TO THE EDITOR:
The key event in the pathogenesis of systemic light chain amyloidosis (AL) is an unstable misfolded secondary or tertiary structure of a monoclonal immunoglobulin (IG) light chain that precipitates in the extracellular compartments.1 The plasma cell disorder underlying AL is likely to lie within the common spectrum of plasma cell diseases, but limited confirmatory data analyzing the genetic architecture of AL are available.2
To address this, we prospectively included 24 newly diagnosed histologically proven AL samples from the National Amyloid Centre, University College London, United Kingdom, before any treatment. AL was confirmed by immunohistochemistry or mass spectrometry. CD138+ cells and peripheral blood were isolated, and DNA was extracted and used in an exome capture protocol and sequenced as previously published.3 This AL data set was compared with previously published monoclonal gammopathy of undetermined significance (MGUS)4 and multiple myeloma (MM)5,6 data sets and filtered similarly to ensure comparability of mutation numbers. Detailed methods and patient’s characteristics may be found in the supplemental Methods and supplemental Table 1, available on the Blood Web site.
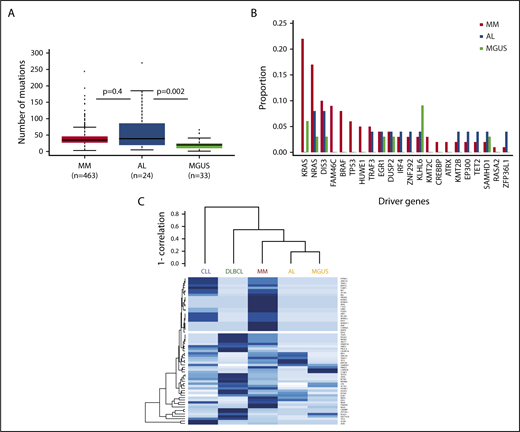
In terms of mutational burden, the median number of exonic, non-IG, nonsynonymous mutations per sample that had a tumor variant allelic fraction greater than 5% was 39 (interquartile range, 5-185), which is more than was seen in MGUS4 (n = 20; interquartile range, 1-41; P = .002), but is not statistically different from MM6 (n = 35; interquartile range, 3-74; P = .4; Figure 1A). When analyzing the individual genes, we identified 1491 genes that were mutated in our data set, with 236 of them being mutated more than once. As expected, there was no unifying mutation in AL. The data set was too small to identify any significantly mutated genes.7 Among the recurrently mutated genes (supplemental Table 2), mutations in IL7R, a gene that encodes for a cell surface receptor known to regulate the V(D)J recombination by altering the accessibility of DNA substrates to the recombinase in pro-B-cells,8 were noted. Thirty-seven percent of samples (n = 9) had a mutation in 1 of the 63 previously described MM driver genes.9 This is less than MM (84.1%)9 and similar to MGUS (36%).4 The number of mutated driver genes per sample ranged from 0 to 5. Among the 63 driver genes, 13 were mutated (Table 1). Most of these mutations were subclonal, with IG-adjusted estimated tumor fractions ranging from 18% to 100% (supplemental Table 3). We identified cases with MM hotspot mutations in NRAS (Q61R and Q61H), but not in KRAS. Even though a trend for fewer KRAS mutations in AL was observed, there was no significant difference in the incidence of driver mutations among MM, MGUS, and AL patients (Figure 1B). Interestingly, there were mutations in some of the other driver genes such as EGR1 (Q95R), DIS3 (D479E, M667L), IRF4 (S332G), and TRAF3 (K99 splicesite). Only the same TRAF3 (K99 splicesite) and DIS3 (D479E, M667L) mutations were seen in the MM data set, supplemental Figure 1A-E.9 In MM, IRF4 mutations are associated with t(11;14), and in this AL data set, the IRF4 mutation (S332G) occurred in a non-t(11;14) patient and was at neither the K123R hotspot seen in MM6 nor the L116R seen in chronic lymphocytic leukemia.10 There were no mutations in the previously reported adverse prognostic genes such as ATM, ATR, ZFHX4, and TP53; nevertheless, mutations in BRCA2 (P1088T, N372H) and the driver gene EP300 (I997V) were seen, suggesting DNA-repair pathway involvement. Copy number analysis did not reveal any copy number changes at these loci, suggesting the absence of biallelic inactivation. Finally, evidence would suggest the presence of NF-κB pathway activation, with not only mutations in the driver genes (such as TRAF3, IRF4) but also mutations in kinases (such as LYN [I165T]), downstream transducing molecules (CARD11 [R1077V]), and inhibitors (NFKBIE splicesite; supplemental Figure 2). We compared the mutational landscape of AL to previously sequenced MM (n = 1273)9 and MGUS (n = 33)4 samples. Overall, there were 101 genes in common between AL, MGUS, and MM. Ninety-three percent of the AL-mutated genes (n = 1386) were shared with either MM or MGUS. Only 7% of these genes (n = 105) had not previously been reported in MM or MGUS (supplemental Figure 3 and Table 4). None of these mutations was recurrent. A gene enrichment analysis of the 105 genes that were mutated in AL only11 did not reveal any specific pathway enrichment, suggesting they are random. There were no mutations in the ribosome subunit genes.12 Among the 68 differentially expressed genes,2 we only identified a mutation in TIAM2 previously reported in MM.2 Of note, we did identify a mutation in the eighth exon of PSMA2 (P223S) that was among the differentially expressed genes found by Abraham and colleagues.13 In a hierarchical clustering approach of driver mutation based on previously published data,4,5,14,15 AL clustered with MGUS and was closely related to MM (Figure 1C). In terms of translocations, we identified 7 t(11;14) encompassing 30% of cases. The breakpoints, located 2 to 600 kb upstream of CCND1, were consistent with those seen in other lymphoid malignancies (supplemental Figure 4). They were all generated via class switch recombination with breakpoints occurring in the IGHA1 (2/7) and IGHM (5/7) switch regions. There was no evidence of other canonical translocations, but we identified a t(1;14) involving IGHG4 and the RCC1 gene in a t(11;14) patient (supplemental Figure 5). There was no evidence of interchromosomal translocations involving MYC; 1 patient had an 8q24 gain, 5′ to MIR1208, suggesting MYC rearrangements also occur in AL. Regarding other cytogenetic abnormalities, there was no difference in the incidence of copy number changes with the exception of del(1p) and del(14q), which was lower than expected in MM (n = 1, 4%, [P = .008] and n = 0 [P < .0001], respectively), but similar to MGUS (supplemental Figure 6).
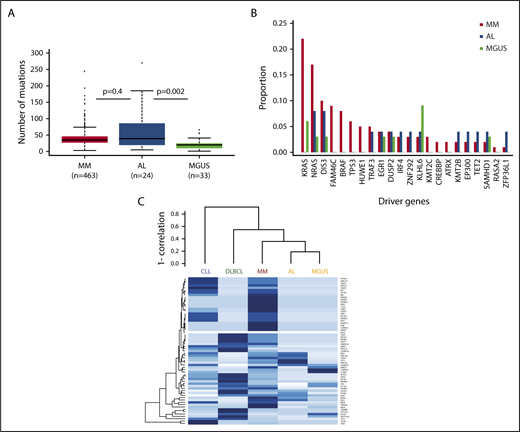
The mutational spectrum of amyloidosis resembles MM and MGUS. (A) Boxplot suggesting AL has more mutations than MGUS, but not a statistically different number than MM. (B) Frequencies of select driver mutations in AL, MGUS, and MM. (C) Clustering of driver mutation frequencies.
The mutational spectrum of amyloidosis resembles MM and MGUS. (A) Boxplot suggesting AL has more mutations than MGUS, but not a statistically different number than MM. (B) Frequencies of select driver mutations in AL, MGUS, and MM. (C) Clustering of driver mutation frequencies.
This is the largest data set of whole-exome sequencing of AL to date and combined, these data suggest the underlying disease in AL resembles other plasma cell disorders such as MGUS and MM. The number of mutations in AL was comparable to MM, suggesting AL is a more complex disease than MGUS. Similar to previously published AL cases that had undergone whole-exome sequencing,2 we failed to identify any unifying mutation. We were nonetheless able to detect MM-defined driver mutations. They occurred at similar frequencies to MGUS and were less common than in MM. They were predominantly subclonal, suggesting they occurred late during disease progression. There was evidence of mitogen-activated protein kinase (MAPK) activation with NRAS mutations (8%), in keeping with previously published data by Rossi and colleagues,16 as well as NF-κB activation (17%) and DNA-repair pathway alterations (12%). Unlike previous reports, none of the NRAS mutated patients had classical CRAB criteria, but 1 had another myeloma-defining event (SFLC ratio > 100, with an involved chain >1000 mg/L).17 These data are consistent with observations from MGUS and smouldering myeloma, where NRAS mutations may be found with lower frequencies than in MM.4,16,18 There was a 93% overlap in mutated genes, indicating a common spectrum of mutations between AL and other plasma cell disorders. Attempts to cluster patients based on MM driver genes placed AL close to MM and MGUS, and not with other lymphoid malignancies.
Overall, AL showed a similar mutation burden to MM, but resembled MGUS in terms of copy number changes and driver gene mutations, suggesting AL lies within the continuous spectrum from MGUS to MM. Given that the similarities with MGUS and MM are outweighing the differences, it is unlikely that the plasma cell biology per se explains the clinical presentation of AL; however, these data support the ongoing use of myeloma-based therapy for this disease.
The online version of this article contains a data supplement.
Acknowledgments
The authors acknowledge the Institute of Cancer Research Tumour Profiling Unit for their support. This work was supported by a Myeloma UK program grant, a Cancer Research UK CTAAC sample collection grant, and a Cancer Research UK Biomarkers and Imaging Discovery and Development grant, as well as funds from the National Institutes of Health Biomedical Research Centre at the Royal Marsden Hospital. E.M.B. was supported by a grant from the Fédération française de recherche sur le myélome et les gammapathies under the auspices of La Fondation de France.
Authorship
Contribution: E.M.B., D.R., G.J.M., B.A.W., and A.D.W. provided conception and design of the study; E.M.B., D.R., S.S., P.P., B.A.W., and A.D.W. provided acquisition of data; E.M.B., C.A., C.P.W., Y.W., S.K.J., M.A.B., N.W., M.F.K., D.C.J., J.R.J., C.P., C.S., T.F., C.D., F.E.D., G.J.M., and B.A.W. provided analysis of data; E.M.B., G.J.M., B.A.W., and A.D.W. provided writing of manuscript; and all authors provided review or revision of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ashutosh D. Wechalekar, National Amyloidosis Centre, University College London (Royal Free Campus), Rowland Hill Street, London NW3 2PF, United Kingdom; e-mail: a.wechalekar@ucl.ac.uk.