

TO THE EDITOR:
Patients with xeroderma pigmentosum (XP) are abnormally sensitive to sunlight as a consequence of an autosomal recessive defect in nucleotide excision repair enzymes.1 Seven complementation groups have been reported, of which the most common is XP-C. The XPC protein is the first player in the repair process, and is necessary to recognize bulky DNA adducts and to recruit other DNA repair enzymes.2 In the absence of photoprotection, this deficiency causes skin tumors with an extremely high frequency.3
In the last 30 years, we established a cohort of 161 patients with XP-C from 142 consanguineous North African families living in France, all exhibiting a founder homozygous XPC c.1643_1644 delTG; p.Val548AlafsX572 (delTG) mutation leading to the complete absence of the XPC protein.4 Here, we report that 13 of these patients (8.07%), 7 men and 6 women, originating from 10 families, developed myelodysplastic syndrome with an excess of blast cells (EB-MDS), acute myeloid leukemia (AML), or T-cell acute lymphoblastic leukemia (T-ALL) at ages ranging from 7 to 29 years (Table 1; supplemental Patient Data, available on the Blood Web site). In these families, 12 additional siblings also suffering from XP-C did not develop an overt MDS or AML during the follow-up period (up to 20 years; Table 1; supplemental Figure 1). Overall, the frequency of MDS and AML in this cohort is several thousand times higher than what was reported in the corresponding French general population (0.04 per 100 000 for 15- to 19-year-olds; 0.19 per 100 000 for 20- to 24-year-olds).5 Using a PubMed search, we compiled all reported cancer cases in patients with XP worldwide between 1958 and 2018. Among 1510 patients with XP, 11 had a hematological malignancy (including 3 patients also in our cohort; supplemental Table 1). Of these, 5 had the North African XPC founder mutation delTG, and 5 others originating from the Mediterranean basin may carry the same founder mutation. The remaining patient had neurological signs suggesting XPA rather than XPC mutation.6
Details on our 13 patients with XP-C with MDS and/or AML/T-cell ALL are shown in Table 1 and supplemental Patient Data. All patients were diagnosed as classical XP because of their early sensitivity to sunlight and skin cancers. Specific DNA repair defect was confirmed in skin fibroblasts available from 9 patients.4 The homozygous founder XPC delTG mutation was identified in the 13 patients or relatives (Table 1). We had previously calculated the XPC delTG as being as old as 1250 years in North Africa, suggesting a common ancestor.4 XP frequency in Tunisia and carrier frequency in Morocco were estimated to be 1/10 000 and 1/250, respectively.7,8
Whole-exome sequencing (WES) of germinal DNA was performed in 6 patients from distinct families, and in both parents in 3 patients. We detected, as expected, a high level of inbreeding (supplemental Table 2) and a Middle East population distribution of variants, except in patient #6, in whom variant distribution was at the boundary between Middle East and European populations (supplemental Figure 2).
The hematological malignancies were diagnosed at a median age of 22 years, and median age at death was 25 years (Table 1). Only patient #11, who was diagnosed AML recently and received an allogeneic hematopoietic stem cell transplant (HSCT), remained alive 3 months after HSCT (supplemental Table 3). Of the 7 patients diagnosed as having MDS-EB, previously known as RAEB, 2 died before transformation, whereas the 5 others progressed to AML (>20% bone marrow blast cells). One of these patients had been treated for an early-progenitor T-ALL, and later an unrelated MDS-EB that progressed to terminal AML (patient #5; Figure 1A). Five other patients presented directly an overt AML at diagnosis. The thirteenth patient (#9) was diagnosed with a T-ALL. When available, bone marrow cytogenetics showed typical MDS/AML-associated abnormalities, with deletions affecting chromosomes 5q, 7q, or 20q and trisomy 8 (Table 1; Figure 1A-B; supplemental Patient Data). Paired leukemic and germline (skin fibroblast cells) WES analysis in 5 patients with MDS/AML identified somatic TP53 deleterious mutations in every case (Table 1; supplemental Table 4). Rare additional somatic myeloid or T-ALL–associated cancer gene mutations were found (Table 1).
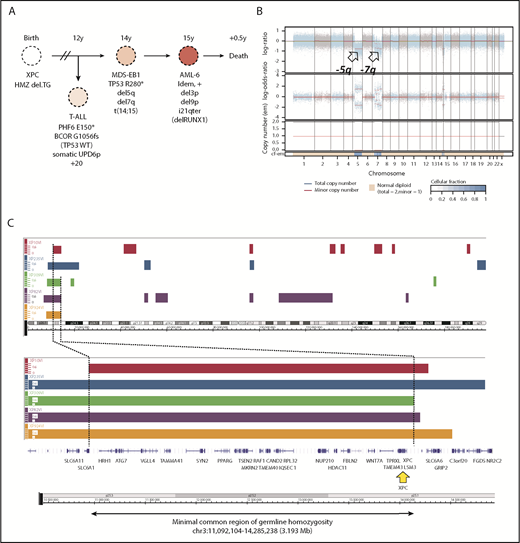
Germline genetics and clonal evolution in patients with XP-C with MDS and leukemia. (A) Bone marrow progression in patient #5 (XP924VI) showing clonal evolution. Chromosomal abnormalities and somatic point mutations are shown. (B) Copy number and allele heterozygosity analysis of the bone marrow MDS-EB1 sample showing deletion 5q and 7q (arrows) in patient #5. WES data were analyzed using the FACETS tool; total copy number log-ratio (logR; upper); allele-specific log-odds-ratio data (logOR; middle); corresponding integer (total, minor) copy number calls (bottom). The estimated cellular fraction (cf) profile is plotted at the bottom. (C) Minimal common region of germinal homozygosity in 3p25 including the XPC gene (yellow arrow), as shown by the analysis of single nucleotide polymorphism array data in the 5 patients with available fibroblast cell DNA (Affymetrix array) and confirmed using WES data (Illumina). Data are shown using the Genome Wide SNP6 Array and the Chromosome Analysis Suite. Loss of heterozygosity segments on chromosome 3 (top); minimal region of homozygosity as mapped by patient XP10VI and XP309VI, on the left and right sides (genomic position on chr3: 11 092 104 and 14 285 238, using hg19 reference, NM_00628.4), respectively (bottom).
Germline genetics and clonal evolution in patients with XP-C with MDS and leukemia. (A) Bone marrow progression in patient #5 (XP924VI) showing clonal evolution. Chromosomal abnormalities and somatic point mutations are shown. (B) Copy number and allele heterozygosity analysis of the bone marrow MDS-EB1 sample showing deletion 5q and 7q (arrows) in patient #5. WES data were analyzed using the FACETS tool; total copy number log-ratio (logR; upper); allele-specific log-odds-ratio data (logOR; middle); corresponding integer (total, minor) copy number calls (bottom). The estimated cellular fraction (cf) profile is plotted at the bottom. (C) Minimal common region of germinal homozygosity in 3p25 including the XPC gene (yellow arrow), as shown by the analysis of single nucleotide polymorphism array data in the 5 patients with available fibroblast cell DNA (Affymetrix array) and confirmed using WES data (Illumina). Data are shown using the Genome Wide SNP6 Array and the Chromosome Analysis Suite. Loss of heterozygosity segments on chromosome 3 (top); minimal region of homozygosity as mapped by patient XP10VI and XP309VI, on the left and right sides (genomic position on chr3: 11 092 104 and 14 285 238, using hg19 reference, NM_00628.4), respectively (bottom).
Collectively, these data strongly suggest a familial predisposition to hematological malignancies that cosegregates with the homozygous founder Mediterranean delTG mutation with an intermediate penetrance. No other clinical phenotypes, personal or family history of cytopenia, or cancer predisposition were reported in these families. Given that MDS/AML are extremely rarely seen in patients with XP with different XPC or other XP gene mutations, and that the delTG XPC mutation leads to the total absence of XPC protein similar to more than 90% of XPC mutations (supplemental Figure 3),9 we searched for an additional predisposing genetic variant that would cosegregate with the MDS/AML phenotype. First, WES of germline DNA performed in 6 patients did not identify any variant in known familial MDS/AML genes, including GATA2, RUNX1, CEBPA, DDX41, TP53, BRCA1, BRCA2, FANC, SBDS, SRP72, ERCC6L2, DNAJC21, MBD4, SAMD9, SAMD9L, telomere genes, and other genes involved in hematopoietic malignancies.10-12 Second, genome-wide search for shared homozygous or heterozygous single nucleotide polymorphisms from filtered variant files only retrieved the delTG XPC mutation (supplemental Methods). Third, single nucleotide polymorphism array analysis of fibroblast DNA detected no copy number variation, but a unique shared homozygosity region that delineated 3.2 Mbp at chromosome 3p25, confirmed using WES analysis, which, as expected, included the XPC gene (Figure 1C). We captured and sequenced this complete region, but none of the rare variants common to all patients was predicted to be deleterious, nor located in genes that would suggest a relevant predisposition. Because the Fanconi anemia gene FANCD2 is located in the vicinity of this region at 3p25,13 we sequenced this gene but did not find any suspect variant, while mitomycin-C testing in skin fibroblasts ruled out a Fanconi anemia pathway deficiency.
TP53 mutations and complex karyotypes involving del5q and del7q are often associated with secondary or therapy-related MDS or AML.14-17 However, with the exception of 3 patients (#5, #7, and #8; supplemental Patient Data), the other patients did not receive chemo- or radiotherapies before their hematological malignancies were discovered. Importantly, if hematological malignancies were caused by genotoxic environmental exposure in the context of XP DNA repair deficiency, the very high frequency of hematological malignancies should have been reported for patients with XP whatever their ethnic and genetic backgrounds, which is not the case.
In conclusion, we report the exceptionally high frequency of hematological, mostly myeloid, malignancies in a subpopulation of patients with XP-C with the same XPC mutation, with this later having been spread in North African families for more than 1000 years. MDS/AML in these patients display early age onset, bone marrow dysplastic features, low-intermediate blast cell counts, somatic TP53 mutations, and complex karyotypes including del5q and del7q. This is reminiscent of MDS/AML associated with aging, genotoxic stress, or posttherapeutic in the DNA repair–proficient population.14,15 Interestingly, we and others have also reported the extremely frequent rate of TP53 mutations in skin tumors that occur in patients with XP,18 suggesting hypermutability and common oncogenesis pathways. Although the complete absence of XPC protein in patients with the Mediterranean delTG XPC mutation rules out a specific role of this particular mutation in MDS/AML development, a distinct genome variation, located or not in the minimal region of homozygosity and still to be identified, may contribute to MDS/AML development in the XP-related, DNA repair-deficient background. Whatever the molecular mechanism, the reported myeloid cancer predisposition should prompt a careful monitoring of blood diseases in delTG XPC homozygous patients who originate from Mediterranean South basin (ie, annual blood examination), to enable early detection of cytopenias or blast cells. Clinicians must have a high degree of MDS/AML suspicion in these patients, and perform bone marrow examinations if blood counts are consistently abnormal.
The online version of this article contains a data supplement.
Acknowledgments
The authors are very thankful to the physicians who took care of some of these patients with XP: C. Blanchet-Bardon, M. F. Avril, S. Hadj-Rabia, and R. Itzykson from Paris, France, and L. Schirmer (Nancy, France), C. Hardy (Vaison-la-Romaine, France), J. F. Stalder (Nantes, France), L. Sutton and J. Lejeune (Tours, France), G. Michel (Marseille), and M. Beylot-Barry (Bordeaux, France). The authors are also thankful to I. Plo (Villejuif, France), D. Stoppa-Lyonnet (Paris, France), and A. Remenieras, M. Lafage-Pochitaloff and I. Arnoux (Marseille, France) for their sequence analysis and helpful discussion. The authors are thankful to J. C. Ehrhart (Villejuif) for critical reading of the manuscript.
Part of this work was supported by the “Association des Enfants de La Lune” (Bellegarde sur Valserine, France) (A.S.), the European Research Council St Grant Consolidator #311660, the ANR-10-IBHU-0002 Saint-Louis Institute program, and the Programme d’Action Intégrée de Recherche CONECT-AML (Collaborative Network on research for children and teenagers with AML) INCa-ARC-LIGUE_11905 (J.S.).
Authorship
Contribution: A.S. and J.S. are the principal investigators; A.S., N.D., V.S., N.A., A.R., V.A., J.-L.S., A.T., C.F.M.M., F.R., L.D.L.R., C.R., F.S.d.F., M.S., T.L., P.K., S.D.B., E.S., and J.S. brought clinical and biological data; S.Q., Y.B., P.D., and M.S., performed bioinformatic analyses; A.S., S.Q., E.S. and J.S. wrote the paper; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alain Sarasin, Laboratory of Genetic Instability and Oncogenesis, UMR 8200 CNRS, University Paris-Sud and Gustave Roussy Institute, 94805 Villejuif, France; e-mail: alain.sarasin@gustaveroussy.fr.

