Key Points
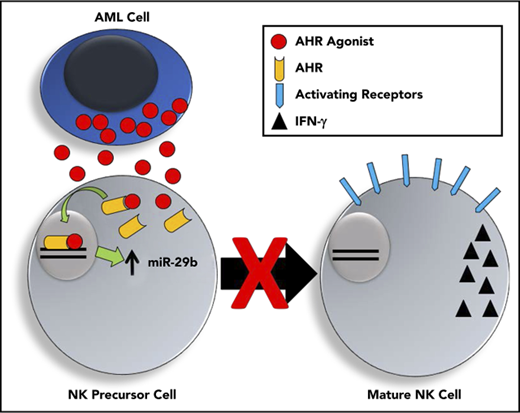
Human and murine AML activate the AHR pathway, which can regulate miR-29b expression and impair NK cell development and function.
AML-induced impairment of NK cell development and function can be reversed with AHR antagonist.

Abstract
Acute myeloid leukemia (AML) can evade the mouse and human innate immune system by suppressing natural killer (NK) cell development and NK cell function. This is driven in part by the overexpression of microRNA (miR)–29b in the NK cells of AML patients, but how this occurs is unknown. In the current study, we demonstrate that the transcription factor aryl hydrocarbon receptor (AHR) directly regulates miR-29b expression. We show that human AML blasts activate the AHR pathway and induce miR-29b expression in NK cells, thereby impairing NK cell maturation and NK cell function, which can be reversed by treating NK cells with an AHR antagonist. Finally, we show that inhibition of constitutive AHR activation in AML blasts lowers their threshold for apoptosis and decreases their resistance to NK cell cytotoxicity. Together, these results identify the AHR pathway as a molecular mechanism by which AML impairs NK cell development and function. The results lay the groundwork in establishing AHR antagonists as potential therapeutic agents for clinical development in the treatment of AML.
Introduction
Acute myeloid leukemia (AML) is a common form of cancer with a relatively poor prognosis and a 5-year survival rate <25%.1 Despite our growing knowledge concerning the molecular pathogenesis of AML, only modest progress has been made with respect to novel treatments that have translated into significantly improved patient outcomes. Recent efforts have thus focused on immunological strategies to mediate a specific and effective clinical response. Natural killer (NK) cells are innate lymphoid cells capable of recognizing and killing leukemic cells in an antigen-independent manner. Earlier work demonstrated the curative potential of donor NK cells following bone marrow transplants from haploidentical killer immunoglobulin receptor (KIR) mismatched donors.2 Despite these initial promising results, these immune-based strategies have not yet achieved broader clinical success. This could be attributable in part to immune suppression by the leukemic cells.3,4
MicroRNAs (miRs) are 18- to 24-nucleotide long RNA strands that are evolutionarily conserved and primarily function to target the complementary sequences found in 3′ untranslated regions of messenger RNAs (mRNAs) and thereby promote dicer-mediated degradation. They have long been implicated as both drivers of cancer as well as diagnostic and prognostic indicators of cancer.5,6 The evolutionarily conserved miR-29 family consists of miR-29a, miR-29b, and miR-29c whose seed sequences are nearly identical to one another.7 miR-29b is transcribed in 2 distinct loci as a cluster with either −29a or −29c on chromosome 7 (mir-29a/b1) or chromosome 1 (miR-29b2/c), respectively. Several transcription factors have already been demonstrated to directly regulate miR-29b expression including repressors such as C-Myc, NF-κB, transforming growth factor β, and PU.1 and inducers such as C/EBPA, GATA-3, MBP-1, STAT1, and Sox2.8-14 miR-29b has been linked to lymphoma, melanoma, leukemia, and prostate, colon, and lung cancer.15-19 In AML specifically, its expression has previously been demonstrated to inhibit expression of the antiapoptotic protein Mcl-1 and promote expression of proapoptotic protein BIM, resulting in leukemic cell death and improved patient survival, whereas decreased miR-29b expression was associated with resistance to apoptosis and more aggressive disease.20
NK cells pass through discrete stages of development in secondary lymphoid tissues beginning with stage 1, a CD34+ multipotent stem cell population, and ending with fully mature cytolytic stage 5 conventional NK cells or stage 6 NK cells that acquire memory to cytomegalovirus infection.21 The stage 3 NK cell developmental intermediates (NKDIs), identified as Lin−CD34−CD117+CD94−NKp80−, are notable for being the first CD34− subset with NK cell potential. These cells are also distinguished from other stages by their expression of the aryl hydrocarbon receptor (AHR) and interleukin-22 (IL-22).22 Of note, group 3 innate lymphoid cells (ILC3s) are similar in phenotype but likely with no NK cell potential.21 For those stage 3 cells that transition to stage 4 NKDIs, the former downregulate AHR and CD117 as they mature to acquire TBET, EOMES, CD94, and the capacity to produce interferon γ (IFN-γ) and kill major histocompatibility complex class I–deficient target cells.21 Stage 5 cells then acquire CD16 and functionally are enriched for cytotoxic mediators such as perforin and granzyme B. Stage 6 includes memory NK cells that are recognized by expression of CD57.21
Aside from their role in graft-versus-leukemia,2 NK cells are potent mediators of cancer surveillance and control.23 However, increasing evidence now exists to demonstrate that AML blasts are able to manipulate and impair the potency of NK cells.3,4,24-27 Recently, AML blasts were shown to impair the differentiation of immature NKDIs.28 In particular, the CD27+CD11b+ NKDI in mice and the CD94+/CD16− (CD56bright) NKDI in humans were shown to be reduced in AML as the result of decreased Tbet and Eomes, 2 transcription factors required for normal maturation of NK cells.28 miR-29b has previously been shown to prevent the translation of both Tbet and Eomes in T cells,29 and when NK cells from miR-29b–deficient mice were evaluated in mice with AML, the NK cell developmental arrest was reversed.28 Furthermore, the CD56bright NKDI (CD94+/CD16−) isolated from AML patients were shown to have significantly higher expression of miR-29b than the same cell isolated from healthy controls, suggesting this mechanism may be replicated in human disease.28 Although the NK cell developmental defect was associated with increased miR-29b expression, the factor or factors regulating miR-29b expression in AML patient NK cells were not known. Here we provide a specific mechanism by which AHR drives alterations in miR expression to impede human NK cell development.
AHR is a basic helix-loop-helix family of ligand-activated transcription factors originally discovered for its induction of the P450 pathway in response to environmental contaminants such as 2,3,7,8-tetrachlorodibenzo-p-dioxin.30 More recently, however, its role in cancer has become increasingly evident.31 AHR’s expression, nuclear translocation, and transcriptional activity have been associated with impaired cancer differentiation and lower patient survival.32 In support of its role in oncogenesis, transgenic mice overexpressing AHR form gastric and hepatic tumors.33,34 In addition to having a key role in benzene-induced AML, AHR has been shown to be a highly regulated pathway in AML, although its exact role in the disease has yet to be determined.35,36 In normal NK cell development, AHR activation also prevents the progression of stage 3 NKDIs to stage 4 NK cells,22,37 whereas AHR antagonists promote stage 3 NK cell differentiation to stage 4 NK cells.22,38 The exact downstream targets of AHR and the mechanism(s) by which this block in NK cell differentiation occurs have yet to be elucidated.
In this report, we demonstrate that AML blasts activate the AHR pathway in NK cells resulting in direct upregulation of miR-29a/b1 and suppression of NK cell development and NK cell function. Further, we show that inhibition of AHR in AML reverses its immune suppressive effect and increases the blast’s susceptibility toward NK cell cytotoxicity.
Material and methods
Tissue collection and isolation
All human tissue was obtained by protocols approved by the Ohio State University Internal Review Board (protocol #2009C0019). Fresh human peripheral blood (PB) was obtained from the American Red Cross (Columbus, OH), and fresh human tonsil tissue was obtained from Nationwide Children’s Hospital. AML patient samples were obtained through the assistance of the Ohio State University Leukemia Tissue Bank. NK cells and developmental intermediates were obtained from negative enrichment followed by fluorescence-activated cell sorting with a BD Biosciences Aria II to purity >98% as previously described.28,39 A sample flow diagram of fluorescence-activated cell sorting and culture immunophenotyping is presented in supplemental Figure 1 (available on the Blood Web site).
Cell culture
NK cells were cultured in media containing Dulbecco’s modified Eagle medium (DMEM)/F-12 (2:1), 1% antibiotic/antimycotic (Life Technologies), 24 mM 2-mercaptoethanol, 20 mg/mL ascorbic acid, 0.05 mg/mL sodium selenite (Sigma), and 10% heat-inactivated human AB serum (Valley Biomedical).39,40 Stage 3 cells were plated on nonirradiated OP9-DL1 murine stromal feeder cells in media supplemented with 20 ng/mL human IL-7 to evaluate differentiation in the presence or absence of AML cells. For coculture experiments, NKDIs were plated in 24-well plates on nonirradiated OP9-DL1 cells, and AML cell lines were added on top in a transwell insert. Media were replaced, and AML cells were refreshed 2× per week, and NKDIs were plated on new stroma 1× per week. Primary AML aphaeresis samples were maintained in RPMI medium containing 20% fetal bovine serum, 1% antibiotic/antimycotic (Life Technologies), human interleukin-3 (hIL-3; 10 ng/mL), hIL-6 100 ng/mL, and stem cell factor (100 ng/mL). The AHR antagonist CH223191 was used at a concentration of 3 μM (Calbiochem), and the AHR agonist 5,11-dihydroindolo[3,2-b]carbazole-6-carboxaldehyde (FICZ) was used at 300 nM (Enzo Life Sciences).22 HepG2 cells were obtained from American Type Culture Collection and cultured in DMEM/F-12 containing 20% fetal bovine serum and 1% antibiotic antimycotic (Life Technologies).
Quantitative real-time polymerase chain reaction (RT-PCR)
mRNA was isolated utilizing the Total RNA Purification Kit Plus (Norgen Biotek Corp.) Reverse transcription of mRNA was performed utilizing the Superscript VILO system (Life Technologies). Quantitative PCR (qPCR) was performed using either predesigned TaqMan probes or primers with SYBR Green Master Mix (Life Technologies) using a ViiA 7 RT-PCR system (Life Technologies). CYP1A141 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)39 (internal control) SYBR Green primers were obtained from Sigma-Aldrich as previously described,28 and miR-29b (000413 human miR-29b) and 001094 RNU44 internal control TaqMan primer/probes were purchased commercially (Life Technologies). Gene expression was normalized to an internal control (ΔCt = Ct gene of interest – Ct internal control). Relative mRNA expression for each gene tested was calculated as 2–ΔΔCt. Primers are listed in Table 1.
miR-29b lentiviral knockdown
Antisense miR-29b lenti-viral construct and controls (System Biosciences, Palo Alto, CA) were prepared, and stage 3 cells were transduced as previously described.42 Cells were then cultured in NKDI culture medium containing 10 ng/mL hIL-15 and hIL-1β for 7 days. Green fluorescent protein (GFP)+ stage 3 cells were then sorted to >98% purity and cultured for 14 days additional days in described medium supplemented with 10 ng/mL hIL-15.
Luciferase constructs and ChIP assays
CYP1A1 promoter, miR-29a/b1, and −29b2/c were cloned from genomic DNA isolated from primary human NK cells using PureLink Genomic DNA (Life Technologies). Standard PCR methods were employed using modified primers previously described for CYP1A143 and miR-29a/b18 and miR29b2/c generating segments with a 5′ KpnI and 3′ HindIII for cloning upstream of luciferase in the pGL3 plasmid backbone (Promega). Plasmids were transfected into HepG2 cells using Lipofectamine 2000 (Life Technologies), and cells were subsequently cultured in DMEM/F-12 with AHR modulating agents as described for 24 hours. Genes were modified using short interfering RNAs (Dharmacon) transfected with Dharmafect. Lysates were then prepared and analyzed using the Dual-Luciferase Reporter Assay (Promega). Chromatin immunoprecipitation (ChIP) was performed against AHR, or rabbit monoclonal isotype control (Cell Signaling), as per the manufacturer’s instructions (Pierce Magnetic ChIP). The presence of the miR-29b promoter was evaluated with primer sequences listed in Table 1, utilizing the RT-PCR methods described previously.
Flow cytometry
All experiments were analyzed with an LSRII flow cytometer (BD Bioscience), followed by FlowJo software analysis (TreeStar). Nonspecific staining was minimized by using the appropriate isotype controls. Antibodies used to sort for NKDIs and analyze NK cells by flow cytometry can be found in Table 2.
NK cell cytotoxicity
Primary human AML cells were maintained in culture with vehicle control (dimethyl sulfoxide [DMSO]) or CH223191 (3 μM) for 5 days prior to cytotoxicity assay. NK cells were obtained from fresh PB from normal donors (American Red Cross) and stimulated overnight with 2.5 ng/mL hIL-2. The following day, primary AML cells were used as targets where indicated in a standard 4-hour chromium release assay as previously described.28,39 Only cytotoxicity assays with <20% spontaneous AML cell death were included in these studies.
Statistical analysis
Statistical analysis of all experiments was performed by Prism GraphPad software. Student t test was used for comparing between 2 groups, and analysis of variance with correction for multiple comparisons was used when appropriate.
Results
AML generates soluble AHR agonists
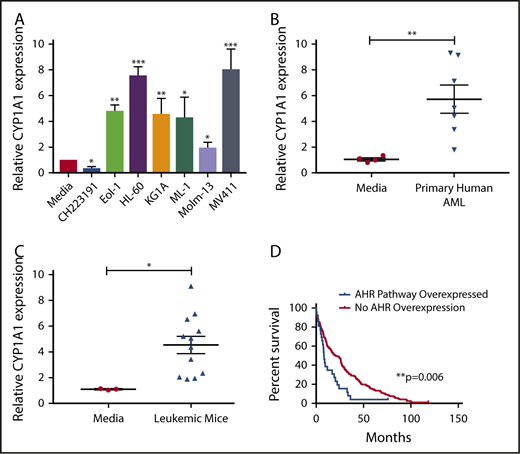
The human hepatocellular carcinoma cell line HepG2 has been used as an effective screening tool for detecting AHR agonists by monitoring expression of downstream promoters, such as that of CYP1A1, which contain AHR binding sites, more commonly referred to as dioxin responsive elements (DREs).44 By incubating HepG2 cells in medium from cultures of AML cell lines (Figure 1A) or primary AML patient samples (Figure 1B), we found that each sample induced CYP1A1 expression, suggesting that AML is capable of activating the AHR pathway of surrounding cells in a paracrine manner. We also evaluated AML blasts isolated from the murine MLL-PTD/FLT3-ITD AML model and saw significant induction of CYP1A1 expression (Figure 1C).28,45
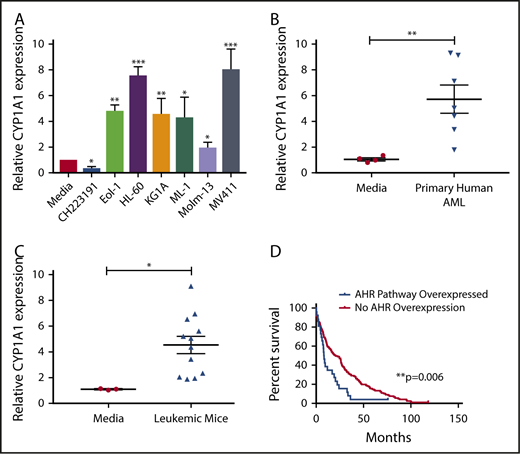
AHR Ligands are produced by AML cells lines and primary AML blasts. Conditioned media were harvested from AML cell lines (A), primary human AML blasts (B), or from murine leukemic cells (C) and used to treat HepG2 liver cells. RNA was isolated from HepG2 cells following 24-hour treatment, and CYP1A1 gene expression was evaluated by qPCR in each cohort as an indicator of AHR pathway activation. Data presented as expression relative to CYP1A1 expression in HepG2 cells treated with media only. A minimum of 2 independent experiments were conducted, representative figure depicted. (D) Kaplan-Meier overall survival curve comparing patients with higher mRNA expression of AHR-regulated genes (CYP1A1, CYP1A2, CYP1B1, UGT1A1, CYP2S1, and AHRR) relative to the mean expression in adult de novo AML. These data were generated through cBioPortal interface, based on data generated by The Cancer Genome Atlas Research Network. Student t test, *P < .05; **P < .01; ***P < .001. Error bars indicate standard deviation (SD).
AHR Ligands are produced by AML cells lines and primary AML blasts. Conditioned media were harvested from AML cell lines (A), primary human AML blasts (B), or from murine leukemic cells (C) and used to treat HepG2 liver cells. RNA was isolated from HepG2 cells following 24-hour treatment, and CYP1A1 gene expression was evaluated by qPCR in each cohort as an indicator of AHR pathway activation. Data presented as expression relative to CYP1A1 expression in HepG2 cells treated with media only. A minimum of 2 independent experiments were conducted, representative figure depicted. (D) Kaplan-Meier overall survival curve comparing patients with higher mRNA expression of AHR-regulated genes (CYP1A1, CYP1A2, CYP1B1, UGT1A1, CYP2S1, and AHRR) relative to the mean expression in adult de novo AML. These data were generated through cBioPortal interface, based on data generated by The Cancer Genome Atlas Research Network. Student t test, *P < .05; **P < .01; ***P < .001. Error bars indicate standard deviation (SD).
To understand the potential clinical significance of these findings, we examined the publicly available The Cancer Genome Atlas Research Network database to stratify AML patients relative to their AHR pathway activation footprint and perform a single variate analysis. In these studies, we evaluated overexpression of a panel of established AHR downstream targets, including CYP1A1, CYP1A2, CYP1B1, UGT1A1, CYP2S1, and AHRR,46,47 and surveyed for overall survival. The analysis showed that patients with evidence of AHR pathway activation had a significantly decreased overall survival (Figure 1D). The same downstream targets were also evaluated in invasive breast cancer, pancreatic adenocarcinoma, lung adenocarcinoma, and glioblastoma using the same single variate analysis and a significant association with survival was not observed (data not shown).
AHR is a direct transcriptional regulator of miR-29b
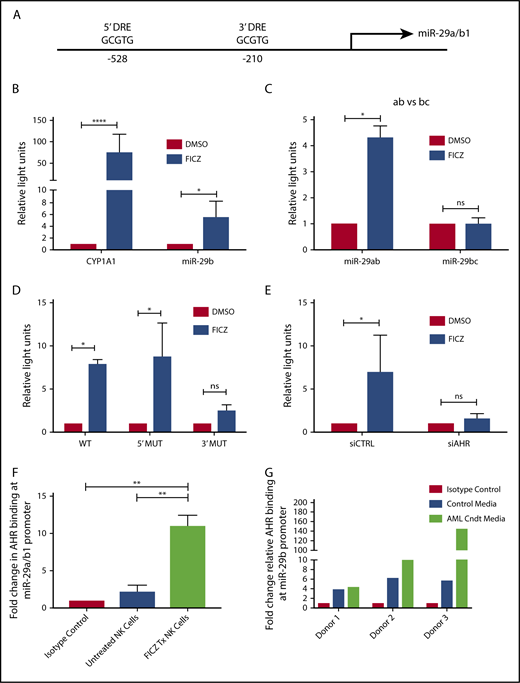
With the discovery that soluble factors in media obtained from AML cell lines and primary AML blasts activated the AHR pathway (Figure 1), and the previous studies demonstrating a role for AHR modulation in NK cell development,22 we postulated the elevation in miR-29b expression in CD94+/CD16− CD56bright NK cells in AML patients may be mediated through AHR activation,28 and that miR-29b could be a direct downstream target of AHR responsible for the observed phenotype. To address this hypothesis, we first inspected the promoter regions of both miR-29a/b1 and –b2/c for DREs (AHR binding sites).9 The evolutionarily conserved DRE, 5′-GCGTG-3′, was originally described based on the CYP1A1 promoter both in humans and in mice.48,49 We found 2 putative DREs at positions −528 and −210 from the transcriptional start site of human miR-29a/b1, referred to as the 5′ and 3′ binding sites, respectively, throughout this paper (Figure 2A).8,9 These putative DREs were also found to be conserved between humans and mice (data not shown). No putative DREs were found in the promoter of miR-29b2/c.
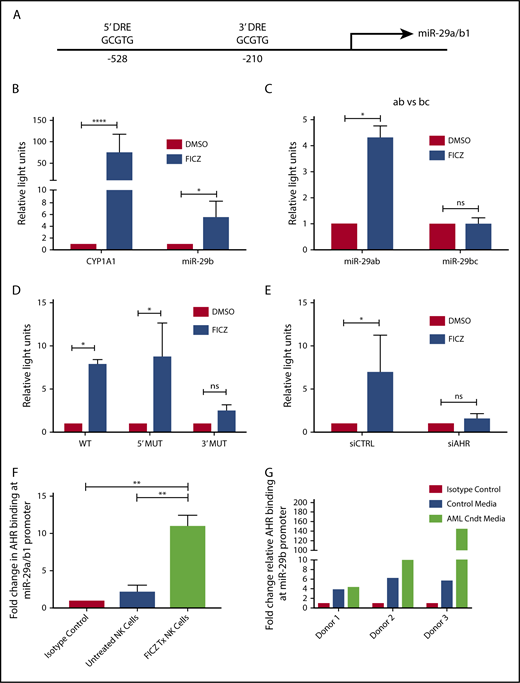
AHR regulates miR-29b. (A) Illustration depicting 2 predicted AHR-binding consensus sequences, also known as DREs, in the miR-29b promoter, highlighted in the red boxes. pGL3 luciferase reporter plasmids were cloned into HepG2 cells with seed sequence for either CYP1A1 (positive control) or miR-29b (B), miR-29a/b1 or miR-29b2/c (C), or miR-29b with the 5′ AHR site or the 3′ AHR binding site within the miR-29b promoter mutated (D). Each group was then treated with DMSO or FICZ (an AHR agonist). (E) pGL3 luciferase reporter plasmids were cloned into the miR-29b promoter in HepG2 cells, and AHR was knocked down in the same cells. The scrambled control (siCTRL) and the siAHR were treated with DMSO or FICZ and luciferase was measured. (F) CD56+/Lineage negative NK cells were isolated from PB and treated with vehicle DMSO vehicle control or FICZ. ChIP was performed with anti-AHR or isotype control antibody. The anti-AHR ChIP product was evaluated for miR-29b promoter expression in by qPCR to validate association between AHR and the miR-29b promoter. Data reported as expression relative to control. (G) Primary PB allogeneic NK cells were cultured with either media only or AML conditioned media, and miR-29b ChIP was performed similar to panel F. Minimum of 2 independent studies performed with representative figures depicted. *P < .05; ***P < .0001. Error bars indicate SD.
AHR regulates miR-29b. (A) Illustration depicting 2 predicted AHR-binding consensus sequences, also known as DREs, in the miR-29b promoter, highlighted in the red boxes. pGL3 luciferase reporter plasmids were cloned into HepG2 cells with seed sequence for either CYP1A1 (positive control) or miR-29b (B), miR-29a/b1 or miR-29b2/c (C), or miR-29b with the 5′ AHR site or the 3′ AHR binding site within the miR-29b promoter mutated (D). Each group was then treated with DMSO or FICZ (an AHR agonist). (E) pGL3 luciferase reporter plasmids were cloned into the miR-29b promoter in HepG2 cells, and AHR was knocked down in the same cells. The scrambled control (siCTRL) and the siAHR were treated with DMSO or FICZ and luciferase was measured. (F) CD56+/Lineage negative NK cells were isolated from PB and treated with vehicle DMSO vehicle control or FICZ. ChIP was performed with anti-AHR or isotype control antibody. The anti-AHR ChIP product was evaluated for miR-29b promoter expression in by qPCR to validate association between AHR and the miR-29b promoter. Data reported as expression relative to control. (G) Primary PB allogeneic NK cells were cultured with either media only or AML conditioned media, and miR-29b ChIP was performed similar to panel F. Minimum of 2 independent studies performed with representative figures depicted. *P < .05; ***P < .0001. Error bars indicate SD.
To determine if the putative DREs were functional, HepG2 cells were transfected with PGL3 luciferase plasmids cloned with the promoter of miR-29a/b1 or CYP1A1 (Figure 2B). Increased luciferase activity was observed with the miR-29ab1 promoter when treated with the AHR agonist FICZ. However, no change in luciferase activity was noted with the miR-29b2/c promoter, suggesting that all AHR regulation of miR-29b occurs at the miR-29a/b1 locus (Figure 2C; supplemental Figure 2). We did not appreciate any significant differences when the 5′ DRE was mutated or deleted; however, all of the FICZ-induced activity was abrogated when the 3′ DRE was altered showing that this downstream site is essentially exclusively used by AHR for regulation of miR-29b (Figure 2D). Silencing AHR by short interfering RNAs also negated the FICZ response with the miR-29a/b1 promoter confirming that AHR was essential for all FICZ induced luciferase activity (Figure 2E).
In order to determine whether AHR is capable of binding the miR-29a/b1 promoter in fresh primary human NK cells, PB CD3−CD56+ human NK cells were treated with vehicle control or FICZ for 24 hours, and then anti-AHR ChIP-PCR was performed with primers flanking the 3′ DRE (Figure 2F). A nearly 10-fold enrichment of the miR-29b 3′ DRE was found in FICZ-treated cells. To furthermore test the ability of AML cells to indirectly modulate miR-29b transcription in NK cells, we cultured allogeneic PB NK cells in AML conditioned media and performed ChIP experiments for the miR-29b promoter. We found that the conditioned media caused more AHR binding at the miR-29b promoter when compared with controls (Figure 2G). Consistent with the data derived from the HEPG2 system, these data show that AHR is a direct transcriptional regulator of miR-29b in primary human NK cells and can be enhanced by AML conditioned media.
miR-29b regulates NK cell development
We next sought to determine if the AHR-miR-29b axis existed within NKDIs. Stage 3 cells have been shown to have the highest relative AHR expression among NKDIs.50-52 NK cell development largely halts when NKDIs are cultured with AHR agonists.50 Indeed, we found that stage 3 cells cultured in the presence of FICZ upregulated miR-29b (Figure 3A), in addition to other downstream AHR targets (data not shown).
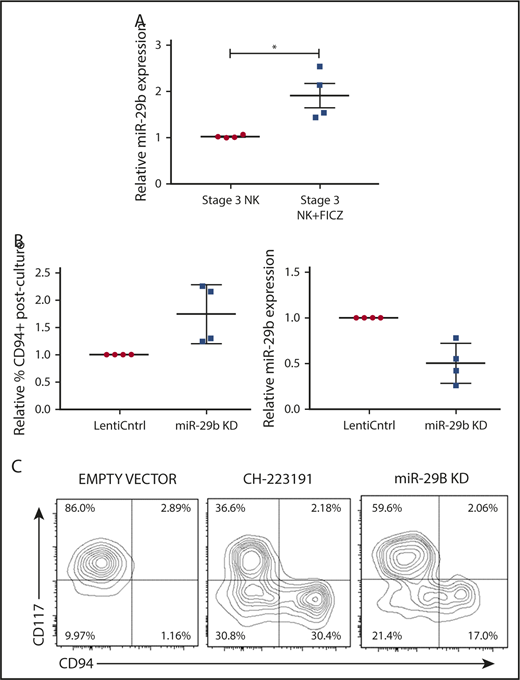
miR-29b is expressed by NKDIs and regulates NK cell development. (A) Freshly isolated tonsillar stage 3 cells were incubated for 24 hours with FICZ or DMSO, and miR-29b expression was determined by quantitative RT-PCR. (B) Stage 3 NKDI cells were cultured ex vivo in the presence of IL-15 and IL-1β and infected with an empty lentiviral vector (empty vector; negative control), cultured with the AHR inhibitor CH223191 (positive control), or infected with the miR-29b lentiviral knockdown (miR-29B KD). In total, 6 independent studies were performed with stage 3 NK cells from 10 primary human donors. (C) A representative figure is depicted. Student t test, *P < .05. Error bars indicate SD.
miR-29b is expressed by NKDIs and regulates NK cell development. (A) Freshly isolated tonsillar stage 3 cells were incubated for 24 hours with FICZ or DMSO, and miR-29b expression was determined by quantitative RT-PCR. (B) Stage 3 NKDI cells were cultured ex vivo in the presence of IL-15 and IL-1β and infected with an empty lentiviral vector (empty vector; negative control), cultured with the AHR inhibitor CH223191 (positive control), or infected with the miR-29b lentiviral knockdown (miR-29B KD). In total, 6 independent studies were performed with stage 3 NK cells from 10 primary human donors. (C) A representative figure is depicted. Student t test, *P < .05. Error bars indicate SD.
Next, we wanted to determine the effect of miR-29b modulation on NK cell development. We virally transduced stage 3 NKDIs freshly isolated from human tonsils with either miR-29b overexpression or knockdown lentiviral vectors. Stage 3 NKDIs transduced with miR-29b overexpression quickly and irreversibly underwent apoptosis in repeated experiments, consistent with prior reports showing that miR-29b induces apoptosis in leukemic cells.53 However, human stage 3 NKDIs transduced with miR-29b knockdown vectors showed consistent progression toward further NK cell maturation (Figure 3B). This pattern of NK cell maturation mirrored the results obtained with parallel studies using the AHR antagonist CH22319154-56 where inhibition of AHR led to more NK cell differentiation (Figure 3C).22 These results suggested that miR-29b expression mediates NK cell maturation.
AML suppression of human NK cell maturation and function is AHR dependent
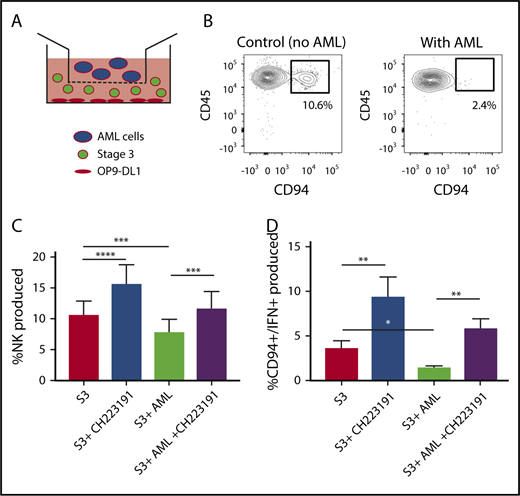
We next wanted to determine the role of AML-derived AHR activation on the developmental potential of immature NK cells. Stage 3 human NKDIs were isolated from tonsils and cultured with the AHR activating AML cell line MV411 in a transwell system for 4 weeks (Figure 4A). We found that in the presence of AML cells, there was a significant reduction in stage 4 CD94+ NK cells. In addition, these NK cells were less functional as there was also a reduction in CD94+/IFN-γ+ NK cells as compared with controls (Figure 4B-C; supplemental Figure 4). However, addition of the AHR antagonist CH223191 to the transwell cultures restored CD94+ NK cell development in the presence of the AML blasts, as well as significantly increased IFN-γ, close to levels obtained without AML blasts (Figure 4D).
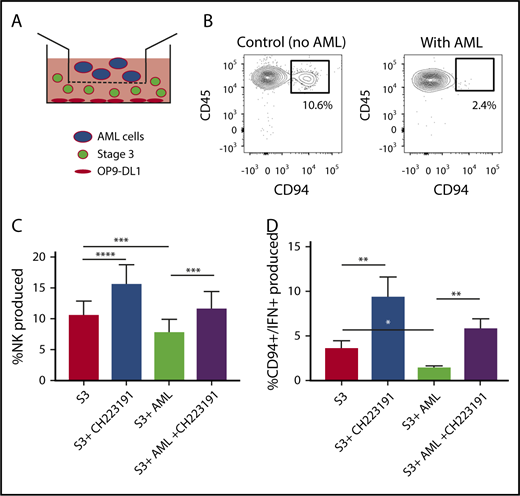
NK cell development is altered by AML cells. (A) Graphical illustration of NK cell development coculture in vitro. (B) Postculture flow cytometry analysis of CD94+ mature NK cells in the presence of AML vs control. (C) Stage 3 NK cells were cocultured via transwell with MV411 AML cells with and without treatment with the AHR inhibitor, CH223191 (CH); n = 14 independent donors. Stage 3 (S3) Lin−CD34−CD117+CD94−NKp80−. (D) Post-coculture, IFN-γ was evaluated following phorbol 12-myristate 13-acetate/ionomycin stimulation; n = 5 independent donors. Student t test, *P < .05; **P < .01; ***P < .001; ****P < .0001. Error bars indicate SD.
NK cell development is altered by AML cells. (A) Graphical illustration of NK cell development coculture in vitro. (B) Postculture flow cytometry analysis of CD94+ mature NK cells in the presence of AML vs control. (C) Stage 3 NK cells were cocultured via transwell with MV411 AML cells with and without treatment with the AHR inhibitor, CH223191 (CH); n = 14 independent donors. Stage 3 (S3) Lin−CD34−CD117+CD94−NKp80−. (D) Post-coculture, IFN-γ was evaluated following phorbol 12-myristate 13-acetate/ionomycin stimulation; n = 5 independent donors. Student t test, *P < .05; **P < .01; ***P < .001; ****P < .0001. Error bars indicate SD.
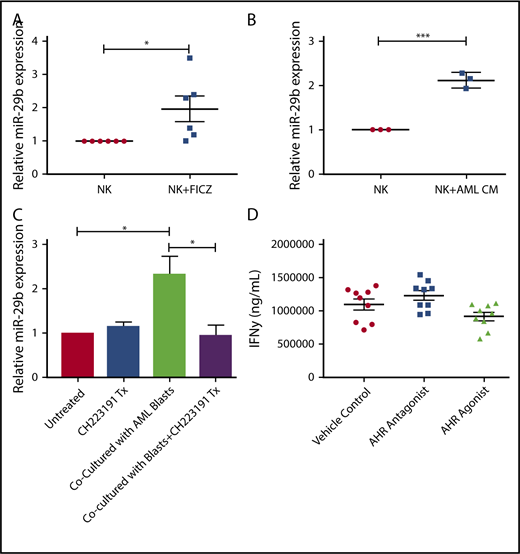
AML regulates miR-29b in mature human NK cells
Given the observation of reduced numbers of stage 4 NKDI cells in PB of AML patients, we sought to determine if activation of the AHR pathway, responsible at least in part for maturation arrest in the stage 3 NKDI, was also operative for the more mature stages 4 and 5 NKDI. We first found that the AHR agonist FICZ could increase miR-29b expression in PB NK cells (Figure 5A), and that treating PB NK cells with AML conditioned media also led to increased miR-29b expression (Figure 5B). To determine if AML was modulating NK cell miR-29b expression through an AHR-mediated pathway, transwell cocultures separating AML blasts from PB NK cells were used. These cocultures were then treated with or without the AHR antagonist CH223191. We observed that coculture of AML blasts with NK cells induced more than a twofold increase in miR-29b expression in the NK cells (Figure 5C). Furthermore, the antagonist CH223191 abrogated the increase in miR-29b, suggesting that this process was dependent on activation of AHR. We treated mature NK cells with both AHR activators and inhibitors and found no significant difference in mature NK cell function, suggesting that AHR does not play a strong direct role in NK cell function once NK cells have matured (Figure 5D).
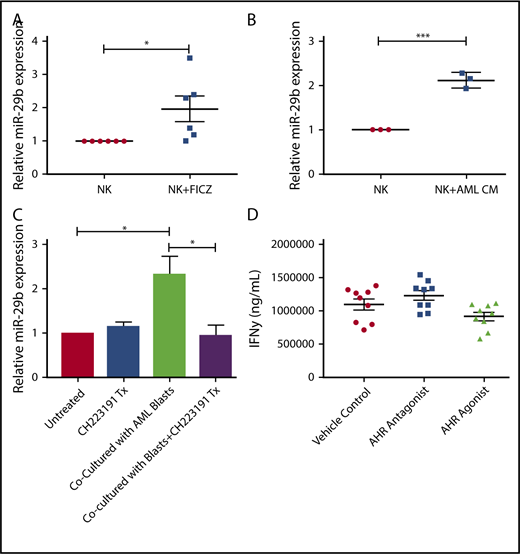
Effect of AHR ligands on miR-29b expression in NK cells. (A-B) CD56+/Lineage negative NK cells were treated with DMSO (vehicle control) or FICZ (AHR agonist), or conditioned media obtained from U937 AML cells for 4 hours, and miR-29b expression was evaluated by RT-PCR. (C) Primary NK cells were cultured alone or in transwell with AML blasts either untreated or treated with CH223191. miR-29b expression was evaluated by qPCR, representative donor depicted, n = 3. (D) NK cells were treated with DMSO (vehicle control), AHR agonist (FICZ), or AHR antagonist (CH223191) for 48 hours, stimulated with IL-12 and IL-18, and evaluated for the production of soluble IFN-γ by enzyme-linked immunosorbent assay (Student t test, *P > .05). Error bars indicate SD.
Effect of AHR ligands on miR-29b expression in NK cells. (A-B) CD56+/Lineage negative NK cells were treated with DMSO (vehicle control) or FICZ (AHR agonist), or conditioned media obtained from U937 AML cells for 4 hours, and miR-29b expression was evaluated by RT-PCR. (C) Primary NK cells were cultured alone or in transwell with AML blasts either untreated or treated with CH223191. miR-29b expression was evaluated by qPCR, representative donor depicted, n = 3. (D) NK cells were treated with DMSO (vehicle control), AHR agonist (FICZ), or AHR antagonist (CH223191) for 48 hours, stimulated with IL-12 and IL-18, and evaluated for the production of soluble IFN-γ by enzyme-linked immunosorbent assay (Student t test, *P > .05). Error bars indicate SD.
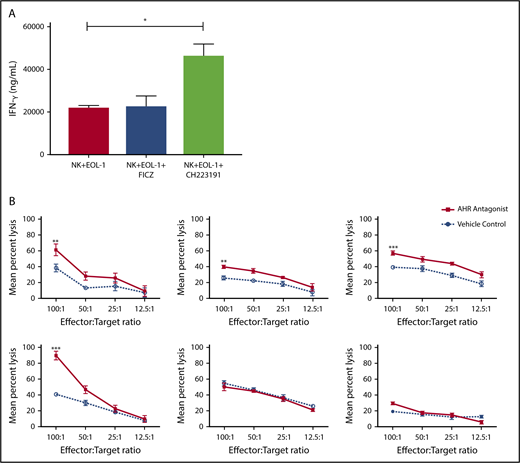
An AHR antagonist reduces AML blast resistance to NK cell–mediated cytotoxicity
AHR expression was previously reported in AML blasts46 and was validated here in primary human AML blasts and AML cell lines (Figure 6A). When primary human AML blasts were cultured in the presence of the AHR antagonist CH223191, there was reduced survival and increased spontaneous apoptosis as compared with vehicle control treated cells (Figure 6B-C). In addition, we found that when AML blasts were preincubated with CH223191 and subsequently cocultured with NK cells, the NK cells produced more IFN-γ compared with when the NK cells were cocultured with blasts preincubated with vehicle control or FICZ (Figure 7A). Likewise, when we tested for NK cell killing of AHR antagonist CH223191-treated primary AML blasts from 10 different patients, we observed increased killing of 8 of the 10 patient-derived blasts compared with controls (Figure 7B). Collectively, these findings suggest that antagonizing constitutive AHR activation in AML blasts lowers their threshold for spontaneous apoptosis and decreases their resistance to NK cell cytotoxicity.

Effect of AHR modulation on AML blasts and NK cell function. (A) AHR expression profiles of AML cell lines and primary AML samples were evaluated by flow cytometry. (B-C) Primary human AML samples were either treated with DMSO (vehicle control) or CH223191 for 48 hours, and cell survival by trypan blue counting and apoptosis by annexin V/propidium iodide (PI) flow cytometric evaluation, representative donor, n = 5 primary donors and 4 AML cell lines (*P = .03).
Effect of AHR modulation on AML blasts and NK cell function. (A) AHR expression profiles of AML cell lines and primary AML samples were evaluated by flow cytometry. (B-C) Primary human AML samples were either treated with DMSO (vehicle control) or CH223191 for 48 hours, and cell survival by trypan blue counting and apoptosis by annexin V/propidium iodide (PI) flow cytometric evaluation, representative donor, n = 5 primary donors and 4 AML cell lines (*P = .03).

Blocking AHR on AML cells increases susceptibility to NK cell IFN-y production and cytotoxicity. (A) EOL-1 cells were treated for 5 days with AHR agonist (FICZ) or AHR antagonist (CH223191) and then washed and cocultured with NK cells in the presence of IL-12 and IL-18 (10 ng/mL) for 48 hours and evaluated for the production of soluble IFN-γ by enzyme-linked immunosorbent assay. (B) Primary AML blasts were cultured for 5 days with AHR antagonist (red, CH223191) or vehicle control (blue, DMSO). Cells were then washed to remove antagonist. Normal donor NK cells were cocultured with the primary AML targets for 4 hours in a standard chromium release assay. Six representative primary AML donors depicted (n = 10 total evaluated). Student t test, *P < .05; **P < .01; ***P < .001. Error bars indicate SD.
Blocking AHR on AML cells increases susceptibility to NK cell IFN-y production and cytotoxicity. (A) EOL-1 cells were treated for 5 days with AHR agonist (FICZ) or AHR antagonist (CH223191) and then washed and cocultured with NK cells in the presence of IL-12 and IL-18 (10 ng/mL) for 48 hours and evaluated for the production of soluble IFN-γ by enzyme-linked immunosorbent assay. (B) Primary AML blasts were cultured for 5 days with AHR antagonist (red, CH223191) or vehicle control (blue, DMSO). Cells were then washed to remove antagonist. Normal donor NK cells were cocultured with the primary AML targets for 4 hours in a standard chromium release assay. Six representative primary AML donors depicted (n = 10 total evaluated). Student t test, *P < .05; **P < .01; ***P < .001. Error bars indicate SD.
Discussion
In this report, we elucidate a mechanism by which AML can evade the innate immune system, and we identify AHR as a potential therapeutic target to reverse this process. First, we show that both primary AML patient blasts and AML cell lines are potent activators of the AHR pathway, and we show that AML patients whose blasts overexpress genes in the AHR pathway have a significantly worse prognosis. Second, through luciferase and ChIP studies we discovered that activated AHR binds directly to the miR-29b promoter and regulates miR-29b expression. Third, we found that AHR activation in stage 3 NKDIs results in increased levels of miR-29b, and coculture with AML cells impaired further NK cell maturation and subsequent function in an AHR-dependent manner. Finally, we found that antagonizing AHR rescues the impaired development of immature NKDIs and even primes AML blasts for mature NK cell activation and killing. Collectively, our data suggest that AML utilizes activation of the AHR pathway to evade eradication by NK cells.
The finding that cancer can activate the AHR pathway has previously been reported in other malignancies. For example, glioblastoma multiform has been found to produce kynurenine (Kyn), an AHR agonist that impairs T cells in the microenvironment.41 Kyn is a tryptophan degradation product generated by tryptophan-2,3-dioxygenase, and the authors demonstrated that tryptophan-2,3-dioxygenase was expressed in the glioblastoma multiform cells as well as in a number of other cancers including Burkitt lymphoma, Ewing sarcoma, and bladder, cervical, colorectal, lung, and ovarian carcinomas.41 Kyn can also be produced through the indoleamine-2,3-dioxygenase 1 and 2 enzymes, which are similarly upregulated in a number of various cancers including colorectal and renal cell carcinoma.57 There have been studies showing that subsets of pediatric and adult AML patients with intermediate risk cytogenetics that have higher expression of indoleamine-2,3-dioxygenase or Kyn have a worse prognosis and worse overall survival, suggesting Kyn may play a role in the pathogenesis of AML.58-60 In our study, we documented the activation of the AHR pathway in AML blasts and its effects on NK cell development and function; however, we did not attempt to determine if activation of the AHR pathway was because of a specific ligand or multiple ligands. Because of the breadth of metabolites, dietary compounds, xenobiotics, and microbiota that have the ability to activate the AHR pathway, there could be a multitude of different ligands acting in concert in AML.61
Because of the extensive range of ligands that can activate the AHR pathway, there is great interest in understanding how targeting AHR and blocking downstream signaling can be translated to the clinical setting. The AHR antagonist, CH223191, has not yet been tested in clinical trials, but these preliminary findings suggest that it or other AHR antagonists with similar potency may be useful in the treatment of AML. Early clinical trials of StemReginin, a separate AHR antagonist, have proved useful in expanding undifferentiated CD34 cells in vitro and subsequently improved hematopoietic engraftment in patients.62 Our studies suggest that AHR antagonists clinically have the potential to restore normal NK cell development, which has been reported to be altered in AML patients,28 as well as make AML blasts more susceptible to NK cell–mediated killing. In addition, there was evidence to suggest primary AML blasts rely on AHR pathway activation for survival, and there may also be direct antitumor efficacy by blocking AHR pathway activation. Thus, future studies investigating the safety profile and potential for utilizing AHR antagonists directly in human therapy appear to be warranted.
The exact role of AHR in the pathogenesis of AML has yet to be defined, and it is still unknown if the downstream transcriptional profile in NK cells and AML blasts is similar. Preliminary studies suggest a role for AHR in mediating major histocompatibility complex I expression in AML blasts, and a putative AHR consensus binding site is located in the promoter region of HLA-A. However, additional mechanistic studies will need to be conducted to further define the functional consequences of AHR blockade in the AML blast population (data not shown). AHR has a broad range of targets in addition to miR-29b, and regulation likely differs in leukemic cells and immune cells. Given the fact that AML patients with overexpression of genes in the AHR pathway have a worse prognosis and our knowledge now of miR-29b as a direct target of AHR, one might assume that increased levels of miR-29b in AML blasts would correlate with worse prognosis. However, multiple studies have recently shown that decreased expression of miR-29b in AML blasts and cell lines actually correlates with increased DNA methylation and overall worse prognosis.20,63 In addition, it is important to note that our single variate approach in our data search is a simplified model of identifying the role of AHR in AML. The interactions are part of a complex network of signals, disease subtype and severity, and many other factors that are not accounted for with this approach and rather were performed more for hypothesis generating motives. In contrast, increased miR-29b actually correlates with increased leukemic apoptosis because of targeting of MCL-1, an antiapoptotic gene.63 Chromatin remodeling, mutations, or other alterations in the cancer cell itself, however, could dissociate AHR signaling in the AML blast from that in the NK cell. These biological differences highlight the importance of evaluating miR regulation in normal and malignant cells, as well as in immune cell subsets, as there are often cell- and tissue-specific differences governing their expression and target genes. This work also suggests the specificity for miR-based therapeutics is critical as these tissue-specific differences can lead to unanticipated off-targeted effects, such as evidenced by the initial miR-34a mimic clinical trials in solid tumors.64 Finally, the importance of defining the AHR signature in other cancers is crucial to delineate which cancer types have the best potential benefit from AHR targeted therapeutic modalities.
Defining targetable mediators of NK cell development in AML is critical for the successful implementation of NK cell–based therapeutics. NK cells are known to play a key role in innate immune surveillance against tumors, and previous work by Ruggeri et al has demonstrated that NK cells have the potential to be curative as a cellular therapy in AML.2 However, there has been a lack of broad clinical success following these early reports, and improvement in our understanding of how AML and the leukemic microenvironment mediate immune suppression may improve our ability to treat such patients successfully.2 This may be of particular importance in the posttransplant setting as NK cells are some of the earliest immune cells to emerge after transplant and can potentially be activated against minimal residual disease.65 Although our studies here do not investigate the early transplant period, it is possible that the leukemic microenvironment may still be a significant challenge to overcome. Therefore, addition of AHR antagonists or specific targeting of miR-29b expression in NKDI may be effective in promoting NK cell maturation and cytotoxic function to eliminate relapse following first remission in AML and/or following myeloablative transplantation. AHR also has been described in other cell types including ILC3 and TH17 cells, and how AHR is modulated in these cell types in the setting of AML and other AHR modulating cancers is an important avenue that has yet to be explored in depth. Further studies evaluating the impact of AHR blockade utilizing humanized in vivo modeling strategies will be critical next steps for therapeutic development and determining antitumor efficacy in a more complex microenvironment.
In summary, our study identifies a new mechanism by which AML is able to suppress NK cell development and function. With the potential for dual efficacy of AHR antagonism through the promotion of NK cell maturation, as well as lowering the susceptibility of AML cells to NK cell–mediated killing, this therapeutic pathway warrants further investigation to move it toward clinical application with the goal of improving treatment of AML patients.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Donna Neuberg for statistical guidance.
This work was supported by an American Association for Cancer Research award (17-20-46-MUND) (B.L.M.-B.) and by National Cancer Institute, National Institutes of Health grants (CA89341, CA095426, CA163205, CA016058, and CA068458 [M.A.C.] and F30CA196244 [S.D.S.]). The authors also gratefully acknowledge the use of the Ohio State University Comprehensive Cancer Center Analytical Cytometry Shared Resource and the AML cells provided by the Ohio State University Comprehensive Cancer Center Leukemia Tissue Bank Shared Resource, which is supported by National Cancer Institute, National Institutes of Health (P30CA016058).
Authorship
Contribution: S.D.S., A.G.F., M.A.C., and B.L.M.-B. designed the project; B.L.M.-B., S.D.S., A.P.N., Luxi Chen, Li Chen, M.H.Z., S.B.C., A.A.-R.T., N.H., M.W., and G.E. performed experiments; T.H., D.M.B., J.N.S., J.Y., X.Z., and A.M. provided mouse colony, patient samples, and research materials; S.D.S., A.G.F., M.A.C., and B.L.M.-B. wrote the manuscript; and all authors critically read and approved the draft manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bethany L. Mundy-Bosse, The Ohio State University, 460 W 12th Ave, Room 882 BRT, Columbus, OH 43210; e-mail: bethany.mundy@osumc.edu.