

Abstract
Hemoglobin and myoglobin are among the most extensively studied proteins, and nitrite is one of the most studied small molecules. Recently, multiple physiologic studies have surprisingly revealed that nitrite represents a biologic reservoir of NO that can regulate hypoxic vasodilation, cellular respiration, and signaling. These studies suggest a vital role for deoxyhemoglobin- and deoxymyoglobin-dependent nitrite reduction. Biophysical and chemical analysis of the nitrite-deoxyhemoglobin reaction has revealed unexpected chemistries between nitrite and deoxyhemoglobin that may contribute to and facilitate hypoxic NO generation and signaling. The first is that hemoglobin is an allosterically regulated nitrite reductase, such that oxygen binding increases the rate of nitrite conversion to NO, a process termed R-state catalysis. The second chemical property is oxidative denitrosylation, a process by which the NO formed in the deoxyhemoglobin-nitrite reaction that binds to other deoxyhemes can be released due to heme oxidation, releasing free NO. Third, the reaction undergoes a nitrite reductase/anhydrase redox cycle that catalyzes the anaerobic conversion of 2 molecules of nitrite into dinitrogen trioxide (N2O3), an uncharged molecule that may be exported from the erythrocyte. We will review these reactions in the biologic framework of hypoxic signaling in blood and the heart.
Introduction
Nitric oxide (NO) is a diatomic gas molecule that is a critical regulator of basal blood vessel tone and vascular homeostasis (antiplatelet activity, modulation of endothelial and smooth muscle proliferation, and adhesion molecule expression).1-5 NO is a paracrine signaling molecule, as it is produced in endothelium and then diffuses to vicinal smooth muscle to activate soluble guanylyl cyclase that produces cGMP, and ultimately produces smooth muscle relaxation. Nitric oxide is subject to rapid inactivation reactions with hemoglobin that greatly limit its lifetime in blood, however recent studies suggest that NO formed from endothelial NO synthases is also oxidized by oxygen or plasma ceruloplasmin to form nitrite.6 Nitrite transport in blood provides an endocrine form of NO that is shuttled from the lungs to the periphery, while limiting the exposure of authentic NO to the scavenging red cell environment. Then during the rapid hemoglobin deoxygenation from artery to vein the nitrite is reduced back to NO. Such a cycle conserves NO in the one electron oxidation state. In this model, the nitrite pool represents the “live payload,” only one electron away from NO.
We have hypothesized that nitrite and the heme-globin family subserve a critical NO signaling function in blood, smooth muscle cells, the cardiomyocyte, and skeletal muscle cells. In these environments, NO is conserved by oxidation, allowing nitrite storage and free nitrite diffusion. As oxygen tensions decrease, nitrite reactions with deoxygenated hemoglobin, myoglobin, neuroglobin, cytoglobin, and other heme proteins may generate NO to regulate physiologic hypoxic signaling.
A brief history of hemoglobin and nitrite
Hemoglobin is certainly the most studied protein in human history, from the structural and biophysical characterization of its allosteric properties to the detailed mechanisms of its transcriptional regulation. In fact, it has been appreciated since the time of Darwin that red cells participated in oxygen binding and transport. In 1875, in the book Insectivorous Plants, Darwin writes: “It is known that the protoplasm of plants exhibits its spontaneous movements only as long as it is in an oxygenated condition; and so it is with the white corpuscles of the blood, only as long as they receive oxygen from the red corpuscles.”7,p60 The ability of sodium nitrite to oxidize hemoglobin and other hemoproteins has also been recognized for almost 150 years.8 John Haldane, from the Physiology Laboratory in Oxford, in describing the chemistry of meat curing, first reported that the reaction of nitrite with deoxyhemoglobin and deoxymyoglobin generated nitric oxide (NO). In his paper “The red colour of salted meat,”9 he writes: “On cutting into pieces of uncooked salted meat, obtained from the butcher, I noticed that the color of the exposed surface was bright red wherever the salt had penetrated, but slowly became dull on exposure to the air. … On extracting the freshly exposed salted part with water the red pigment was found to be quite soluble and to give a spectrum … possessing two absorption bands at about the position of the oxyhaemoglobin bands, but not nearly so well defined. This spectrum was found to be identical with that of nitric oxide hemoglobin and the behavior of the pigment in all other respects showed that it was nothing else but pure NO-hemoglobin.” Haldane concluded that “NO-hemoglobin is formed by the action of nitrite on hemoglobin in the absence of oxygen, and in the presence of reducing agents.”9 This is the first account that we are aware of describing deoxyhemoglobin-mediated reduction of nitrite to NO, leading to the formation of iron-nitrosyl-hemoglobin.
In 1937, Brooks presented his detailed analysis of the deoxyhemoglobin-nitrite reaction in “The action of nitrite on haemoglobin in the absence of oxygen.” Using spectroscopic analysis he concluded that 1 mol protonated nitrite (HNO2; nitrous acid) and 2 mol ferrous deoxyhemoglobin (HbFe2+) generate 1 mol methemoglobin (HbFe3+) and 1 mol iron-nitrosyl-hemoglobin (HbFe2+-NO).10 Mechanistically, he reasoned that nitrous acid must react with deoxyhemoglobin to form NO and methemoglobin (Equation 1). The NO produced in this reaction would then bind to a second deoxyhemoglobin to form iron-nitrosyl-hemoglobin (Equation 2).
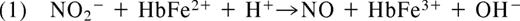
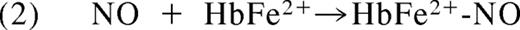
As early as 1977, Tomoda and colleagues proposed that that the reaction of nitrite and Hb might be allosterically modulated. They observed that inositol hexaphosphate (IHP) significantly decreases the rate of hemoglobin oxidation by nitrite (Tomoda et al,11 Tomoda and Yoneyama12 ), a result we confirmed many years later. Because IHP lowers hemoglobin oxygen affinity, the authors suggested that hemoglobin oxidation by nitrite was favored by higher oxygen affinity states of hemoglobin.11,12 Later, in 1981 Doyle et al reported that the reaction of nitrite and deoxyhemoglobin was linearly dependent on hydrogen ion in the pH range of 6.0 to 8.0, however the observed order of proton dependence was 0.88 rather than the ideal 1.0.13 The authors interpreted this unexpected deviation to imply that nitrous acid may not be the sole reactant with deoxyhemoglobin, suggestive of other proton effects on hemoglobin allosteric conformations. This observation has now been attributed to the redox Bohr effect on nitrite reduction.14
Modern laboratory methodologies, such as least squares analysis of spectroscopic experiments, electron paramagnetic resonance spectroscopy, molecular mutation of hemoglobin molecules, and in vitro and in vivo physiologic assays, have allowed for a more expansive understanding of this simple second-order reaction and its physiologic significance. Such analysis of the deoxyhemoglobin-nitrite reaction has confirmed that under purely anaerobic conditions it generates equimolar quantities of methemoglobin and iron-nitrosyl-hemoglobin, as characterized in equation 1.14-16 This reaction also possesses 3 unique properties that we will review in some detail.14 (1) Under truly anaerobic conditions, when excess nitrite is reacted with 99.9% deoxyhemoglobin, the reaction deviates from a second-order rate law and appears to exhibit zero-order kinetics in terms of deoxyheme concentration, such that the reaction rate remains constant as deoxyheme is consumed. (2) Closer inspection of the instantaneous rate reveals that the reaction is composed of a lag (slow) phase, followed by a fast phase, and then slows down again. Thus the reaction proceeds in a sigmoidal process, analogous to the hemoglobin-oxygen binding affinity. (3) In the presence of an oxygen leak, the reaction rate increases dramatically and appears to exhibit second-order dependence. Thus the reaction appears to be catalyzed by oxygen.
It is now clear that all of these unusual properties, such as the unusual effects of IHP, proton, and oxygen on reaction rate and the sigmoidal deviation from second-order kinetics, all occur secondary to the allosteric nature of the hemoglobin reaction with nitrite. Nitrite, therefore, reacts not with a single static molecule of deoxyhemoglobin, but rather with the allosterically different T and R conformational states of the hemoglobin tetramer. This unique chemistry provides a mechanism for hemoglobin-based sensing of dropping oxygen and rising protons coupled to the generation of a vasodilator signal, suggesting a role for this reaction in hypoxic vasodilation and hypoxic NO signaling.
Short review of allostery in hemoglobin oxygen transport
The primary function of hemoglobin (Hb) is gas transport. If blood did not contain red cells and Hb, only approximately 200 μM oxygen could be dissolved and transported. The hemoglobin concentration in red blood cells (RBCs) is approximately 20 mM in heme (5 mM or so in Hb tetramer) so that a hematocrit of 0.5 (50%) increases oxygen-carrying capacity by approximately 50-fold. Hemoglobin is able to deliver oxygen effectively due to cooperativity of oxygen binding. After initial binding or release of oxygen, binding or release of subsequent oxygen molecules is increased. Thus Hb has a high oxygen affinity at high oxygen tensions and a low oxygen affinity at low oxygen tensions, manifested by the characteristic sigmoidal oxygen binding curve.17 Cooperative binding of oxygen has been explained in terms of a 2-state allosteric model where there is a high oxygen affinity R-state and a low oxygen affinity T-state, each defined by the quaternary state of the Hb tetramer.18 Allostery is proposed to be accomplished through translation of motion of the heme iron upon oxygen binding or release. The heme motion results in breakage or formation of intersubunit salt bridges of the hemoglobin tetramer so the αβ dimers rotate approximately 15 degrees with respect to each other.19,20 Although a 2-state model cannot quantitatively explain all of the Hb functions, it is widely accepted as a solid working model for most thermodynamic and kinetic behavior.21
Hemoglobin would not be capable of adequately transporting oxygen without the presence of 2,3-biphosphoglycerate (BPG) in the RBC. BPG shifts the hemoglobin P50 from approximately 2.5 mmHg (extremely hypoxic) to approximately 25 mmHg. BPG is a heterotrophic allosteric effector, which means that by binding to Hb it affects oxygen binding at distant sites. Other heterotrophic effectors that increase the Hb P50 include chloride, phosphate, carbon dioxide, and protons. Thus, lowering the pH (increasing proton concentration) lowers Hb oxygen affinity and promotes oxygen release. Likewise, in the phenomenon known as the Bohr effect, oxygen release from Hb promotes proton uptake and vice versa. The mechanism of the Bohr effect according to the 2-state allosteric model is that, like other negative heterotrophic effectors, protons stabilize the T quaternary state. This has also been called the redox Bohr effect as the T-state stabilization is associated with an increase in heme redox potential (increase in E1/2).
These classic allosteric properties of the hemoglobin tetramer—cooperative oxygen binding, R-to-T 2-state allosteric transitions, proton and redox Bohr effects, and T-state stabilizing effects of BPG—all come into play in the reaction of nitrite with hemoglobin. An examination of the allosteric properties of hemoglobin in the nitrite reduction reaction reveals that the conversion of nitrite to NO is also under allosteric control.
Allosterically regulated nitrite reduction: reaction kinetics, allosteric autocatalysis, redox potential, ligand accessibility, and the redox Bohr effect
Reaction kinetics
When excess nitrite is added to completely deoxygenated Hb, the hemoglobin is oxidized forming methemoglobin and NO as described in Equation 1. A simple second-order reaction predicts that the rate of the reaction should be proportional to the concentration of deoxyhemoglobin. Thus, as the reaction proceeds (and deoxyhemoglobin is converted to methemoglobin or binds NO to form iron-nitrosyl-hemoglobin) the reaction rate is predicted to become slower, so that a plot of deoxyhemoglobin versus time would produce an exponential decay curve. Surprisingly, as shown in Figure 1A,15 the curve clearly deviates from the exponential, suggesting a more complicated reaction.14,16 Upon closer inspection, it is observed that the reaction rate (given by the slope of the curve in Figure 1A) speeds up and then slows down, producing a sigmoidal shape. This is clearly illustrated when the instantaneous rate itself is plotted versus time as in Figure 1B.14,15,22

The sigmoidal “apparent zero order” behavior of the nitrite reaction with deoxyhemoglobin. Deoxyhemoglobin (50 μM) reaction with nitrite (10 mM) at pH 7.4 and 37°C. (A) Time-resolved absorption spectra were deconvoluted to determine the percentage of each species as a function of time. Deoxyhemoglobin is observed to form equal amounts of methemoglobin and iron-nitrosyl-hemoglobin. Deviation from first-order behavior is evident in the curve for decay of deoxyhemoglobin, having a sigmoidal shape. In experiments with excess nitrite, the spectrum of nitrite bound to methemoglobin (nitrite-methemoglobin) should be included. (B) The instantaneous rate of the reaction shown in panel A where the negative of the slope of the decay curve for deoxyhemoglobin is plotted as a function of time. The figure is reproduced from Grubina et al15 with permission.
The sigmoidal “apparent zero order” behavior of the nitrite reaction with deoxyhemoglobin. Deoxyhemoglobin (50 μM) reaction with nitrite (10 mM) at pH 7.4 and 37°C. (A) Time-resolved absorption spectra were deconvoluted to determine the percentage of each species as a function of time. Deoxyhemoglobin is observed to form equal amounts of methemoglobin and iron-nitrosyl-hemoglobin. Deviation from first-order behavior is evident in the curve for decay of deoxyhemoglobin, having a sigmoidal shape. In experiments with excess nitrite, the spectrum of nitrite bound to methemoglobin (nitrite-methemoglobin) should be included. (B) The instantaneous rate of the reaction shown in panel A where the negative of the slope of the decay curve for deoxyhemoglobin is plotted as a function of time. The figure is reproduced from Grubina et al15 with permission.
Allosteric autocatalysis
The kinetics of the nitrite/deoxyhemoglobin reaction as depicted in Figure 1 have been explained in terms of allosteric autocatalysis.14 R-state Hb reduces nitrite faster than T-state Hb.14,16 As the reaction of excess nitrite with Hb proceeds during this anaerobic reaction, R-state Hb is stabilized by the formation of methemoglobin and iron-nitrosyl-hemoglobin (Equations 1 and 2). Note that NO bound to the heme of hemoglobin, like other heme ligands oxygen and carbon monoxide, increases the ligand affinity of other hemes on the tetramer (increases R-state). Thus, although there are fewer vacant hemes to react with nitrite as the reaction proceeds, these remaining hemes react faster with nitrite as they are part of R-state rather than T-state Hb tetramers. Autocatalysis is achieved through each nitrite-heme reaction producing 2 R-state stabilizing hemes: one oxidized and one bound to NO. Thus, the loss of available hemes, which tends to slow down the reaction, is counterbalanced by the increasing reaction rate of the remaining individual hemes. This results in the nearly linear, but rather sigmoidal, kinetic progress curve as shown in Figure 1A. The function of Hb as an allosteric nitrite reductase is illustrated conceptually and mathematically in Figure 2.
![Figure 2. (A) Allosteric autocatalysis. Reaction of nitrite with deoxyhemoglobin exhibits R-state catalysis. Tetrameric T-state deoxyHb reduces nitrite to NO, generating a met-heme (3+) and an iron-nitrosyl-heme (Fe+2-NO), on the same or different Hb tetramers, which stabilize the tetramer(s) in the R state. Increasing R-state character is associated with a higher bimolecular rate constant for nitrite reduction. As a result, ferrous deoxyhemes on these R-state stabilized tetramers react with nitrite faster than those on T-state stabilized tetramers, thereby exponentially propagating nitrite reduction and R-state stabilization. This process therefore represents a unique allosteric autocatalytic reaction mechanism. Please note that we are showing the bimolecular rate constant, not the overall reaction rate, and that in the case of hemoglobin, the overall hemoglobin rate constant is not constant, but changes with the T-to-R allosteric transition. The overall rate constant is dependent on the intrinsic reactivity of nitrite with heme and is highest in R state. The actual rate of a second-order reaction is determined by the concentration of deoxyheme multiplied by the concentration of nitrite and multiplied by the bimolecular rate constant. Panel A is reproduced from Grubina et al22 with permission. (B) Distribution of R and T ligand populations modulates nitrite reduction. The fractions of R and T oxygen-liganded species are plotted as a function of oxygen saturation. The quaternary state (R or T) is indicated and the number of ligands bound to each tetramer is indicated by the subscript, so that R3 indicates an R-state conformation with 3 oxygen ligands bound. The fraction of each species was calculated using the MWC Perutz 2 state model with the value of c set at 0.015, where c is the ratio of equilibrium-binding constants for T (taken as 1/77 mm Hg) and R states. The ratio L = T0/R0 was taken as 105. The fractions of some intermediate species are so small that they do not appear on the graph. (C) Contribution of quaternary states to nitrite reaction rate. The reaction rates of nitrite with deoxygenated Hb are plotted as a function of oxygen saturation for cases where the product of the nitrite and Hb concentrations are 10−6 M2. At each oxygen saturation the rate of the reaction was calculated as [nitrite] × {kt(4[T0] + 3[T1] + 2[T2] + [T3]) + kR(4[R0] + 3[R1] + 2[R2] + [R3]), where the square brackets refer to Hb concentrations. Here, kR/kT was rounded to 100 and kT was set to 0.2 M−1s−1. The contribution by R-state and T-state molecules was obtained by calculating the products of kR and kT separately (so, for example, the R-state contribution is [nitrite] × kR(4[R0] + 3[R1] + 2[R2] + [R3])).](/view-large/figure/6940258/zh80180824250002.jpeg)
(A) Allosteric autocatalysis. Reaction of nitrite with deoxyhemoglobin exhibits R-state catalysis. Tetrameric T-state deoxyHb reduces nitrite to NO, generating a met-heme (3+) and an iron-nitrosyl-heme (Fe+2-NO), on the same or different Hb tetramers, which stabilize the tetramer(s) in the R state. Increasing R-state character is associated with a higher bimolecular rate constant for nitrite reduction. As a result, ferrous deoxyhemes on these R-state stabilized tetramers react with nitrite faster than those on T-state stabilized tetramers, thereby exponentially propagating nitrite reduction and R-state stabilization. This process therefore represents a unique allosteric autocatalytic reaction mechanism. Please note that we are showing the bimolecular rate constant, not the overall reaction rate, and that in the case of hemoglobin, the overall hemoglobin rate constant is not constant, but changes with the T-to-R allosteric transition. The overall rate constant is dependent on the intrinsic reactivity of nitrite with heme and is highest in R state. The actual rate of a second-order reaction is determined by the concentration of deoxyheme multiplied by the concentration of nitrite and multiplied by the bimolecular rate constant. Panel A is reproduced from Grubina et al22 with permission. (B) Distribution of R and T ligand populations modulates nitrite reduction. The fractions of R and T oxygen-liganded species are plotted as a function of oxygen saturation. The quaternary state (R or T) is indicated and the number of ligands bound to each tetramer is indicated by the subscript, so that R3 indicates an R-state conformation with 3 oxygen ligands bound. The fraction of each species was calculated using the MWC Perutz 2 state model with the value of c set at 0.015, where c is the ratio of equilibrium-binding constants for T (taken as 1/77 mm Hg) and R states. The ratio L = T0/R0 was taken as 105. The fractions of some intermediate species are so small that they do not appear on the graph. (C) Contribution of quaternary states to nitrite reaction rate. The reaction rates of nitrite with deoxygenated Hb are plotted as a function of oxygen saturation for cases where the product of the nitrite and Hb concentrations are 10−6 M2. At each oxygen saturation the rate of the reaction was calculated as [nitrite] × {kt(4[T0] + 3[T1] + 2[T2] + [T3]) + kR(4[R0] + 3[R1] + 2[R2] + [R3]), where the square brackets refer to Hb concentrations. Here, kR/kT was rounded to 100 and kT was set to 0.2 M−1s−1. The contribution by R-state and T-state molecules was obtained by calculating the products of kR and kT separately (so, for example, the R-state contribution is [nitrite] × kR(4[R0] + 3[R1] + 2[R2] + [R3])).
(A) Allosteric autocatalysis. Reaction of nitrite with deoxyhemoglobin exhibits R-state catalysis. Tetrameric T-state deoxyHb reduces nitrite to NO, generating a met-heme (3+) and an iron-nitrosyl-heme (Fe+2-NO), on the same or different Hb tetramers, which stabilize the tetramer(s) in the R state. Increasing R-state character is associated with a higher bimolecular rate constant for nitrite reduction. As a result, ferrous deoxyhemes on these R-state stabilized tetramers react with nitrite faster than those on T-state stabilized tetramers, thereby exponentially propagating nitrite reduction and R-state stabilization. This process therefore represents a unique allosteric autocatalytic reaction mechanism. Please note that we are showing the bimolecular rate constant, not the overall reaction rate, and that in the case of hemoglobin, the overall hemoglobin rate constant is not constant, but changes with the T-to-R allosteric transition. The overall rate constant is dependent on the intrinsic reactivity of nitrite with heme and is highest in R state. The actual rate of a second-order reaction is determined by the concentration of deoxyheme multiplied by the concentration of nitrite and multiplied by the bimolecular rate constant. Panel A is reproduced from Grubina et al22 with permission. (B) Distribution of R and T ligand populations modulates nitrite reduction. The fractions of R and T oxygen-liganded species are plotted as a function of oxygen saturation. The quaternary state (R or T) is indicated and the number of ligands bound to each tetramer is indicated by the subscript, so that R3 indicates an R-state conformation with 3 oxygen ligands bound. The fraction of each species was calculated using the MWC Perutz 2 state model with the value of c set at 0.015, where c is the ratio of equilibrium-binding constants for T (taken as 1/77 mm Hg) and R states. The ratio L = T0/R0 was taken as 105. The fractions of some intermediate species are so small that they do not appear on the graph. (C) Contribution of quaternary states to nitrite reaction rate. The reaction rates of nitrite with deoxygenated Hb are plotted as a function of oxygen saturation for cases where the product of the nitrite and Hb concentrations are 10−6 M2. At each oxygen saturation the rate of the reaction was calculated as [nitrite] × {kt(4[T0] + 3[T1] + 2[T2] + [T3]) + kR(4[R0] + 3[R1] + 2[R2] + [R3]), where the square brackets refer to Hb concentrations. Here, kR/kT was rounded to 100 and kT was set to 0.2 M−1s−1. The contribution by R-state and T-state molecules was obtained by calculating the products of kR and kT separately (so, for example, the R-state contribution is [nitrite] × kR(4[R0] + 3[R1] + 2[R2] + [R3])).
From a chemical kinetic standpoint, this reaction is unique, and has been described as an allosteric autocatalytic reaction.14 Like most autocatalytic reactions, it has a slow or lag phase followed by a fast phase. However, in this case, the autocatalytic or fast phase is driven by an allosteric transition of the hemoglobin tetramer from the T to the R state.
Redox potential and ligand accessibility
The dependence of the rate of nitrite reduction on Hb quaternary state predicts that hemes within partially oxygenated Hb tetramers such as R3 (where the Hb molecule is in the R-state and has 3 oxygen molecules bound and one vacant heme) will react faster than T-state hemes, and so the reaction will be faster at partial oxygen tensions. Indeed, it has been shown that the rate of Hb-mediated nitrite reduction is fastest when the Hb is approximately 50% saturated with oxygen (Figure 2B,C).14,23 Calculations have shown that the intrinsic bimolecular rate constant for nitrite reduction by R-state hemes, kR, is approximately 60 times greater than the bimolecular rate constant for nitrite reduction by T-state hemes, kT.14,24 The observed rate of the reaction will be given by
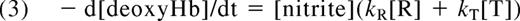
where [R] and [T] represent the concentration of R-state and T-state vacant hemes, respectively.
The bimolecular rate constant for the R-state hemoglobin tetramer (kR) could be higher than the rate constant for the T-state tetramer (kT) due to differences in the redox potential of the heme allowing more facile electron transfer or due to differences in binding or coordination of nitrite in the heme pocket (ie, hydrogen bonding or heme pocket geometry). These 2 factors can be summarized as (1) a lower redox potential of R-state compared with T-state hemes or (2) a tertiary structure around the heme that is more conducive to nitrite binding and subsequent reaction.
R-state Hb is known to have a lower redox potential than T-state Hb, meaning that it is oxidized easier, that is, is a better reductant.25,26 Thus, when nitrite binds to a heme on R-state hemoglobin the equilibrium distribution of electrons would increase the probability that electron transfer and reduction of nitrite would occur.
The lower redox potential and potentially greater tendency to bind nitrite are not mutually exclusive effects in increasing kR; however, they may contribute to a different extent. Recently, the half-life of the reaction of nitrite with deoxyhemoglobin and modified hemoglobins developed as potential blood substitutes was shown to decrease (reaction getting faster) as the redox potential increased.27 Based on these data, the authors concluded that ligand affinity, rather than redox potential, is the major determinant of the nitrite-deoxyhemoglobin reaction rate in these samples. In another study, the rate that deoxygenated normal adult hemoglobin, HbA0, reacts with nitrite was compared with the rate that sickle cell hemoglobin, HbS, reacts with nitrite.22 The ligand affinity of solution phase (unpolymerized) HbS is the same as HbA0,28-31 so that any differences in the nitrite-deoxyhemoglobin reaction between these hemoglobin variants could be due to redox potential (note that we cannot exclude unknown differences in nitrite binding that are different from oxygen binding). The redox potential of HbS is lower than HbA0.32-36 Consistent with this fact, the rate of the solution phase deoxyHbS reaction with nitrite was faster than the reaction of deoxyHbA0 with nitrite (Table 1).22 This difference must be due to the lower redox potential of HbS, implicating a role for redox potential in determining the higher value of kR compared with kT. It should be noted that polymerized HbS was found to react more slowly with nitrite than HbA0, which could have important implications in the pathology of sickle cell disease.22 A complete characterization of the reaction rate constants for nitrite reduction by different hemoglobin variants and allosteric states that have been measured is presented in Table 1. These standard measurements for hemoglobin species may provide insight into the physiologic function of the heme-globins in NO signaling.
Proton and the redox Bohr effect
Equation 1 predicts that as the pH is lowered by 1 unit the rate of the nitrite-deoxyhemoglobin reaction should increase 10-fold. However, the observed increase in the rate is slightly less (approximately 9-fold; see historical note on Doyle's observation of a proton dependence of 0.88). One explanation put forth for this discrepancy was that the Hb conformation is involved.13 Indeed, the deviation is explained by the redox Bohr effect, where a decrease in pH stabilizes the Hb T state and raises the heme redox potential.14 As indicated in equation 3, as the proportion of T-state compared with R-state Hb increases, the nitrite-deoxyhemoglobin reaction slows down because kT is much less than kR. It is important to note that between the 2 opposing factors that affect the nitrite-deoxyhemoglobin reaction when pH is lowered, the direct effect of increased proton concentration in forming nitrous acid increases the reaction rate to a much greater extent than the slowing effect attributable to the Bohr effect.
Oxidative denitrosylation
A major challenge to the nitrite reductase hypothesis is the question of how NO could escape from a red blood cell after formation. This is particularly problematic after NO binds to deoxyhemoglobin, because the dissociation rate constant of NO from ferrous Hb is quite slow (10−3 to 10−5 s−1).38,39 As the dissociation rate constant of NO from a ferric heme is much faster (1 s−1),38 oxidation of iron-nitrosyl-hemoglobin should lead to relatively rapid release of NO. This has, in fact, been demonstrated for several oxidants such as peroxynitrite and ferricyanide.40-42 Recently, it has been shown that intermediate products formed in the reaction of nitrite with oxyhemoglobin (note that this is a reaction with oxyhemoglobin rather than deoxyhemoglobin), oxidize the heme of iron nitrosyl hemoglobin and thereby release NO.15 Under partially oxygenated conditions, nitrite reacts with both oxyhemoglobin and deoxyhemoglobin, but the deoxyhemoglobin reaction predominates due to inhibition of the oxyhemoglobin autocatalytic phase.15 Thus, this process of oxidative denitrosylation both channels nitrite to the reductive deoxyhemoglobin reaction and produces NO from iron-nitrosyl-hemoglobin.
Anaerobic N2O3 generation by the catalytic nitrite reductase/anhydrase chemistry of deoxyhemoglobin
One of the most substantial challenges to our hypothesis that hemoglobin is a physiologically relevant mammalian nitrite reductase is that NO produced from nitrite in the red blood cell could not escape scavenging because of secondary rapid reactions with oxygenated hemoglobin, which would destroy the NO via formation of nitrate (NO3−).43,44 One proposal that would overcome this challenge is that there is some intermediate or alternate species formed in the nitrite-deoxyhemoglobin reaction that can diffuse out of the red blood cell and then decompose into NO or some other species with NO-like bioactivity.44,45 One possible intermediate species that fits this description is N2O3, which homolyzes into NO and NO2· or directly nitrosates thiols.44-46
That nitrite and hemoglobin can indeed react to form N2O3 and the mechanism for this process (Figure 3) have recently been discovered.46 First nitrite reacts with deoxyhemoglobin and a proton to form NO and methemoglobin according to Equation 1: NO2− + HbFe+2 (deoxyhemoglobin) + H+ → NO (nitric oxide) + HbFe+3 + OH−.

The N2O3-forming reaction of nitrite and hemoglobin may regulate export of NO from the erythrocyte. Hemoglobin deoxygenation (purple) occurs preferentially at the submembrane of the red blood cell as it traverses the arteriole. Nitrite reacts with deoxygenated hemoglobin to make methemoglobin and NO. Methemoglobin binds nitrite to form an adduct with some Fe+2-NO2 character (Hb-NO2•). This species reacts quickly with NO, forming N2O3, which can diffuse out of the red cell, later forming NO and effecting vasodilation and/or forming nitrosothiols (SNO). Low-molecular-weight nitrosothiols may contribute to exportable vasodilatory activity. The Hb abbreviation indicates ferrous deoxyhemoglobin (Fe+2). Figure is reproduced from Basu et al46 with permission.
The N2O3-forming reaction of nitrite and hemoglobin may regulate export of NO from the erythrocyte. Hemoglobin deoxygenation (purple) occurs preferentially at the submembrane of the red blood cell as it traverses the arteriole. Nitrite reacts with deoxygenated hemoglobin to make methemoglobin and NO. Methemoglobin binds nitrite to form an adduct with some Fe+2-NO2 character (Hb-NO2•). This species reacts quickly with NO, forming N2O3, which can diffuse out of the red cell, later forming NO and effecting vasodilation and/or forming nitrosothiols (SNO). Low-molecular-weight nitrosothiols may contribute to exportable vasodilatory activity. The Hb abbreviation indicates ferrous deoxyhemoglobin (Fe+2). Figure is reproduced from Basu et al46 with permission.
Nitrite also binds to methemoglobin with a dissociation constant of 1 mM, or much lower under some conditions.46,47 The nitrite-methemoglobin species that forms is found to silence the high spin paramagnetic absorbance, observed by electron paramagnetic resonance (EPR) spectroscopy. This EPR silencing is also observed in bacterial nitrite reductases and is indicative of an unusual electronic configuration that, based on density functional theory calculations, appears to possess FeII-NO2• character.46 The presence of a radical NO2• makes it likely that this species will react extremely quickly with NO to form N2O3 (Equation 4 and Figure 3; note that Equation 4 represents overall stoichiometry of Figure 3 reaction series):
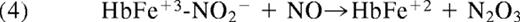
This fast reaction pathway provides 2 avenues for NO escape. It creates a radical-radical reaction pathway for NO formed from equation 1, which can partially compete with the inactivating reaction of the NO with hemoglobin, and it also forms the diffusible, lipophilic, and more stable N2O3 (relative to NO). Given the dimensions of the red blood cell (a biconcave disc), one expects some N2O3, with a lifetime of 1 ms and a diffusion coefficient of 1000 μm2/s, to diffuse out of the erythrocyte. In addition, it is quite possible that different isomers of N2O3 have different and perhaps longer lifetimes further facilitating potential export.48,49
The need for speed: radical-radical reaction pathway to form N2O3 limits NO autocapture by hemoglobin
We certainly appreciate that NO escape after nitrite reduction in the red blood cell is the major challenge to our theory that hemoglobin and myoglobin are functional nitrite reductases.50 However, the evidence that there is an “interaction” between deoxygenated hemoglobin and myoglobin to vasodilate, inhibit cytochrome c oxidase, inhibit cellular oxygen consumption, generate cGMP, form NO gas, and form N2O3 is robust.23,37,46,51-55 Our recent work strongly suggests that N2O3 formation can explain these signaling events despite NO scavenging by hemoglobin. The reason that this pathway is possible is because nitrite binding to methemoglobin produces an intermediate with NO2 radical properties. NO can then react with this intermediate at near radical-radical reaction rates that compete with NO scavenging reactions with viscinal ferrous hemes.46 We fully appreciate that the higher concentration of ferrous hemes in the red cell than the nitrite-methemoglobin intermediate requires compartmentalization of the formed NO with the nitrite-methemoglobin intermediate. We hypothesize that a membrane nitrite reductase metabolome composed of nitrite channels, such as the anion exchange protein or Rh complex, and deoxyhemoglobin, methemoglobin, and carbonic anhydrase, would increase the submembrane concentration of the reactants (proton, nitrite, methemoglobin, and deoxyhemoglobin).24
Physiologic effects of nitrite reactions with hemoglobin
Large doses of nitrite given as a treatment for cyanide poisoning clearly produce hypotension in humans.56 Consistent with this observation, high micromolar pharmacologic concentrations of nitrite were shown to vasodilate isolated in vitro aortic rings by Furchgott and Bhadrakom in 1953.57 Nitrite was later shown by Murad and Ignarro to activate guanylate cyclase and vasodilate aortic rings (Furchgott and Bhadrakom57 ; Mittal et al58 ; Ignarro et al59 ; and Ignarro and Gruetter60 ). However, the high micromolar to millimolar concentrations of nitrite necessary to achieve these effects contrasted with the low nanomolar concentrations of authentic NO needed to vasodilate aortic rings. This apparent low potency of nitrite, and the low levels of nitrite present in human blood, led to a premature dismissal of nitrite as a regulator of blood flow in vivo. Apparently supporting this inclination, studies published by Lauer et al demonstrated that nitrite had no vasodilator activity when infused into the forearm circulation of 3 healthy volunteers, even at concentrations of 200 μM.61 These studies appeared to close the door on any role for nitrite in the regulation of physiologic blood flow.62,63
Before the publication of these negative studies, our group had measured arterial and venous levels of nitrite across the human forearm, and had noted that arterial levels were higher than venous levels.64 When healthy volunteers were given inhaled NO gas, the levels of plasma nitrite increase and this was associated with vasodilation or protection from ischemia-reperfusion injury at a site distant from the pulmonary circulation.65,66 These observations suggested that nitrite was metabolized from artery to vein in the human circulation and that increased levels in plasma during NO gas breathing resulted in transport of NO bioactivity in an endocrine fashion from the pulmonary vasculature to the peripheral organs. To directly test whether nitrite was vasodilatory in vivo at physiologic concentrations, we infused nitrite into the forearm brachial artery of 28 healthy volunteers and, to our surprise, observed substantial vasodilation, even without exercise stress. Nitrite was potent in humans, increasing blood flow by 170% at 200 μM in the forearm circulation, by 22% at 2.5 μM, and produced vasodilation during exercise stress even at levels of 900 nM.51 The potent in vivo vasodilating effects of nitrite have now been confirmed by several investigators.67-73
Why was nitrite more potent in vivo than in isolated aortic rings bioassays? One clue to the mechanism of activation in vivo was the artery-to-venous formation of iron-nitrosyl-hemoglobin (HbFe+2-NO) during the intra-arterial infusion of nitrite in the brachial artery, suggesting that nitrite was being reduced to NO rapidly within one half circulatory time.51 This amount of NO formed from artery to vein was inversely correlated with decrease in oxyhemoglobin saturation, so that as hemoglobin deoxygenated more NO was formed. The mechanism of nitrite-dependent vasodilation appeared to be consistent with nitrite conversion to NO during physiologic hypoxia, in a process tightly coupled to hemoglobin deoxygenation.51,74 The suggestion that nitrite is a hypoxic vasodilator has been challenged by other groups62 ; however, this was recently directly tested by Maher et al in healthy human volunteers.75 Using radionuclide plethysmography, the authors measured venous blood flow and using strain gauge plethysmography they measured arterial blood flow. They found that under normal physiologic conditions the nitrite potently vasodilated the venous circulation. After Maher et al exposed healthy volunteers to 12% oxygen, which reduced arterial hemoglobin oxygen saturation to approximately 85%, the potency of nitrite increased significantly in the arterial circulation but was unchanged in the venous circulation.75
The physiologic observations that nitrite infusions are tightly coupled to NO-hemoglobin formation are consistent with the classic reaction between nitrite and deoxyhemoglobin to form NO as described in Equation 1. We have hypothesized that this reaction possesses the sensor and effector properties necessary for hypoxic vasodilation. The reaction requires deoxyhemoglobin and a proton, providing oxygen and pH sensor chemistry, respectively, and generates NO, a potent vasodilator. Hypoxic vasodilation is a conserved physiologic response to hypoxia that matches blood flow and oxygen delivery to tissue metabolic demand; this process has been characterized for more than 100 years since the initial description by Roy and Brown in 1879.76,77 In mammalian species, the set point for hypoxic vasodilation occurs as the hemoglobin desaturates from 60% to 40%, around the intrinsic P50 of hemoglobin (40-20 mmHg).78 Nitrite appears to meet several basic criterion necessary for an effector of hypoxic vasodilation: the effector (1) must be naturally occurring, (2) must be metabolized or generated in response to tissue hypoxia, and (3) must potently vasodilate in response to hypoxia at an oxygen partial pressure of approximately 20 to 40 mmHg. Aortic ring studies show that nitrite–red blood cell–dependent vasodilation is initiated at an oxygen tension around the hemoglobin P50 (PaO2 of 40 mmHg for rat erythrocytes and 30 mmHg for human erythrocytes).23 Consistent with this, we have observed that this vasodilation occurs as hemoglobin unloads oxygen to 50% saturation, and that this vasodilation is consistent with the biophysical observation of a nitrite reductase activity of hemoglobin allosterically linked to its P50 (Figures 2,4).14,15,23

Biochemistry of nitrite-hemoglobin hypoxic vasodilation along the A1 to A5 arterioles. There exists a steady state anatomic location within the circulation from artery to vein that has the greatest concentration of R3 tetramers (orange line bottom panel), which possess the maximal nitrite reductase activity. At this location, there would always exist an equilibrium rate constant for nitrite reduction (red line top panel and green line bottom panel) and an equilibrium concentration of nitrite and deoxyhemes (maximized in R3 tetramer). As soon as one red cell moves downstream a new one would replace it, thus preserving the concentration of nitrite and R3 hemoglobin at that anatomic position. Thus, there will be an increased nitrite reductase rate and increased NO concentration surrounding the blood vessel. The NO concentration should increase in a bell curve distribution from artery to vein according to the predicted rate for nitrite reduction. The anatomic position of this equilibrium NO concentration will be responsive to tissue metabolism and oxygen consumption by moving the R-to-T transition upstream or downstream. Note that the rate of a second-order reaction is determined by the product of the concentration of 2 reactants and the bimolecular rate constant. In this case, the nitrite concentration changes only a little as hemoglobin deoxygenates, the deoxyhemoglobin concentration increases dramatically (blue line top panel, brown line bottom panel), whereas the bimolecular rate constant decreases dramatically (red line top panel, green line bottom panel) as hemoglobin goes from the R-to-T conformation. So the product of bimolecular rate constant and deoxyheme concentration peaks from 60% to 40% hemoglobin oxygen saturation when the most R3 tetramers are present. Figure was modified and reproduced from Gladwin et al24,43 with permission.
Biochemistry of nitrite-hemoglobin hypoxic vasodilation along the A1 to A5 arterioles. There exists a steady state anatomic location within the circulation from artery to vein that has the greatest concentration of R3 tetramers (orange line bottom panel), which possess the maximal nitrite reductase activity. At this location, there would always exist an equilibrium rate constant for nitrite reduction (red line top panel and green line bottom panel) and an equilibrium concentration of nitrite and deoxyhemes (maximized in R3 tetramer). As soon as one red cell moves downstream a new one would replace it, thus preserving the concentration of nitrite and R3 hemoglobin at that anatomic position. Thus, there will be an increased nitrite reductase rate and increased NO concentration surrounding the blood vessel. The NO concentration should increase in a bell curve distribution from artery to vein according to the predicted rate for nitrite reduction. The anatomic position of this equilibrium NO concentration will be responsive to tissue metabolism and oxygen consumption by moving the R-to-T transition upstream or downstream. Note that the rate of a second-order reaction is determined by the product of the concentration of 2 reactants and the bimolecular rate constant. In this case, the nitrite concentration changes only a little as hemoglobin deoxygenates, the deoxyhemoglobin concentration increases dramatically (blue line top panel, brown line bottom panel), whereas the bimolecular rate constant decreases dramatically (red line top panel, green line bottom panel) as hemoglobin goes from the R-to-T conformation. So the product of bimolecular rate constant and deoxyheme concentration peaks from 60% to 40% hemoglobin oxygen saturation when the most R3 tetramers are present. Figure was modified and reproduced from Gladwin et al24,43 with permission.
There are interesting kinetic versus equilibrium considerations for nitrite reduction that are likely to have physiologic implications. As illustrated in Figure 4, in the arterial circulation blood hemoglobin is fully oxygenated (R state) and with rapid deoxygenation will first generate R tetramer with loss of one oxygen (R3 tetramers; R and T denote the oxy and deoxy tetrameric conformation, and the subscripted number denotes liganded oxygens) prior to conversion to T state. These R3 tetramers have a high reactivity with nitrite (high bimolecular rate constant) and although there are fewer available hemes to bind nitrite the rate of nitrite reduction will be maximal as they kinetically deoxygenate around the P50 (50% saturation with oxygen). This kinetic model is illustrated in Figure 4 by the rise in R3 tetramer during early deoxygenation and the peak rate of nitrite reduction at the P50. This biochemistry allows for rapid nitrite conversion to NO as red cells deoxygenate along the arteriolar vascular tree. Indeed, in experimental systems, the faster one deoxygenates the red cell in the presence of nitrite the faster vasodilation is observed.23 Note that in the arterial vasculature, measurements of tissue oxygen tension and hemoglobin oxygen saturation using modern methodologies suggest that much of the oxygen delivery occurs within the resistance arterioles, especially in the case of skeletal muscle.80 Thus, in these microvascular beds, the anatomic site of hypoxic sensing is proximal to the site of resistive control (arterioles and muscularized capillaries).
If one examines blood flow in these arterioles, it is notable that the vessels are filled with a constant column of flowing blood. As soon as one red cell moves downstream, a new one would replace it, thus preserving the concentration of nitrite and R3 hemoglobin at that anatomic position. Therefore, at any point within such a vessel, there exists a steady state concentration of R3 tetramers, which possess a maximal nitrite reductase activity. At this anatomic location, there would always exist an equilibrium rate constant for nitrite reduction and an equilibrium concentration of nitrite and R3 tetramer. Thus, there will be an increased nitrite reductase rate and increased NO concentration surrounding the blood vessel (Figure 4). The anatomic position of this equilibrium NO concentration will be responsive to tissue metabolism and oxygen consumption by moving the R-to-T transition upstream or downstream.
In venous blood, there are more available deoxyhemes to reduce nitrite to NO, but their reactivity is reduced as the hemoglobin will be largely in the T state. As blood and nitrite pool in the venous circulation deoxygenates, the nitrite in venous blood will bind to T-state tetramers and slowly convert to NO. Whereas the kinetic rate of nitrite reduction is slower, because of the low reactivity (bimolecular rate) of the T-state tetramer, the concentration of nitrite that can be reduced is greater, because of the greater concentration of deoxyhemes available to bind and reduce nitrite. Therefore, equilibrium venous conditions produce higher quantities of NO from nitrite reduction, albeit more slowly, and potentially with more associated NO autocapture (scavenging). This effect might produce slow venodilation and may be harnessed therapeutically for preload reduction in the treatment of heart failure.75
It is important to note that based on the remarkable potency of NO, very little nitrite must be reduced and escape the red blood cell. Consider that the EC50 (the concentration that dilates a vessel 50%) of NO is 5 nM and EC20 is less than 0.5 nM, whereas the concentration of nitrite in a red cell is 250 nM. Therefore a 20% vasodilation requires only 1/1000 molecules of NO or N2O3 to escape. It is also important to recognize in this context that blood flow is proportional to the change in vessel radius to the fourth power. A minimal increase in radius has marked effects on flow so that blood flow regulation occurs below the EC20 of NO.
These studies in aggregate suggest that nitrite represents a naturally occurring circulating small molecule that is converted to NO and induces vasodilation with increasing potency during hypoxia, consistent with a proposal that nitrite represents a physiologic effector of hypoxic vasodilation and hypoxic signaling. Further physiologic studies in animal models and humans with and without disease will be required to validate this thesis.
Role of myoglobin and the heme-globin family as functional nitrite reductases
Ongoing studies are now exploring the role of other heme-globins, such as myoglobin (which can be knocked out), as functional nitrite reductases that regulate the cellular response to hypoxia.37,54 In tissues such as the heart, liver, brain, and aorta the nitrite levels (0.5-10 μM)81,82 are greater than in blood (0.12-0.13 μM)83 and NO is known to regulate several hypoxic cell signaling responses including mitochondrial respiration and biogenesis,84-88 expression of hypoxia inducible factor-1 (HIF-1α),89,90 and angiogenesis.91-93 In these tissues, the reduction of nitrite may provide a mechanism by which NO is generated during hypoxia, when NO synthase becomes oxygen limited. Oxygen is a requisite substrate for the production of NO by nitric oxide synthase (Km = 100 μM; note exact value for NOS Km for oxygen is highly variable in the published literature),94 so in normoxic conditions, oxygen is used by NOS for NO generation. As oxygen tension decreases and oxygen becomes limiting for NOS-dependent NO formation, the nitrite reservoir can be reduced back to NO by reactions with deoxyhemoglobin and deoxymyoglobin. When oxygen concentration decreases to a value around the P50 of myoglobin (3.1 μM), myoglobin deoxygenates and reduces existing nitrite to bioavailable NO.37 This myoglobin pathway would ensure a constant supply of NO production along a large gradient of oxygen tensions in the tissue. This myoglobin-dependent reduction of nitrite would further be enhanced when tissue pH drops, accelerating the reaction.37
Myoglobin has interesting properties as a nitrite reductase. Although it will reduce nitrite only at very low oxygen tensions, because of its high oxygen affinity (low P50), it is a monomeric protein with a very low redox potential.16 In fact, its redox potential is similar to the R-state hemoglobin molecule. Because of the low heme redox potential, it will reduce nitrite approximately 60 times faster than T-state hemoglobin.16 As myoglobin is a monomer without allosteric behavior (consider it “locked” in the R state), the reaction of nitrite with deoxymyoglobin is a second-order reaction with a bimolecular rate constant of 12 M−1s−1 (37°C, pH 7.4) that yields equimolar amounts of metmyoglobin and iron-nitrosyl-myoglobin.14,37 Interestingly, both neuroglobin and cytoglobin have similar low heme redox potentials and high oxygen affinity, suggesting similar properties as myoglobin in terms of nitrite reduction.95,96
One subcellular function that is known to be regulated by NO is mitochondrial respiration. The binding of NO to the heme aa3 site on cytochrome c oxidase (complex IV) to inhibit respiration is well characterized, and is thought to be involved in the physiologic regulation of oxygen gradients, particularly during hypoxia.84,86,87,97 Our group found that the reduction of nitrite by deoxymyoglobin generates bioactive NO that is capable of inhibiting mitochondrial respiration both in isolated mitochondria and in heart homogenates.37 Consistent with a role for myoglobin in regulating hypoxic cellular respiration, Rassaf et al recently compared the effects of nitrite on cardiac function in wild-type and myoglobin knockout mice.54 Using magnetic resonance imaging (MRI) spectroscopy, they found that nitrite inhibited cardiac energetics only in the wild-type mice, reducing the delta energy for ATP hydrolysis and reducing whole-organ oxygen consumption. They hypothesized that this function may underlie cellular hibernation in response to cellular hypoxia.
Many studies now show that nitrite has potent biologic activity in limiting injury and cell death after ischemia-reperfusion stress. This effect is particularly potent in the heart, where concentrations of nitrite as low as 200 nM in plasma reduce cardiac infarction by 50% in mouse, rat, and dogs.43,71,98,99 Inhalation of NO in humans after orthotopic liver transplantation increases the plasma levels of nitrite by approximately 1 μM and limits liver infarction.66 The mechanism of bioactivation of nitrite in the ischemic organ is the subject of intense current study, with groups proposing a central role for xanthine oxidoreductase,100 the mitochondrial electron transport chain,101,102 and NO synthase,103 as well as nonenzymatic acidic disproportionation.104-106 Our studies and those of Rassaf and colleagues quantifying NO generation from nitrite in cardiac tissue and the inhibition of mitochondrial respiration suggest that more than 70% of these effects are attributable to the nitrite reductase activity of myoglobin.37,54 In studies using myoglobin knockout mice and wild-type controls, the Rassaf and Schrader groups have now found that the protective effects of nitrite after myocardial infarction in mice requires myoglobin (Hendgen-Cotta et al107 ). It is likely that multiple overlapping enzymatic pathways for nitrite reduction in tissue allow for graded reduction of nitrite to NO at different oxygen tensions in different organs.43
While these data specifically describe a role for myoglobin as a nitrite reductase that regulates mitochondrial respiration, they also highlight the potential for other hemoproteins, such as neuroglobin and cytoglobin, to be involved in hypoxic NO generation. Further investigation is needed to compare the relative efficiency of different heme proteins as nitrite reductases and characterize the physiologic processes they modulate.
Experimental evidence for an “interaction” between nitrite and deoxyhemoglobin and deoxymyoglobin
Despite current acceptance that nitrite is a potent vasodilator and that the nitrite reaction with deoxyhemoglobin and deoxymyoglobin generates NO, the hypothesis that the heme-globins are functional nitrite reductases remains controversial.50 The central experimental evidence supporting the nitrite reductase activity of the heme-globins is the “interaction” between hemoglobin or myoglobin and nitrite to produce a signaling event distinct from the signal from each species alone. For example, if one looks at the recent study by Yu et al55 evaluating inhaled NO and nitrite infusions to limit the hypertensive effects of hemoglobin-based blood substitutes, the authors clearly show that hemoglobin infusions into mice and sheep produce hypertension. Nitrite infusions at the concentrations used (approximately 10 μM) produced no blood pressure changes. But then the investigators infused hemoglobin and nitrite together, which completely eliminated the hypertensive effects. Although you might on first impression think this is a “neutral effect,” in fact it represents a highly significant physiologic “interaction.” In our aortic ring studies, we observed the same phenomenon under hypoxic conditions.52 Under normoxic conditions, oxyhemoglobin inhibited nitrite-dependent vasodilation, but under hypoxic conditions, deoxyhemoglobin did not appear to inhibit vasodilation. In actuality, there was a significant “interaction” between deoxyhemoglobin and nitrite that vasodilated the aortic ring preparation.
Note that nitrite is unique in this behavior. Authentic NO is inhibited by both oxyhemoglobin and deoxyhemoglobin to a similar extent.52 We propose that in the case of deoxyhemoglobin, NO is both scavenged and generated, whereas with oxyhemoglobin NO is only scavenged. The fact that there is a major “interaction” must be explained. There are only a few possibilities: (1) NO is scavenged only by oxyhemoglobin (this is not true, and we confirm this experimentally),52 (2) nitrite is scavenged only by oxyhemoglobin (this is not true; the rate of reaction of nitrite with deoxy is actually faster than oxy in the lag phases of both reactions),15 or (3) nitric oxide is produced and scavenged, increasing the relative NO concentration. With nitrite and hemoglobin, the NO production is offsetting scavenging.
Another important point to consider is that in biology hemoglobin or myoglobin is always present in the blood and cardiomyocyte, respectively. If one performs aortic ring vasodilation experiments with acetylcholine, the addition of only 100 to 10 000 μM heme (hemoglobin or myoglobin) inhibits all endothelial-dependent vasodilation. There is no dilation and no measurable NO formation, it is all scavenged. Nitrite appears unique in its ability to react with hemoglobin and generate NO signaling that can escape inhibition.
This interaction between nitrite and heme-globins to generate NO and NO signaling is also seen in hemoglobin and myoglobin experiments using mitochondria as a sensitive NO sensor. NO formed from nitrite reduction will bind to cytochrome c oxidase and inhibit respiration. Incubation of low concentrations of nitrite, less than 20 μM, does not inhibit respiration at low oxygen tensions. Similarly, incubation of hemoglobin or myoglobin does not inhibit respiration. But incubations of both together significantly inhibit respiration, indicating a significant interaction that produces an NO signal.37
In recent studies, we have confirmed that this interaction occurs in vivo in the heart using myoglobin knockout mouse models.107 In these experiments, the infusion of nitrite increases NO generation, tissue iron-nitrosyls, tissue S-nitrosothiols, and tissue cGMP levels; inhibits cellular respiration at low oxygen; and potently inhibits myocardial infarction after prolonged ischemia and reperfusion. All of these effects do not occur in the myoglobin knockout mouse, supporting an NO-generating and -signaling interaction between myoglobin and nitrite.
Alternative erythrocyte NO delivery hypotheses
The paradigm for hypoxic vasodilation by red blood cells was first advanced in 1995 with the hypothesis that the R-to-T transition of deoxygenating hemoglobin is coupled to the release of ATP from the erythrocyte, which by binding to purinergic receptors in endothelium causes vasodilation.108-111 This mechanism is supported by the observations of increasing concentrations of ATP in venous blood after hypoxia or physiologic acidemia, the in vitro release of ATP by hypoxic or acidic erythrocytes, and the retrograde propagation of vasodilation from the capillaries to precapillary arterioles after ATP/purinergic receptor/eNOS signaling. We have also observed ATP-dependent vasodilation mediated by red cells in our aortic ring bioassay studies, suggesting that this pathway complements the nitrite reductase pathway.23
A second mechanism was proposed that S-nitrosated hemoglobin (SNO-Hb) releases S-nitrosothiols during hemoglobin deoxygenation with subsequent vasodilation.112-115 Although we support the principle advanced by Stamler and colleagues that the red cell transduces hypoxic NO bioactivity (Jia et al112 ; Stamler et al113 ; Gow and Stamler114 ; Gow et al115 ), their proposed mechanisms have been challenged by multiple laboratories and the reader is encouraged to read these studies and formulate an independent assessment.23,41,116-132
We would propose that experiments with cysteine 93 transgenic mice can be used to test the SNO-hemoglobin hypothesis, and that nitrite experiments in the myoglobin knockout mouse can test the nitrite-reductase hypothesis. A cysteine 93 transgenic mouse has recently been engineered and physiologic experiments reveal no apparent phenotype in blood pressure control, pulmonary or systemic hypertension, exercise endurance, or hypoxic signaling and, perhaps most importantly, no effect of the cysteine substitution on red cell vasodilation of aortic rings.132 The cells vasodilate via release of ATP, independent of the presence or absence of the cysteine 93. In contrast, investigators in Germany have found that the myoglobin knockout mouse exhibits impaired cardiac nitrite-dependent signaling, supporting the nitrite reductase hypothesis.54 We have recently performed in vitro and in vivo nitrite studies in the myoglobin knockout mouse and wild-type control and found that myoglobin is essential for nitrite reduction to NO, nitrite-dependent inhibition of mitochondrial respiration, and nitrite-dependent cytoprotection after ischemia and reperfusion injury (myocardial infarction model).107
It has been suggested that SNO-Hb is a necessary intermediate in nitrite-dependent vasodilation. Although a role for SNO-Hb as an intermediate that limits NO autocapture is appealing in theory, this idea has not withstood the crucible of biochemical and molecular testing. For example, mutation of the cysteine 93 to alanine or modification of the cysteine with NEM actually increases the nitrite reductase rate and increases nitrite-dependent vasodilation.14,23 Furthermore, the bovine myoglobin preparations used in studies of myoglobin-nitrite reduction do not have cysteine 93,37 fish hemoglobins do not have cysteine 93,133,134 and the biopure HbOC product has pegylated cysteine 93,55 yet all these molecules transduce NO signaling via nitrite reduction effectively.
Conclusions
Nitrite transport in blood provides an endocrine form of NO that is shuttled from the lungs to the periphery, while limiting the exposure of authentic NO to the scavenging red cell environment. Then during the rapid hemoglobin deoxygenation from artery to vein the nitrite is reduced to NO, which can either form exportable N2O3, bind as iron-nitrosyl-hemoglobin (which is also released by oxidative denitrosylation), or become oxidized to nitrate. NO formed from erythrocytic135 or endothelial NO synthases is also oxidized in the red cell to nitrate or reacts with plasma ceruloplasmin to form nitrite.6 Such a cycle conserves NO in the one electron oxidation state. In this model, the nitrite pool represents the “live payload,” only one electron away from NO and N2O3.
We propose that nitrite subserves a critical NO signaling function in all heme-rich environments, such as blood, smooth muscle cells, the cardiomyocyte, and skeletal muscle cells. In these environments, NO is conserved by sequential oxidations, allowing nitrite storage and free nitrite diffusion. As oxygen tensions decrease, the bioactive nitrite pool generates N2O3 and NO to regulate signaling in the heme-rich environment.
Acknowledgments
This work was supported by National Institutes of Health (NIH, Bethesda, MD) grants HL058091 and HL078706 and by the Division of Intramural Research of the National Heart, Lung, and Blood Institute (Bethesda, MD).
National Institutes of Health
Authorship
Contribution: M.T.G. and D.B.K.-S. wrote the paper.
Conflict-of-interest disclosure: M.T.G. and D.B.K.-S. are named as coinventors on an NIH government provisional patent application for the use of nitrite salts in the treatment of cardiovascular diseases.
Correspondence: Mark T. Gladwin, Pulmonary and Vascular Medicine Branch, National Heart, Lung, and Blood Institute, Critical Care Medicine Department, Clinical Center, National Institutes of Health, Building 10-CRC, Rm 5-5140, 10 Center Drive, Bethesda, MD 20892-1662; e-mail: mgladwin@nih.gov; or Daniel B. Kim-Shapiro, Department of Physics, Wake Forest University, Winston-Salem, NC 27109-7507; e-mail: shapiro@wfu.edu.

