutant allele burden for MPL W515L (mean mutant allele burden 17% ± 11%,) and MPL W515K (mean mutant allele burden 66% ± 26%, P < .001). (B) Genotyping of single erythroid colonies from 5 ET patients, 4 with MPL W515L and 1 with MPL S505N; a single homozygous colony was identified in patient 3.
Although none of 24 patients in the PT-1 cohort carrying the MPL W515L mutation had a mutant allele burden of greater than 50% at trial entry, this does not exclude the presence of a subclone homozygous for the mutation.27 We therefore genotyped 354 single erythroid colonies cultured from 4 hydroxyurea–treated ET patients with MPL W515L mutations (Figure 2B). In 1 of the 4 patients, a single homozygous colony was found of 94 colonies analyzed; the other 3 patients had only heterozygous colonies, with or without wild-type colonies. Taken together with the mutant allele quantitation, our data suggest that ET patients carrying the MPL W515K allele commonly harbor clones in which the ratio of wild-type to mutant alleles is reduced (for example, as a result of mitotic recombination or deletion of the wild-type allele), but that such clones are less common in patients with the MPL W515L allele. Of the 5 IMF patients with W515L mutations, 3 showed predominance of the mutant allele by direct sequencing (data not shown), indicating that subclones with reduced or lost wild-type allele can occur with this mutation, and may be associated with the development of myelofibrosis.
Three patients in the PT-1 cohort harbored an MPL S505N mutation, with mutant allele burdens of 62%, 47%, and 17%. Further samples were available from 1 patient (mutant allele burden 17%) which demonstrated the absence of the mutation in buccal cells (data not shown), and the presence of both wild-type and heterozygous erythroid colonies (Figure 2B), indicative of an acquired mutation.
Discussion
In this paper we report the existence of 4 mutant MPL alleles in patients with a JAK2 V617F− MPD. No mutations were detected outside exon 10 despite the analysis of platelet cDNA, which excludes the possibility that such mutations might be restricted to the megakaryocyte lineage. Our results are consistent with and extend 2 previous studies using genomic DNA from 14 adults with an MPD28 and 9 children with ET,29 both of which failed to identify MPL mutations outside exon 10. Our results demonstrate that mutations outside MPL exon 10 are not a common cause of V617F− ET but do not exclude the existence of such mutations in a small minority of patients. Changes elsewhere in MPL have recently been reported, although it is not yet clear if they are acquired mutations or inherited polymorphisms.30 Previous studies have reported the W515L and W515K allele in IMF17,19 but only the W515L allele in ET.17 Here we demonstrate the presence of the W515K allele in patients with ET and also report 2 different mutations capable of generating a W515K substitution. We also report 2 patients with ET in whom an MPL S505N allele, previously reported as an inherited mutation,23,31 was detected in granulocytes but was absent from buccal cells. Moreover in one of the patients the mutation was only present in a minority of erythroid colonies, and the other patient had a normal platelet count over several years prior to presentation with ET, making somatic mosaicism unlikely. Taken together, these findings strongly suggest that the MPL S505N allele can occur as both an inherited and acquired mutation. Mutations in the KIT gene have been reported in patients with both sporadic and familial mast cell proliferations. However the alleles involved are different, with inherited mutations not seen in acquired disease and vice versa.32-41 We believe this is the first example of a single allele being associated with both acquired and inherited forms of an MPD.
In IMF our results demonstrate the presence of MPL mutations in 7% of all patients and 15% of JAK2 V617F− patients. These data are similar to previous reports describing a prevalence of 5.5% or 8.5% in all patients17,19 and 9% in JAK2 V617F− patients.16 In ET the prevalence of MPL mutations was similar in our retrospective and prospective cohorts. In our retrospective group of patients we found the prevalence of MPL mutations to be 3.4% overall and 6.4% in JAK2 V617F− patients. In the prospective PT-1 cohort, the prevalence was 4.1% overall and 8.5% in JAK2 V617F− patients. These results are somewhat higher than the 1.3% overall prevalence previously reported,17 but are in keeping with data presented in abstract form.42
Mutant allele burden was measured in the PT-1 cohort using whole blood obtained at trial entry. ET patients with the W515K allele had significantly higher allele burdens than those with the W515L allele. This result is reminiscent of the observation that most PV patients with a JAK2 V617F mutation have homozygous erythroid colonies,27 whereas such clones are rarer in patients with a JAK2 exon 12 mutation.6 JAK2 exon 12 mutations appear to signal more strongly than V617F mutations6 but it is not clear whether the difference between the W515L and W515K mutations reflects quantitative or qualitative alterations in signaling. Interestingly a peripheral blood allele burden of more than 50% was found in 0 of 24 ET patients with the W515L allele, but in 3 of 5 IMF patients. An allele burden of more than 50% implies the existence of one or more additional events giving rise to homozygous or hemizygous mutant clones. Our results would therefore be consistent with the concept that patients labeled as having IMF may be presenting in an accelerated phase of a previously undiagnosed MPD.43 Patients with a longer disease duration, or in whom additional mutations may have increased genomic instability would be expected to have a greater probability of undergoing a second event.
The development of allele-specific PCR assays for each MPL mutation allowed analysis of samples from the PT1 cohort together with an assessment of the clinical and laboratory features associated with MPL mutations. This large cohort includes ET patients in all risk categories, with centralized review of endpoints, comprehensive follow-up, and the participation of a large number of secondary and tertiary centers. As such, these results are likely to be of general relevance to ET patients with MPL mutations. Compared with JAK2 V167F+ patients, those carrying an MPL mutation exhibited lower hemoglobin levels and higher platelet counts at diagnosis, higher serum erythropoietin levels and reduced bone marrow erythroid and overall cellularity. Moreover it was possible to grow thrombopoietin-independent megakaryocyte colonies but not erythropoietin-independent erythroid colonies from ET patients with an MPL mutation, an observation consistent with previous data from patients with IMF.25,26 Taken together, these features suggest that patients with MPL mutations have a lower drive toward erythroid differentiation, accompanied by a more isolated thrombocytosis. There were no consistent differences in other histologic features including megakaryocyte morphology and reticulin grade. Compared with JAK2 V617F− patients, those carrying an MPL mutation were older at diagnosis with reduced bone marrow cellularity that remained significant when patient age was taken into account. However, it was not possible to identify other clinical, histologic, or additional laboratory features that allowed the MPL-mutant subgroup to be distinguished from the remaining JAK2 V617F− patients. Furthermore, MPL-mutant patients did not exhibit altered rates of thrombosis, major hemorrhage, transformation, or death compared with V617F+ or V617F− patients.
In this paper, we describe 44 patients with MPL mutations, more than doubling the number thus-far reported in peer-reviewed journals. Within the PT-1 cohort, however, the number of clinical events in each category was small and statistical analyses were associated with large confidence intervals. Our data should therefore be interpreted acknowledging the possibility of false negative results. Our analysis also included a large number of hypothesis tests, and to minimize the risk of false positive results, we applied a threshold of P less than or equal to .01 for statistical significance.
Known mutations in MPL and JAK2 account for 57% of the PT-1 patients presented here. Comparisons between this group and the remaining 43% who lack an identified mutation did not reveal any significant differences in the prevalence of splenomegaly, abnormal cytogenetics, myelofibrotic transformation, or acute myeloid leukemia. There were also no differences between the mutation-negative and mutation-positive groups in histologic features other than cellularity. Taken together these findings suggest that mutation-negative patients do have a myeloproliferative disorder, the molecular basis for which remains obscure.
Combining our retrospective and prospective cohorts, we identified MPL mutations in 36 patients with ET, only one of whom carried a JAK2 V617F mutation. This is in marked contrast to the only previous report in which 2 of 4 ET patients with an MPL mutation carried a JAK2 V617F mutation,17 and suggests that the frequency of such double positive patients is much less than previously thought. We also identified MPL mutations in 8 IMF patients, none of whom harbored a JAK2 V617F mutation. This observation is not significantly different from previous studies in which 4 of 1617 or 4 of 1819 patients with an MPL mutation carried a JAK2 V617F mutation. If the prevalence of patients with both MPL and JAK2 mutations does prove to be higher in IMF compared with ET, this would be consistent with the concept that IMF represents patients presenting in an accelerated phase of a preexisting MPD.43
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank the patients and staff of the United Kingdom MPD clinics who have contributed samples to this study, and Addenbrooke's Hematology Disorders Sample Bank for processing and managing patient samples. We are also grateful to Dr Brian Huntly for constructive comments on the manuscript, and to Pat Collins for her assistance with managing the trephine slides and PT-1 follow-up data.
This work was supported by the United Kingdom Medical Research Council, the Leukaemia Research Fund, and the Kay Kendal Leukaemia Fund, along with the Leukemia & Lymphoma Society of America.
Authorship
Contribution: P.A.B. designed and applied MPL mutation detection and pyrosequencing assays, processed and genotyped hematopoietic colonies, performed statistical analysis, and cowrote the manuscript; P.J.C. carried out JAK2 V617F mutation and statistical analyses; L.M.S. carried out JAK2 V617F mutation analysis and processed and genotyped hematopoietic colonies; A.J.B. carried out JAK2 V617F mutation analysis and managed patient samples; W.N.E., D.B., and B.S.W. reviewed the trephine biopsies; J.T.R. and H.C.H. provided patient samples and clinical information; R.B. assisted with design and interpretation of pyrosequencing assays; K.W. and G.B. provided statistical support for the PT-1 trials; C.N.H. is principal coinvestigator of the PT-1 trials and provided patient samples and clinical information; A.R.G. is principal coinvestigator of the PT-1 trials, designed experiments, and co-wrote the manuscript. All authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Prof Tony Green, Department of Haema-tology, Cambridge Institute for Medical Research, Hills Road, Cambridge CB2 2XY, United Kingdom; e-mail: arg1000@cam.ac.uk.

