

Key Points
Myelofibrosis OCs are derived from the neoplastic clone.
The bone resorption capacity of myelofibrosis OCs is impaired.

Abstract
Bone marrow (BM) sclerosis is commonly found in patients with late-stage myelofibrosis (MF). Because osteoclasts (OCs) and osteoblasts play a key role in bone remodeling, and MF monocytes, the OC precursors, are derived from the neoplastic clone, we wondered whether decreased OC numbers or impairment in their osteolytic function affects the development of osteosclerosis. Analysis of BM biopsies from 50 MF patients showed increased numbers of multinucleated tartrate-resistant acid phosphatase (TRAP)/cathepsin K+ OCs expressing phosphorylated Janus kinase 2 (JAK2). Randomly microdissected TRAP+ OCs from 16 MF patients harbored JAK2 or calreticulin (CALR) mutations, confirming MF OCs are clonal. To study OC function, CD14+ monocytes from MF patients and healthy individuals were cultured and differentiated into OCs. Unlike normal OCs, MF OCs appeared small and round, with few protrusions, and carried the mutations and chromosomal abnormalities of neoplastic clones. In addition, MF OCs lacked F-actin–rich ring-like structures and had fewer nuclei and reduced colocalization signals, compatible with decreased fusion events, and their mineral resorption capacity was significantly reduced, indicating impaired osteolytic function. Taken together, our data suggest that, although the numbers of MF OCs are increased, their impaired osteolytic activity distorts bone remodeling and contributes to the induction of osteosclerosis.
Introduction
The hallmark of primary and secondary myelofibrosis (MF) is progressive bone marrow (BM) fibrosis.1 In addition to BM fibrosis, up to 70% of MF patients develop osteosclerosis, a well-established indicator of poor prognosis.2,3 Whereas the distribution and degree of BM osteosclerosis have been sporadically studied,4,5 the cause of osteosclerosis in patients with MF has not been identified.
Bone resorption by monocyte-derived osteoclasts (OCs) and bone reconstitution by mesenchymal stromal cell (MSC)–derived osteoblasts (OBs) are tightly regulated processes essential for bone remodeling.6-8 Unlike hereditary osteosclerosis, in which intrinsic defects in OC function tip the bone remodeling balance toward increased bone formation,9 MF osteosclerosis is thought to be induced by overstimulated MSC-derived OBs10,11 or impaired bone resorption, caused by overproduction of the OC inhibitor osteoprotegerin.12,13 However, clinical and laboratory data do not support those hypotheses. In MF, the number of monocytes, the OC precursors, is increased,14-16 whereas the number of MSCs, the OB precursors, is decreased or similar to that of healthy individuals.17,18 Furthermore, in patients undergoing BM transplantation, a reduction in OC numbers precedes reversal of osteosclerosis.19 Also, response to the Janus kinase 2 (JAK2) inhibitor ruxolitinib is accompanied by improvement or stabilization of BM fibrosis and osteosclerosis.20 In addition, the levels of cytokines that counteract the effect of osteoprotegerin and enhance monocyte-to-OC differentiation are significantly high.17,21
Because the number of clonal monocytes, the OC precursors, is increased in MF, and the number of monocyte-derived, morphologically and functionally distinct fibrocytes that induce BM fibrosis in primary MF is significantly increased,17 we postulated that either a reduction in OC numbers or an impairment in OC function hampers osteolysis and tips the bone remodeling balance toward increased bone formation.
Study design
BM biopsies and peripheral blood were obtained from MF patients and healthy donors after acquiring institutional review board–approved informed consent. In situ OC numbers were quantitated after tartrate-resistant acid phosphatase (TRAP) and cathepsin K immunohistochemical staining. BM OC clonality was assessed using phosphorylated JAK2 immunostaining and mutation analysis of JAK2 and CALR genes in microdissected single cells. CD14+ cell-derived OCs were grown in the presence of macrophage colony-stimulating factor and receptor activator of nuclear factor κB ligand (RANKL), and their shape and size, F-actin ring formation, nuclei number, cytogenetic abnormalities, mutant allele burden, cell fusion capacity, and bone resorption activity were analyzed (supplemental Methods, available on the Blood Web site).
Results and discussion
Using fluorescence immunohistochemistry and whole-tissue imaging analysis of BM biopsies from MF patients and healthy individuals, we detected significantly higher numbers of multinucleated TRAP/cathepsin K+ OCs along the bone perimeter of MF patients (n = 50) compared with normal controls (n = 3; median, 36.93 vs 7.04 cells per 100 mm; P = .027; Figure 1A-B). We also found that the numbers of OCs were increased in MF BM with high-grade fibrosis (Figure 1C), confirming previous reports in which morphological22,23 or histochemical studies24 detected large numbers of OC in BM biopsies of patients with high-grade, but not low-grade, BM fibrosis.25
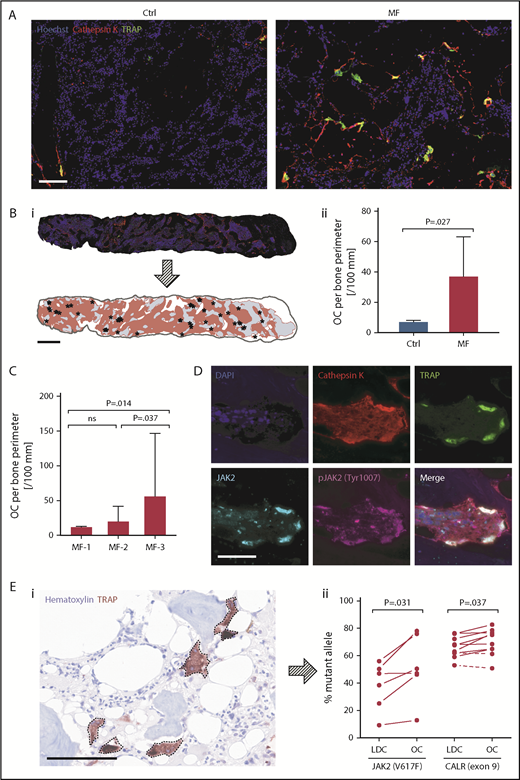
In situ analysis of normal and MF BM biopsy OCs. (A) Representative images of BM tissue from healthy controls (Ctrl; n = 3) and MF patients (n = 50) stained for TRAP and cathepsin K, with Hoechst 33258 as nuclear counterstain. As shown, few OCs are detected in normal BM biopsies, whereas abundant numbers of OCs are detected in the BM of MF patients. (B) Representative BM biopsy image (i) and OC quantitation (ii) in tissue sections from normal Ctrl and MF patients. Depicted BM section was stained for TRAP (green) and cathepsin K (red), with Hoechst 33258 as nuclear counterstain (blue). Whole-tissue image was assembled from 81 spectrally unmixed fields taken at a magnification of ×200, and a phenotype map of specimen was generated by a pattern-recognition algorithm. Red areas represent cellular BM, gray areas outline bone, and asterisks denote individual OCs. Bars represent median with 95% confidence interval (CI). (C) Comparison of OC numbers and BM fibrosis grade. As shown, MF patients with advanced-stage fibrosis (MF-3; n = 27) have higher numbers of OCs compared with patients with MF-1 (n = 4) and MF-2 (n = 13). BM fibrosis was independently graded using the European consensus criteria by pathologists who analyzed the patients’ BM biopsy specimens. Bars represent median with 95% CI. (D) A representative image of BM biopsy sections obtained from MF patients with a JAK2V617F mutation (n = 5). BM biopsy sections were immnuostained with antibodies against TRAP, cathepsin K, and total and phosphorylated (p) JAK2 protein. The cells’ nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images depict multiple OCs along thickened bone trabecula expressing high levels of pJAK2. At the time of biopsy, the patients’ peripheral blood JAK2V617F allele burden was 75.65%. (E) Analysis of randomly microdissected single OCs. (i) Representative image of BM biopsy sections obtained from CALR-mutated MF patients (n = 10) stained with anti-TRAP antibodies and hematoxylin QS nuclear counterstain. Dashed lines denote individual TRAP+ cells that were captured from the BM tissue sections by immunoguided laser microdissection. (ii) JAK2V617F (n = 6) and CALR exon 9 mutation (n = 10) allele burden in circulating low-density cells (LDC) and OCs randomly microdissected from the BM of mutation MF patients harboring 1 of those mutations. As shown, most single microdissected OCs expressed a higher mutant allele burden than LDC. Dashed lines denote samples with a decrease in allele burden. γ correction was not applied. Bars represent 100 μm (A,D,Ei) and 1 mm (Bi). ns, not significant.
In situ analysis of normal and MF BM biopsy OCs. (A) Representative images of BM tissue from healthy controls (Ctrl; n = 3) and MF patients (n = 50) stained for TRAP and cathepsin K, with Hoechst 33258 as nuclear counterstain. As shown, few OCs are detected in normal BM biopsies, whereas abundant numbers of OCs are detected in the BM of MF patients. (B) Representative BM biopsy image (i) and OC quantitation (ii) in tissue sections from normal Ctrl and MF patients. Depicted BM section was stained for TRAP (green) and cathepsin K (red), with Hoechst 33258 as nuclear counterstain (blue). Whole-tissue image was assembled from 81 spectrally unmixed fields taken at a magnification of ×200, and a phenotype map of specimen was generated by a pattern-recognition algorithm. Red areas represent cellular BM, gray areas outline bone, and asterisks denote individual OCs. Bars represent median with 95% confidence interval (CI). (C) Comparison of OC numbers and BM fibrosis grade. As shown, MF patients with advanced-stage fibrosis (MF-3; n = 27) have higher numbers of OCs compared with patients with MF-1 (n = 4) and MF-2 (n = 13). BM fibrosis was independently graded using the European consensus criteria by pathologists who analyzed the patients’ BM biopsy specimens. Bars represent median with 95% CI. (D) A representative image of BM biopsy sections obtained from MF patients with a JAK2V617F mutation (n = 5). BM biopsy sections were immnuostained with antibodies against TRAP, cathepsin K, and total and phosphorylated (p) JAK2 protein. The cells’ nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images depict multiple OCs along thickened bone trabecula expressing high levels of pJAK2. At the time of biopsy, the patients’ peripheral blood JAK2V617F allele burden was 75.65%. (E) Analysis of randomly microdissected single OCs. (i) Representative image of BM biopsy sections obtained from CALR-mutated MF patients (n = 10) stained with anti-TRAP antibodies and hematoxylin QS nuclear counterstain. Dashed lines denote individual TRAP+ cells that were captured from the BM tissue sections by immunoguided laser microdissection. (ii) JAK2V617F (n = 6) and CALR exon 9 mutation (n = 10) allele burden in circulating low-density cells (LDC) and OCs randomly microdissected from the BM of mutation MF patients harboring 1 of those mutations. As shown, most single microdissected OCs expressed a higher mutant allele burden than LDC. Dashed lines denote samples with a decrease in allele burden. γ correction was not applied. Bars represent 100 μm (A,D,Ei) and 1 mm (Bi). ns, not significant.
To determine whether MF OCs are derived from the neoplastic clone, we first analyzed BM biopsies of MF patients who carry the JAK2V617F mutation (n = 5). Using fluorescence immunohistochemistry, we detected phosphorylated JAK2 in multinucleated bone-lining OCs in MF but not normal BM (Figure 1D; supplemental Figure 1), suggesting that, as in MF patients’ diagnostic BM aspirates, JAK2 was constitutively phosphorylated in the patients’ BM OCs. Then, to further delineate these findings, we analyzed single microdissected TRAP+ OCs from BM sections of 6 MF patients with JAK2V617F and 10 with CALR exon 9 mutations. We found that the mutant burden in randomly microdissected OCs (Figure 1Ei) was higher than in the patients’ circulating low-density cells (median difference, 4.86% and 7.49%, respectively; Figure 1Eii). Taken together, these data suggest that, compared with normal BM, MF patients’ BM harbors large numbers of clonal OCs.
Because OCs induce osteolysis, and their numbers are increased in the BM of patients with MF, we wondered why osteosclerosis, rather than osteolysis, is commonly found in MF. To answer this question, we incubated circulating CD14+ monocytes in culture conditions that favor OC formation and investigated the morphological and functional properties of MF OCs (Figure 2A top arrow). Using phase-contrast microscopy, we compared the morphological features of cultured monocyte–derived OCs from 12 MF patients with those of monocyte-derived OCs from 3 healthy donors. In all cultures, MF OCs were rounder and had fewer protrusions (Figure 2Bi) and smaller diameters than healthy donors’ OCs (median, 94.5 vs 108.9 µm per cell; P = .009; Figure 2Bii). Then, using confocal fluorescence imaging, we found that, unlike normal OCs, MF OCs lacked F-actin–rich ring-like structures, a characteristic feature of resorptive cells (Figure 2Ci), and as described in a previous report,24 MF OCs harbored a lower number of nuclei than normal OCs (median, 4.05 vs 5.9 nuclei per cell; P = .02; Figure 2Cii).
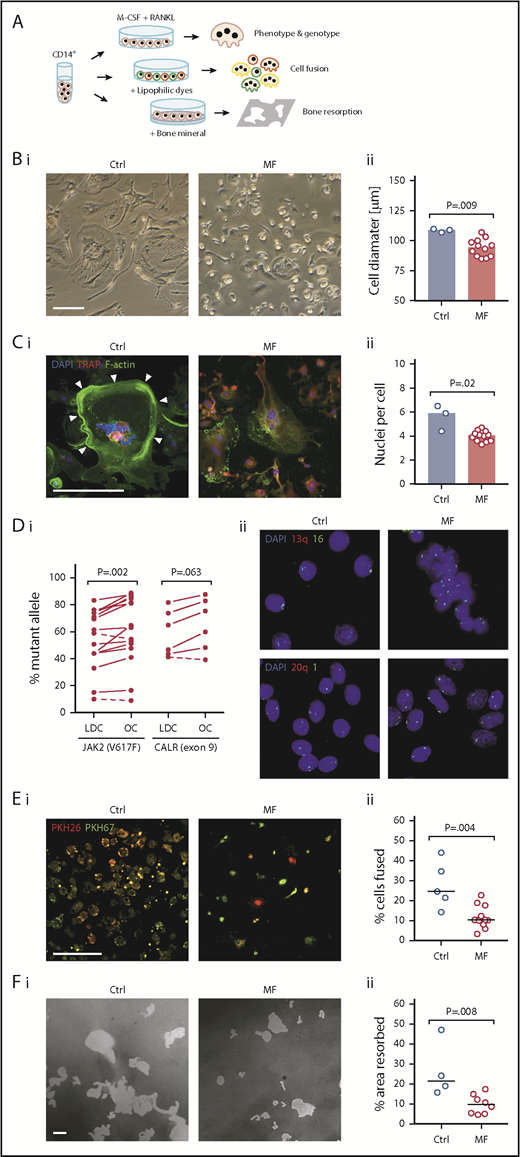
Characterization of monocyte-derived cultured OCs. (A) Schematic diagram of OC characterization studies. Top arrow: fractionated CD14+ monocytes were cultured in the presence of macrophage colony-stimulating factor (M-CSF) and RANKL. Cultured OCs were analyzed after 3 weeks. Middle arrow: cell fusion assay was carried out by culturing CD14+ monocytes labeled with either red or green lipophilic dyes at a 1:1 ratio. Bottom arrow: bone resorption assay was performed after CD14+ monocytes were cultured for 3 weeks in the OC culture assay on top of a bone mineral–coated surface. All experiments were performed in duplicate. Average values are depicted. (B) Morphological analysis of cultured OCs. (i) Representative phase-contrast images of mature OCs derived from CD14+ monocytes of healthy donors (Ctrl; n = 3) and MF patients (n = 12). Compared with Ctrl OCs, MF OCs are smaller and rounder, with almost no protrusions. (ii) Quantitative analysis of OC diameter. Dots represent average values for 500 cells from each patient. Bars denote median. (C) Assessment of OC nuclei. (i) Representative images of Ctrl’s (n = 3) and MF patients’ (n = 12) OCs stained with F-actin dye, anti-TRAP antibody, and 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain. Compared with normal OCs, MF OCs are characterized by fewer nuclei and abnormal F-actin ring (arrowheads) formation. (ii) Quantitation of cultured OCs shows a reduced number of nuclei in MF OCs. Dots represent average values for 500 cells from each patient. Bars denote median. (D) Clonal analysis of MF OCs. (i) Allele burdens of JAK2V617F (n = 15) and CALR exon 9 mutation (n = 6) in low-density cells (LDC) and OCs cultured from MF patients carrying a JAK2V617F or CALR exon 9 mutation. As shown, OCs carried a higher allele mutant burden than circulating LDC. Dashed lines denote samples with a decrease in allele burden. (ii) Fluorescence in situ hybridization analyses confirm that MF OCs are derived from the neoplastic clone. Shown are representative images (×100 magnification) of cultured OCs from MF patients with 13q and 20q deletions (n = 4). Individual chromosomes were stained using 1, 16, 13q, and 20q probes, and nuclei were counterstained using DAPI. Nuclei of MF OCs lack 1 long arm of chromosome 13 or 20 (nuclei showing only 1 red dot). Chromosomes 16 and 1 were used as internal controls (2 green dots). (E) Cell fusion capacity of OC-forming monocytes. (i) Representative images of OCs cultured from 2 populations of CD14+ monocytes from Ctrl (n = 5) and MF patients (n = 12). Membranes of live cells were stained using lipophilic dyes PKH26 and PKH67. A smaller number of cells harboring both dyes (yellow), indicating decreased cell fusion, is seen in MF patient–derived cells. (ii) Quantitation of cells with colocalization signals. Horizontal lines denote median. (F) Bone mineral resorption capacity of cultured OCs. (i) Representative images of OCs grown on top of bone mineral–coated surface from Ctrl (n = 4) and MF patients (n = 8). Cultured MF OCs exhibit a significant decrease in the total resorbed area (white). (ii) Quantitation of the resorbed mineral-coated surface areas. Horizontal lines denote median. No γ correction was applied. Bars, 200 µm.
Characterization of monocyte-derived cultured OCs. (A) Schematic diagram of OC characterization studies. Top arrow: fractionated CD14+ monocytes were cultured in the presence of macrophage colony-stimulating factor (M-CSF) and RANKL. Cultured OCs were analyzed after 3 weeks. Middle arrow: cell fusion assay was carried out by culturing CD14+ monocytes labeled with either red or green lipophilic dyes at a 1:1 ratio. Bottom arrow: bone resorption assay was performed after CD14+ monocytes were cultured for 3 weeks in the OC culture assay on top of a bone mineral–coated surface. All experiments were performed in duplicate. Average values are depicted. (B) Morphological analysis of cultured OCs. (i) Representative phase-contrast images of mature OCs derived from CD14+ monocytes of healthy donors (Ctrl; n = 3) and MF patients (n = 12). Compared with Ctrl OCs, MF OCs are smaller and rounder, with almost no protrusions. (ii) Quantitative analysis of OC diameter. Dots represent average values for 500 cells from each patient. Bars denote median. (C) Assessment of OC nuclei. (i) Representative images of Ctrl’s (n = 3) and MF patients’ (n = 12) OCs stained with F-actin dye, anti-TRAP antibody, and 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain. Compared with normal OCs, MF OCs are characterized by fewer nuclei and abnormal F-actin ring (arrowheads) formation. (ii) Quantitation of cultured OCs shows a reduced number of nuclei in MF OCs. Dots represent average values for 500 cells from each patient. Bars denote median. (D) Clonal analysis of MF OCs. (i) Allele burdens of JAK2V617F (n = 15) and CALR exon 9 mutation (n = 6) in low-density cells (LDC) and OCs cultured from MF patients carrying a JAK2V617F or CALR exon 9 mutation. As shown, OCs carried a higher allele mutant burden than circulating LDC. Dashed lines denote samples with a decrease in allele burden. (ii) Fluorescence in situ hybridization analyses confirm that MF OCs are derived from the neoplastic clone. Shown are representative images (×100 magnification) of cultured OCs from MF patients with 13q and 20q deletions (n = 4). Individual chromosomes were stained using 1, 16, 13q, and 20q probes, and nuclei were counterstained using DAPI. Nuclei of MF OCs lack 1 long arm of chromosome 13 or 20 (nuclei showing only 1 red dot). Chromosomes 16 and 1 were used as internal controls (2 green dots). (E) Cell fusion capacity of OC-forming monocytes. (i) Representative images of OCs cultured from 2 populations of CD14+ monocytes from Ctrl (n = 5) and MF patients (n = 12). Membranes of live cells were stained using lipophilic dyes PKH26 and PKH67. A smaller number of cells harboring both dyes (yellow), indicating decreased cell fusion, is seen in MF patient–derived cells. (ii) Quantitation of cells with colocalization signals. Horizontal lines denote median. (F) Bone mineral resorption capacity of cultured OCs. (i) Representative images of OCs grown on top of bone mineral–coated surface from Ctrl (n = 4) and MF patients (n = 8). Cultured MF OCs exhibit a significant decrease in the total resorbed area (white). (ii) Quantitation of the resorbed mineral-coated surface areas. Horizontal lines denote median. No γ correction was applied. Bars, 200 µm.
Given their abnormal morphological features, we wondered whether, similar to MF BM OCs, cultured MF OCs originate from the neoplastic clone. To answer this question, we cultured peripheral blood monocytes of MF patients whose BM cells harbored a JAK2V617F mutation (n = 15), CALR exon 9 mutation (n = 6), or fluorescence in situ hybridization–detectable chromosomal aberration (n = 4). As expected, we found that, similar to OCs dissected from BM tissue, cultured MF OCs carried JAK2V617F or CALR exon 9 mutant allele burdens that were higher than those of the same patients’ low-density cells (median difference, 16% and 4.43%, respectively; Figure 2Di), suggesting that a vast majority of cultured OCs originated from the neoplastic clone. To confirm this observation, we used fluorescence in situ hybridization that detected 13q and 20q deletions in MF OCs derived from patients with those chromosomal abnormalities, but not in normal OCs (n = 4; Figure 2Dii). Taken together, these data suggest that most MF OCs are derived from the neoplastic clone.
Because multinucleated OCs are formed by cell fusion, and F-actin ring formation is associated with efficient bone resorption,6 we first assessed the cell fusion capacity of OC-forming cells (Figure 2A middle arrow) using lipophilic dyes. Similar to their abnormal morphological features, MF OCs (n = 12) exhibited a significantly lower colocalization signal than normal OCs (n = 5; median, 10.3% vs 24.6%; P = .004; Figure 2E), suggesting that, compared with normal OC-forming cells, MF OC-forming cells undergo fewer cell fusion events.
Then, to assess the osteolytic function of OCs, we cultured CD14+ monocytes in the OC-forming culture conditions on top of a bone-mimicking surface. After 3 weeks, when mature OCs were formed, we quantitated the bone mineral resorption capacity of the OCs (Figure 2A bottom arrow). As predicted, we found that the bone mineral resorption capacity of MF OCs (n = 8) was significantly lower than that of normal OCs (n = 4; median, 9.65% vs 21.5%; P = .008; Figure 2F). Taken together, these data suggest that the bone resorption function of MF OCs is significantly impaired.
In conclusion, a vast majority of the OCs of MF patients are generated by neoplastic monocytes after fewer-than-normal fusion events. The morphological features of MF OCs are abnormal, and their bone resorption capacity is significantly impaired. In the absence of adequate bone resorption, the bone remodeling balance is likely tipped toward bone regeneration. As a result, osteogenesis prevails, and osteosclerosis ensues. Additional studies aimed at identifying the molecular mechanisms causing those abnormalities are warranted.
Presented in abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9 December 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Hanns A. Pielenz Foundation and performed in part in the Flow Cytometry and Cellular Imaging Core Facility, which is supported by the National Cancer Institute, National Institutes of Health (P30 CA016672), and in the Molecular Cytogenetics Facility, which is funded by the Center for Genetics and Genomics.
Authorship
Contribution: I.V. designed and carried out experiments, analyzed data, and wrote the manuscript; T.M. designed and carried out experiments and analyzed data; A.S.M. performed fluorescence in situ hybridization analysis; C.C.Y. and L.C. contributed samples for in situ studies; S.V. directed the project and supervised the study; and Z.E. conceived and supervised the study and wrote the manuscript. All authors provided critical feedback and helped shape the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zeev Estrov, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 428, Houston, TX 77030; e-mail: zestrov@mdanderson.org.


