Key Points
Higher allelic burden at day 21 of post-HCT is associated with higher risk of relapse and mortality.
Longitudinal tracking of AML patients receiving HCT is feasible and provides clinically relevant information.

Abstract
Next-generation sequencing (NGS) has been applied to define clinically relevant somatic mutations and classify subtypes in acute myeloid leukemia (AML). Persistent allelic burden after chemotherapy is associated with higher relapse incidence, but presence of allelic burden in AML patients after receiving allogeneic hematopoietic cell transplantation (HCT) has not been examined longitudinally. As such, we aimed to assess the feasibility of NGS in monitoring AML patients receiving HCT. Using a targeted gene panel, we performed NGS in 104 AML patients receiving HCT using samples collected at diagnosis, pre-HCT, and post-HCT at day 21 (post-HCTD21). NGS detected 256 mutations in 90 of 104 patients at diagnosis, which showed stepwise clearances after chemotherapy and HCT. In a subset of patients, mutations were still detectable pre-HCT and post-HCT. Most post-HCT mutations originate from mutations initially detected at diagnosis. Post-HCTD21 allelic burdens in relapsed patients were higher than in nonrelapsed patients. Post-HCTD21 mutations in relapsed patients all expanded at relapse. Assessment of variant allele frequency (VAF) revealed that overall VAF post-HCTD21 (VAF0.2%-post-HCTD21) is associated with an increased risk of relapse (56.2% vs 16.0% at 3 years; P < .001) and worse overall survival (OS; 36.5% vs 67.0% at 3 years; P = .006). Multivariate analyses confirmed that VAF0.2%-post-HCTD21 is an adverse prognostic factor for OS (hazard ratio [HR], 3.07; P = .003) and relapse incidence (HR, 4.75; P < .001), independent of the revised European LeukemiaNet risk groups. Overall, current study demonstrates that NGS-based posttransplant monitoring in AML patients is feasible and can distinguish high-risk patients for relapse.
Introduction
Advent and use of next-generation sequencing (NGS) have identified common somatic mutations and defined genetic subtypes of acute myeloid leukemia (AML).1-3 A subset of common somatic mutations detected at baseline has shown to be prognostic and has been incorporated into the recent risk-stratification systems.4-6 Unlike most solid tumors, longitudinal sample collection of bone marrow (BM) or peripheral blood has become a part of routine clinical practice in AML, which makes it practical to track mutations longitudinally without additional invasive biopsy procedures. Taking advantage of serial samples, there has been great interest in developing assays to measure a trace amount of AML cells after chemotherapy or stem cell transplantation using multicolor flow cytometry, quantitative polymerase chain reaction, or, more recently, NGS.7-11 Using NGS on serial samples collected at diagnosis and at the time of morphologic complete remission (CR), Klco et al demonstrated that persistent allelic burden at morphologic CR is common and higher allelic burden at morphologic CR is associated with an increased risk of relapse.12 More recently, studies using NGS on serial samples also observed similar patterns and reported that residual allelic burden at CR is associated with higher risk of relapse and mortality in AML patients.10,11,13 However, residual allelic burdens associated with clonal hematopoiesis of indeterminate potential such as DNMT3A, TET2, and ASXL1 during remission were observed to have limited power to predict overall survival (OS) and relapse risk.10,11,14
Currently, allogeneic hematopoietic cell transplantation (HCT) is a potentially curative treatment option in AML.15-19 HCT reduces risk of relapse and improves relapse-free survival and OS in intermediate- and adverse-risk AML in first CR.17,20,21 However, clinical outcomes after allogeneic HCT still vary among patients who achieved first CR.8,22-24 Apart from cytogenetics and a number of clinical variables identified at diagnosis, molecular markers that can be assessed either at diagnosis or after chemotherapy or HCT provide additional prognostic information, and reveal clonal hierarchy in relapsing patients. Mutation profiling on serial samples showed that some mutations persist even after therapy, leading to clonal expansion during remission, and eventually lead to relapse of leukemia.25-27 As some mutations persist at morphologic CR, a subset of mutations may still be detected after allogeneic HCT and could increase the risk of relapse and reduce OS.28,29 As such, association between allelic burden detected post-HCT and transplant outcome is a topic of interest.
To the best of our knowledge, no studies have explored clonal changes and allogeneic HCT outcome on serial samples taken before and after allogeneic HCT using NGS in a systematic manner. We hypothesized that allelic burden measured pre- and post-HCT by NGS could be prognostically relevant, particularly in terms of HCT outcome. Current study aims to investigate the feasibility of posttransplant monitoring using NGS and its prognostic implications on HCT outcomes in AML patients.
Materials and methods
Patient cohorts and acquisition of samples
One hundred four AML patients who received HCT between January 2004 and November 2015 were included in this study. The major inclusion criteria were age <65 years and maintenance of CR before HCT after standard 3+7 induction and consolidation therapy. Patients in CR with incomplete or partial hematologic recovery and patients diagnosed with acute promyelocytic leukemia were excluded. Detailed chemotherapy and transplantation procedures are provided in supplemental Materials and methods (available on the Blood Web site). In total, 529 samples from 104 patients were collected. Samples were obtained at the time of initial diagnosis, pre-HCT (before conditioning therapy), post-HCT (day 21, 90, 180, and yearly thereafter), and at relapse. At time of diagnosis, pre-HCT (before conditioning therapy), day 21 post-HCT, BM samples from all 104 patients were available. T-cell fractions (CD3+) from 80 diagnostic samples were fractionated using FACSAria III. Among 23 relapsed patients, samples from 20 patients were collected. For samples taken at later time points, 38 (36 from BM and 2 from peripheral blood [PB]), 20 (18 from BM and 2 from PB), and 4 (3 from BM and 1 from PB) samples were available for day 90, 180, and yearly thereafter. Lastly, 57 donor samples were collected and subject for targeted sequencing. This study was approved by the institutional ethics review board at Chonnam National University Hwasun Hospital and Soon Chun Hyang University Bucheon Hospital, Korea.
NGS and variant calling
The list of targeted genes was compiled from large-scale mutation-profiling studies as well as our previous studies on hematologic malignancies (supplemental Table 1).1,2,27,30,31 For the 3 patients who did not have a mutation shared between initial diagnosis and posttransplant relapse sample within our targeted gene panel, we performed whole-exome sequencing using the SureSelect Human All Exon Kit v4 (Agilent, Santa Clara, CA). Mean on-target coverage of targeted sequencing and whole-exome sequencing were 1725.6X and 220.2X, respectively (supplemental Table 2). Read processing and variant calling procedure were performed similar to our previous study after minor modifications27 ; detailed descriptions are provided in supplemental Materials and methods. Of 273 detected mutations at 1 or more sampling time points, 256 mutations were detected at diagnosis, 15 at relapse, and 1 at pre-HCT. The remaining 1 mutation was detected in a donor sample and transferred to a recipient. The minimum variant allele frequency (VAF) in variant calling procedure to be called was 0.02 for targeted sequencing and 0.05 for the whole-exome sequencing in at least 1 of the serial samples. Once mutations were called significant, allelic burden from all samples from the patient was traced, regardless of meeting the minimum VAF.
Statistical analysis
The day of the stem cell infusion was defined as day 0. OS was analyzed using the Kaplan-Meier method and the groups were compared using the log-rank test. Relapse incidence was calculated using the Gray method considering competing events. The prognostic impact of risk factors for OS and relapse incidence was determined using the Cox proportional hazard model and the Fine-Gray proportional hazard regression model, respectively. P < .05 was considered significant. A detailed description is provided in supplemental Materials and methods.
Results
Patient characteristics and treatment outcomes
In total, 104 patients diagnosed with AML were included in this study. Median age of enrolled patients was 42 years (range, 15-63 years). The revised European LeukemiaNet (ELN) risk group categorized 31 patients (29.8%) as favorable, 47 (45.2%) as intermediate, and 26 (25.0%) as adverse (Figure 1A). Myeloablative conditioning was performed in 85 patients (81.7%) and peripheral blood stem cells were used as a source of stem cell in 97 patients (93.3%). VAF 0.2% at day 21 after HCT (VAF0.2%-post-HCTD21) grouped 88 patients (84.6%) as the low VAF group and 16 (15.4%) as the high VAF group. The detailed patient characteristics and treatment outcomes are summarized in Table 1.

Distribution of risk groups and mutation dynamics of 104 AML patients. (A) Distribution of 104 patients in each risk group defined by the revised ELN recommendations for AML. Thirty-one patients were favorable risk, 47 were intermediate risk, and 26 were adverse risk. (B) Bar plot shows mutational status of genes frequently detected at diagnosis. Color indicates sampling time point. Mutation status at diagnosis is colored red, pre-HCT blue, and post-HCT green. (C) Reduction of allelic burden of 256 mutations detected at diagnosis from initial diagnosis to pre-HCT is described. (D) Further clearance of persistent allelic burden from pre-HCT to post-HCT (day 21) is shown. (E) Summary of mutational status of 256 mutations from initial AML clones until 21 days after allogeneic HCT.
Distribution of risk groups and mutation dynamics of 104 AML patients. (A) Distribution of 104 patients in each risk group defined by the revised ELN recommendations for AML. Thirty-one patients were favorable risk, 47 were intermediate risk, and 26 were adverse risk. (B) Bar plot shows mutational status of genes frequently detected at diagnosis. Color indicates sampling time point. Mutation status at diagnosis is colored red, pre-HCT blue, and post-HCT green. (C) Reduction of allelic burden of 256 mutations detected at diagnosis from initial diagnosis to pre-HCT is described. (D) Further clearance of persistent allelic burden from pre-HCT to post-HCT (day 21) is shown. (E) Summary of mutational status of 256 mutations from initial AML clones until 21 days after allogeneic HCT.
Landscape of somatic mutations in 104 AML patients at diagnosis
In total, we detected 256 somatic mutations from 90 of 104 AML patients (86.5%) at the time of diagnosis (Figure 1B; supplemental Figure 1; supplemental Table 3). The median number of mutations per patient was 2 (range, 0-9). The 256 mutations consisted of 144 nonsynonymous single-nucleotide variants (SNVs), 53 frameshift insertions, 20 stop-gain SNVs, 17 nonframeshift insertions, 9 synonymous SNVs, 7 frameshift deletions, 3 splicing mutations, and 3 nonframeshift deletions. The most frequent mutation was FLT3–internal tandem duplication (FLT3-ITD; n = 31 of 104, 29.8%; supplemental Table 4). Other frequently mutated genes include DNMT3A (25.0%), NPM1 (22.1%), CEBPA (14.4%), IDH2 (14.4%), NRAS (10.6%), and PTPN11 (10.6%).
Clonal dynamics of allelic burden from diagnosis, pre-HCT to post-HCT
We first examined the changes in mutation from diagnosis to pre-HCT at the time of CR. From diagnosis of AML to pre-HCT, we observed a significant reduction of VAF (mean reduction rate, 93.12%; P < .001; Figure 1C). When assessing their mutation status pre-HCT, mutations were persistent in >1 patient pre-HCT in DNMT3A (n = 17 of 26; 65%), IDH2 (n = 7 of 15; 47%), and FLT3-ITD (n = 6 of 31; 19%). In general, mutations in genes involved in DNA methylation (IDH1, IDH2, TET2, and DNMT3A) were persistent pre-HCT compared with other mutations where mean reduction rate was 80.5% (VAFs ≥ 2% pre-HCT in 27 of 54 mutations; 50%). For mutations affecting other biological pathways, only 15 of 202 mutations (7.4%) were detected at over 2% VAF pre-HCT where mean VAF reduction rate was 96.5%. Noticeably, only 7 of 54 mutations (13%) in genes involved in DNA methylation showed complete clearance pre-HCT, whereas complete clearance rate was 47.0% for the rest of mutations (95 of 202 mutations).
We next examined the changes in mutations detected post-HCTD21. When tracing remaining mutations pre-HCT (142 of 256 mutations) that originated from initial AML clones, HCT had a significant impact on remaining VAFs, clearing an additional 110 mutations (average VAF of 4.71% pre-HCT and 0.83% post-HCTD21; Figure 1D). At post-HCT, mutations were frequently detected in DNMT3A (n = 8 of 26; 31%), NPM1 (n = 3 of 23; 13%), and FLT3-ITD (n = 4 of 31; 13%). In addition, 9 cleared mutations pre-HCT were detected post-HCTD21.
Our results showed that both chemotherapy and HCT have significantly reduced allelic burden. Among the 256 mutations with detected allele frequency over 2% at initial diagnosis, only 42 mutations (16.4%) remained at over 2% allele frequency pre-HCT. In fact, only 142 of 256 mutations (55.5%) were detected pre-HCT (42 + 49 + 51 in Figure 1E) whereas 114 of the initial mutations were eradicated. At post-HCTD21, 41 mutations (28.9%) were detected including 9 mutations that were cleared pre-HCT. Among those 41 mutations, 19 mutations were detected at 0.2% or higher VAF. Other than mutations originated from the initial AML clone, an additional 4 mutations were detectable post-HCTD21. Altogether, 23 mutations from 16 patients were detected post-HCTD21, including 14 mutations persistent through chemotherapy and HCT; 5 mutations cleared pre-HCT, but reappearing post-HCTD21; 2 mutations from a relapse clone; 1 mutation from a nonleukemic clone; and 1 mutation from donor marrow.
Allelic burden detected post-HCTD21 and its association with posttransplant relapse
When tracing the 23 mutations from 16 patients who had post-HCTD21 VAF > 0.2% from all available serial samples, 20 mutations were detected prior to HCT (either at diagnosis or pre-HCT) and 2 acquired (or selected) at relapse (Figure 2A-B). A remaining mutation seems to be of donor origin (DNMT3A-R882C; supplemental Figure 2). From available samples taken at 3 months, we were able to track 6 of 23 mutations and observed that 4 mutations (2 DNMT3A, 1 FLT3, and 1 STAG2 mutations) were cleared by 3 months post-HCT. For the remaining 2 mutations (both in DNMT3A), 1 seems to be a donor origin (VAF = 0%, 8.18%, 3.42%, 3.72% in pre-HCT, donor, post-HCTD21, and 3 months post-HCT samples) and another seems to be from a nonleukemia clone (VAF = 0.27% and 8.26% in diagnosis and pre-HCT samples).
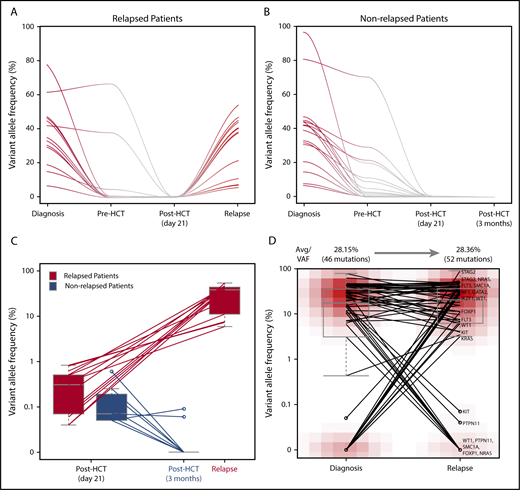
Dynamics of remaining allelic burden post-HCTD21and clonal association between initial AML and post-HCT relapse AML clone. Trace of mutations that are present at diagnosis and post-HCT (day 21) in (A) relapsed and (B) nonrelapsed patients. (C) Changes of remaining allelic burden from post-HCT (day 21) to relapse and 3 months post-HCT. Two patients relapsed at day 21 are excluded in this figure. (D) Comparison of allelic burdens in the initial AML clone and the post-HCT relapse AML clone.
Dynamics of remaining allelic burden post-HCTD21and clonal association between initial AML and post-HCT relapse AML clone. Trace of mutations that are present at diagnosis and post-HCT (day 21) in (A) relapsed and (B) nonrelapsed patients. (C) Changes of remaining allelic burden from post-HCT (day 21) to relapse and 3 months post-HCT. Two patients relapsed at day 21 are excluded in this figure. (D) Comparison of allelic burdens in the initial AML clone and the post-HCT relapse AML clone.
Among 16 patients, 7 patients did not relapse. Mutations from 2 patients seem to originate either from the donor (DNMT3A-R882C) or a nonleukemic clone (DNMT3A-V296M). Mutations present post-HCTD21 from the remaining 5 nonrelapsed patients were in DNMT3A (n = 3), ASXL1 (n = 1), or IDH2 (n = 1; supplemental Figure 2). For patients with available sample at relapse or at 3 months follow-up after HCT, we compared their allelic burden post-HCTD21 as well as the temporal dynamics. For relapsed patients, allelic burden present post-HCTD21 was higher than that of nonrelapsed patients and they all expanded at relapse (Figure 2C). On the other hand, post-HCTD21 mutations detected at initial diagnosis from nonrelapsed patients were cleared by 3 months after HCT, and if not, then by 6 months in patients with samples taken at later time points.
Among 104 patients, 23 patients (22.1%) relapsed, where samples from 20 patients were available and sequenced. For patients with available relapse samples, we investigated the clonal relationships between the original and relapse AML clone (Figure 2D). VAFs of mutations detected in the initial AML clone and post-HCT relapse clone were comparable (28.2% vs 28.4%). Among the 61 mutations detected from longitudinal monitoring in 20 relapsed patients, 37 were stable (60.6%), whereas 9 were cleared (14.8%) and 15 acquired (or selected) at relapse (24.6%). Seventeen patients carried at least 1 mutation shared between the initial AML clone and the posttransplant relapse clone within the targeted gene panel. For the remaining 3 patients, whole-exome sequencing confirmed that the post-HCT relapse clone of 2 patients shares at least 1 mutation with the initial AML clone (supplemental Tables 3 and 5). Altogether, for 19 of 20 relapsed patients, their post-HCT relapse clones share at least 1 exonic mutation with the initial AML clone.
Pre- and post-HCT VAF and its association with OS and relapse incidence after HCT
We next assessed whether the presence of mutational burden pre- or post-HCT was associated with OS and relapse incidence. Using recursive partitioning, we observed that patients with higher VAFs post-HCT (ie, ≥0.2%) had increased relapse incidence and worse OS. At post-HCTD21, the 3-year OS rate was significantly lower in patients with high VAF0.2%: 36.5% ± 12.3% and 67.0% ± 5.1% for high and low VAF0.2%, respectively (P = .006; Figure 3A). Three-year relapse incidence increased significantly with high VAF0.2%-post-HCTD21: 56.2% ± 12.4% vs 16.0% ± 3.9% (P < .001; Figure 3B). Using the same method, we obtained 2.0% as an optimal allelic burden cutoff. According to the pre-HCT VAF by 2.0%, we still observed patients with higher VAF at pre-HCT show worse survival. However, 3-year OS rates were 49.4% ± 9.7% and 67.3% ± 5.5% in patients with high and low VAF2% with marginal statistical difference (P = .14; supplemental Figure 3). Relapse incidence was not associated with the level of VAF pre-HCT (P = .44; supplemental Figure 3; supplemental Table 6).

Association between allelic burden and risk of OS and relapse risk. (A) OS and (B) relapse incidence depending on the presence of allelic burden at day 21 after allogeneic HCT. (C) OS and (D) relapse incidence depending on the presence of allelic burden in 47 patients classified as intermediate risk based on 2017 ELN recommendations. (E) OS and (F) relapse incidence depending on the presence of allelic burden in 26 patients classified as poor risk based on 2017 ELN recommendations. Number of patients at risk for every 2 years is shown below each Kaplan-Meier curve.
Association between allelic burden and risk of OS and relapse risk. (A) OS and (B) relapse incidence depending on the presence of allelic burden at day 21 after allogeneic HCT. (C) OS and (D) relapse incidence depending on the presence of allelic burden in 47 patients classified as intermediate risk based on 2017 ELN recommendations. (E) OS and (F) relapse incidence depending on the presence of allelic burden in 26 patients classified as poor risk based on 2017 ELN recommendations. Number of patients at risk for every 2 years is shown below each Kaplan-Meier curve.
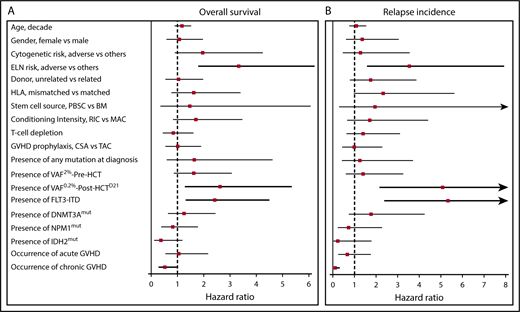
In univariate analysis, we found the VAF0.2%-post-HCTD21, FLT3-ITD, and ELN adverse-risk group to be associated with increased risk of relapse and poor OS, whereas occurrence of chronic graft-versus-host disease (GVHD) was a favorable factor for relapse incidence and OS. Multivariate analyses confirmed that VAF0.2%-post-HCTD21 (hazard ratio [HR], 3.07; 95% confidence interval [CI; 1.48-6.38]; P = .003) and the ELN adverse-risk group (HR, 3.68 [1.94-6.95]; P < .001) were independent prognostic factors for OS (Table 2). These 2 factors were also associated with higher relapse incidence: VAF0.2%-post-HCTD21 (HR, 4.75 [2.00-11.29]; P < .001) and ELN adverse-risk group (HR, 2.84 [1.13-7.13]; P = .027). Chronic GVHD was a favorable factor for relapse incidence (HR, 0.10 [0.03-0.32]; P < .001) but was associated with higher nonrelapse mortality (HR, 4.40 [1.27-15.29]; P = .020). When stratifying each ELN risk group, VAF0.2%-post-HCTD21 could further stratify the patients in intermediate- and adverse-risk group with respect to both OS and relapse incidence (Figure 3C-F).
Discussion
The present study aims to investigate the prognostic value of posttransplant NGS monitoring on serial samples in AML patients receiving allogeneic HCT. Assessment of clonal dynamics from diagnosis, pre-HCT to post-HCT, showed stepwise reduction of allelic burden via chemotherapy and HCT. Although most mutations were eradicated post-HCT, some mutations from initial AML clones were still detectable pre-HCT and post-HCT at day 21. Consequently, high VAF0.2%-post-HCTD21 defined the high-risk AML patients for mortality and relapse. Multivariate analyses showed that high VAF0.2%-post-HCTD21 and the ELN adverse-risk group were independent prognostic factors. VAF0.2%-post-HCTD21 further stratified the revised 2017 ELN intermediate- and adverse-risk groups. On the other hand, residual allelic burdens were not shown to affect HCT outcome when patients were in first remission prior to HCT, agreeing with Rothenberg-Thurley et al.32
Our results showed that longitudinal somatic mutation profiling on serial samples allowed assessment of clonal dynamics throughout the course of treatment including HCT. In particular, posttransplant NGS monitoring on serial samples taken after induction chemotherapy and after allogeneic HCT reveals stepwise clearance of allelic burden from the initial AML clone. First, although the majority of mutation burden is cleared pre-HCT, only 44.5% show complete clearance of somatic mutations (Figure 1E). Consistent with Klco et al,12 persistent mutations pre-HCT were enriched in genes associated with DNA methylation, such as DNMT3A, TET2, and IDH1/2. When traced further, HCT successfully eradicated the remaining VAF after chemotherapy, clearing a similar number of mutations as induction chemotherapy (Figure 1D). When reduction rates from both treatments were combined, we observed that HCT was more effective in reducing allelic burdens that are more persistent even after chemotherapy if not completely cleared (supplemental Figure 4).
In previous studies in same domain, allelic burden of mutations in DNMT3A, TET2, and ASXL1 (DTA) were excluded.10,11 In our data, although dynamics of DTA mutations were significantly different when compared with non-DTA mutations (repeated measures by general linear model; P < .001), we observed that conditioning regimen ablated both residual leukemia clones and host hematopoiesis including age-related clonal hematopoiesis (supplemental Figure 5). As such, we have not masked allelic burdens of DTA mutations as we believe that residual allelic burden after chemotherapy and after HCT are distinct.
Assessment of mutation dynamics demonstrated that allelic burden detected post-HCT had mostly originated from the initial leukemia clone (Figure 2A-B). Among 45 detected mutations (including 4 not detected at initial diagnosis) post-HCTD21, 11 mutations were cleared pre-HCT, but reappeared post-HCTD21. Among 11 mutations, 5 mutations from 5 patients were over 0.2% post-HCTD21, where 4 of 5 patients relapsed, demonstrating the prognostic value of posttransplant NGS. For relapsed patients, these mutational burdens detected post-HCT all expanded at relapse (Figure 2C). On the other hand, remaining allelic burdens were cleared by 6 months in nonrelapsed patients. Except for 1 patient, all relapsed AML clones carried at least 1 exonic mutation from the initial AML clone, suggesting the clonal origin of post-HCT relapse (Figure 2D).
The clinically relevant aspect of the current study is that monitoring of overall allelic burden can provide a prognostic indicator for long-term survival following HCT. In our cohort, patients with high VAF0.2%-post-HCTD21 showed increased relapse incidence and mortality compared with patients with low allelic burden <0.2% post-HCTD21 (Table 1; Figures 3-4). Identifying prognostic factors following HCT is important as the timing of intervention is critical for patients with impending relapse.33-35 With respect to posttransplant survival, day 21 of post-HCT is a critical time point to provide any therapeutic intervention early when impending relapse is highly suspected. The median time to relapse in patients with high VAF0.2%-post-HCTD21 was 57 days, which implies prompt therapeutic action should be delivered in a timely manner before clinically full-blown relapse follows.

Risk factors associated with OS and relapse incidence. Forest plots showing the HRs for (A) OS and (B) cumulative incidence of relapse on clinical and genetic variables. Significant variables are shown in bold. CSA, cyclosporine; MAC, myeloablative conditioning; PBSC, PB stem cell; RIC, reduced-intensity conditioning; TAC, tacrolimus.
Risk factors associated with OS and relapse incidence. Forest plots showing the HRs for (A) OS and (B) cumulative incidence of relapse on clinical and genetic variables. Significant variables are shown in bold. CSA, cyclosporine; MAC, myeloablative conditioning; PBSC, PB stem cell; RIC, reduced-intensity conditioning; TAC, tacrolimus.
Posttransplant NGS monitoring at day 21 is a valuable tool that stratifies prognosis in the patients after HCT, irrespective of baseline AML risk groups without additional biopsy procedure (Figure 4; Table 2; supplemental Table 6). Incorporation of mutation profiles into a prognostic stratification system in AML has refined its classification, improving stratification according to their long-term outcomes.36-41 Our study confirmed that baseline ELN risk classification has prognostic significance with regard to relapse incidence and OS (supplemental Figure 6; Table 2; supplemental Table 6). More importantly, in the respective ELN intermediate or adverse groups, VAF post-HCTD21 could further stratify the patients in terms of both relapse incidence and OS, implying that NGS-based longitudinal monitoring provides further prognostic value (Figure 3C-F). This finding demonstrated that monitoring allelic burden of mutations detected at initial diagnosis longitudinally is a very useful prognostic marker in AML for survival and relapse risk in addition to their baseline risk group at initial AML diagnosis. However, future study of comparing the results from NGS and multicolor flow cytometry using a much larger cohort in serial samples would further confirm the practicability of NGS in monitoring whether AML patients received HCT in a prospective manner. In the current study, we were not able to compare these 2 alternative technologies due to unavailability of flow cytometry data at day 21 post-HCT. In particular, for a very low level of allelic burden post-HCT, it would likely be more informative to perform NGS multiple times in deeper coverage. And this is certainly feasible as most patients carry <15 mutations within exon regions at initial diagnosis of AML.42
In summary, posttransplant NGS-based mutation profiling provides deeper understanding on mutation dynamics throughout the course of AML and its clinical relevance. First, it revealed dynamics of the AML clone throughout the sampling time points. We observed significant reduction in allelic burden after chemotherapy and HCT although some mutations were present after HCT. More importantly, presence of allelic burden at day 21 post-HCT can estimate the risk of posttransplant relapse and mortality. Overall, our data and analysis demonstrated that NGS-based monitoring of AML patients receiving allogeneic HCT provides valuable information and needs to be combined with the baseline mutational profile and clinical evaluation to predict posttransplant outcome and mortality.
The sequencing data reported in this article have been deposited in the European Nucleotide Archive (accession number PRJEB27619).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Aaron D. Schimmer for insightful discussion and advice on the manuscript. All authors also thank 3 anonymous reviewers for valuable comments and suggestions on this manuscript.
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) as funded by the Ministry of Science, ICT and Future Planning (NRF-2015R1A2A1A10054579 and NRF-2017R1C1B5017389), the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (1720160). Z.Z. acknowledges funding from the Natural Science and Engineering Council of Canada (NSERC) (RGPIN-2017-06743) and from the National Natural Science Foundation of China (NSFC) (81672923). D.D.H.K. acknowledges funding from the Leukemia & Lymphoma Society of Canada (New Idea Award) and the Princess Margaret Cancer Foundation. T.K. was supported by a scholarship from the NSERC (PGS-D). The biospecimens used for this study were provided by the Biobank of Chonnam National University Hwasun Hospital, a member of the Korea Biobank Network.
Authorship
Contribution: T.K., J.H.M., J.-S.A., H.-J.K., Z.Z., and D.D.H.K. designed the study; J.-S.A., Y.-K.K., S.-S.L., S.-Y.A., S.-H.J., D.-H.Y., J.-J.L., S.H.C., J.-y.L., M.-G.S., Y.J.L., S.K.S., S.-K.P., and H.-J.K. collected samples and performed experiments; T.K., M.S.T., and Z.Z. analyzed the sequencing data; T.K., J.H.M., Z.Z., and D.D.H.K. interpreted the data and performed statistical analyses; and T.K., J.H.M., H.-J.K., Z.Z., and D.D.H.K. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zhaolei Zhang, Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, 160 College St, Rm 608, Toronto, ON, Canada, M5S 3E1; e-mail: zhaolei.zhang@utoronto.ca; Hyeoung-Joon Kim, Genome Research Center for Hematopoietic Disease, College of Medicine, Chonnam National University, 322 Seoyang-ro, Hwasun-eup, Hwasun-gun, Jeollanam-do, Republic of Korea, 58128; e-mail: hjoonk@chonnam.ac.kr; and Dennis Dong Hwan Kim, Princess Margaret Cancer Centre, University of Toronto, 610 University Ave, Toronto, ON, Canada, M5G 2M9; e-mail: dr.dennis.kim@uhn.ca.