

Metabolic pathways intersect with many processes important in hematology, including thrombosis, innate and adaptive immunity, malignant transformation, and stem cell function. These pathways are increasingly recognized as potentially targetable for therapeutic intervention. In this issue of Blood, identify the energy sensor AMP-activated protein kinase (AMPK) in platelets as a regulator of thrombosis.1
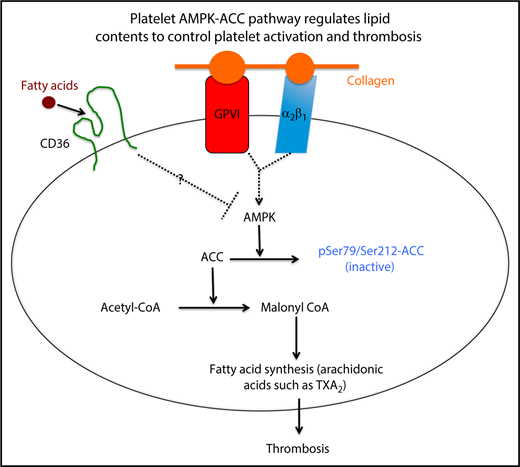
A simplified model showing how the platelet AMPK-ACC pathway integrates signaling through surface receptors for fatty acids and collagen into a metabolic response that promotes thrombosis by enhancing synthesis of thromboxane.
A simplified model showing how the platelet AMPK-ACC pathway integrates signaling through surface receptors for fatty acids and collagen into a metabolic response that promotes thrombosis by enhancing synthesis of thromboxane.
Platelet hyperreactivity is a key factor that promotes arterial thrombosis, and our laboratory and others have described how exogenous factors, including dyslipidemia and hyperglycemia, influence platelet activation. However, the impact of endogenous lipid metabolism on platelet function remains largely unexplored. Lepropre et al demonstrated that platelet AMPK, by phosphorylating acetyl coenzyme A (acetyl-CoA) carboxylase (ACC), regulates endogenous lipid synthesis, including the arachidonic acid–derived eicosanoid thromboxane A2 (TXA2), thereby contributing to thrombus formation (see figure).
AMPK is a ubiquitously expressed serine/threonine kinase normally activated by low cellular energy state due to its unique ability to sense intracellular AMP levels; as ATP becomes depleted, AMP levels rise and AMPK is activated.2 On activation, it has multiple targets, and the fundamental effect is to suppress metabolic processes that consume energy, such as lipogenesis, while stimulating pathways for energy production, such as fatty acid oxidation and mitochondrial oxidative phosphorylation. The major substrate for AMPK to achieve these effects is ACC, which converts acetyl-CoA to malonyl CoA, a limiting step in synthesis of fatty acids.3 AMPK, by phosphorylating ACC1/2 on serine residues (Ser79/Ser212), inhibits its activity and thereby puts a brake on fatty acid synthesis (see figure). To explore the in vivo role of the AMPK-ACC pathway in platelets, Lepropre et al used a double knock-in (DKI) mouse model in which the AMPK phosphorylation sites on ACC1/2 were mutated to prevent inactivation. They showed that this resulted in shorter tail bleeding times and enhanced carotid artery thrombosis in a FeCl3–induced injury model. This was associated with increased platelet phospholipid content and enhanced generation of TXA2, a potent platelet agonist, in response to collagen stimulation. The effects of AMPK-ACC inactivation appeared to be specific to collagen as DKI platelets showed normal activation to agonists for CLEC-2 and to G protein–coupled receptors for ADP and TXA2. Surprisingly, DKI platelets also showed normal activation to a small peptide agonist for GPVI, suggesting that there may be a requirement for additional collagen receptors, perhaps integrin α2β1 in AMPK activation.
Platelet hyperreactivity is often observed in patients with diseases associated with thrombotic risk, such as atherosclerosis, hyperlipidemia, diabetes, obesity, cancer, and chronic systemic vasculitis. This may be an important contributing factor to increased risk of myocardial infarction and stroke. Understanding how these conditions influence platelet activation will provide key knowledge for designing novel therapeutic interventions. Previous work focused on signaling through platelet surface receptors known to be components of innate immune system, including toll-like receptors and scavenger receptors. These receptors continuously scan the intravascular environment to detect endogenous and exogenous danger-associated signals. One of these receptors, the type II scavenger receptor CD36,4 interacts with specific danger signals, including oxidized low-density lipoprotein, to promote platelet activation and thrombosis by generating reactive oxygen species and activating redox sensing/signaling events,5 clearly implicating cellular metabolism in platelet function. Lepropre et al have now identified another essential metabolic sensor, AMPK, as a regulator of platelet reactivity. This raises the intriguing possibility of “rescuing” the abnormality in platelet metabolism as a therapeutic antithrombotic strategy in certain high-risk conditions. In fact, atherogenic conditions are associated with suppressed AMPK activities in various tissues,6 and many studies indicate that AMPK activating agents bring potential beneficial effects in other cell types.2
An important question not fully addressed is whether platelet bioenergetics are affected by the AMPK-ACC pathway. Lepropre et al detected no obvious alterations in oxygen consumption or glycolysis using the Seahorse metabolic flux assay. However, this should be interpreted with caution as washed platelets used in these assays are a very artificial condition for metabolic measurements. Another interesting question raised here is whether signaling through platelet surface receptors is intrinsically coupled to intracellular metabolic status. For example, CD36 facilitates long-chain fatty acid translocation across the plasma membrane.7 In myocytes, membrane CD36 binds and suppresses AMPK activity, whereas fatty acid binding releases AMPK for activation, leading to fatty acid oxidation.8 Because CD36 is highly expressed on the platelet surface, it will be important to test whether CD36, AMPK, and fatty acid metabolism are interconnected to coordinate platelet activation, especially under atherogenic conditions (see figure). Altogether, this interesting paper uncovers a novel metabolic regulatory mechanism in the platelets, which affects thrombosis in vivo, further demonstrating an important link between metabolism and thrombosis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.


