Key Points
SLAMF7highCD16– monocytes increased in the PB of patients with MF in correlation with JAK2V617F mutation.
Anti-SLAMF7 antibody suppressed monocyte-derived fibrocyte differentiation and could be a potent therapeutic agent for MF.

Abstract
Monocyte-derived fibrocytes recently garnered attention because the novel pathogenesis of myelofibrosis (MF), and suppression of fibrocyte differentiation by serum amyloid P remarkably improved MF. We previously revealed that human fibrocytes highly expressed signaling lymphocytic activation molecule F7 (SLAMF7) compared with macrophages and that SLAMF7high monocytes in the peripheral blood (PB) of MF patients were significantly elevated relative to those in healthy controls (HCs). In this study, we evaluated SLAMF7high monocyte percentage in the PB of HCs, myeloproliferative neoplasm (MPN) patients with MF, and MPN patients without MF by using a cross-sectional approach. We found that MPN patients with MF who harbored JAK2V617F had a significantly elevated SLAMF7high monocyte percentage, which correlated positively with the JAK2V617F allele burden. In addition, the serum concentration of interleukin-1ra (IL-1ra) was significantly correlated with the SLAMF7high monocyte percentage and JAK2V617F allele burden. These findings suggest that both SLAMF7high monocytes and IL-1ra could be useful noninvasive markers of MF onset. Furthermore, the JAK2V617F allele burden of SLAMF7high monocytes was significantly higher than that of SLAMF7low monocytes and could be a potential target of elotuzumab (Elo), an anti-SLAMF7 antibody used for treating multiple myeloma. Elo independently inhibited differentiation of fibrocytes derived not only from HCs but also from MF patients in vitro. Elo also ameliorated MF and splenomegaly induced by romiplostim administration in humanized NOG mice. In conclusion, an increase of SLAMF7high monocytes with higher JAK2V617F allele burden was associated with the onset of MF in MPN patients harboring JAK2V617F, and Elo could be a therapeutic agent for MPN patients with MF who harbor JAK2V617F.
Introduction
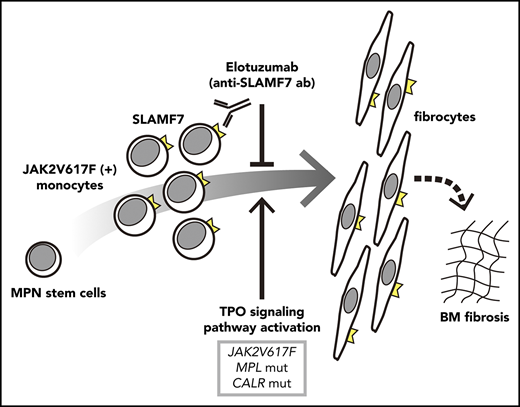
Primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (MF), and post-essential thrombocythemia MF are characterized by bone marrow (BM) fibrosis, megakaryocytic hyperplasia, anemia, and splenomegaly. The majority of patients with PMF harbor JAK2, calreticulin (CALR), or myeloproliferative leukemia (MPL) (thrombopoietin [TPO] receptor) mutations as driver mutations,1 which activate the TPO signaling pathway that plays a certain role in the proliferation and differentiation of a myeloproliferative neoplasm (MPN) clone. TPO is a key regulator of megakaryocyte and platelet production,2 and it enhances the proliferation of hematopoietic stem cells.3 The TPO signaling pathway is significantly associated with the development of BM fibrosis. Mice transplanted with TPO-overexpressing BM cells presented with symptoms such as MF and splenomegaly.4,5 MPLW515L and MPLW515A, the mutations that activate MPL, also cause BM fibrosis in mice.6,7 Romiplostim (Rom), a TPO-receptor (TPO-R) agonist, induces BM fibrosis in rats and some patients with immune thrombocytopenic purpura.8 We found that the administration of 1 mg/kg of Rom once per week induced an MF-like phenotype in C57BL/6J mice within 2 to 3 weeks, and we considered them as the mouse MF model.9
MF occurrence can be attributed to various pathogenic mechanisms. A recent study revealed that the neoplastic clone of fibrocytes is essential in the pathogenesis of PMF and serum amyloid P (also known as PRM-151), which suppressed fibrocyte differentiation10 and significantly improved survival and BM fibrosis in a murine xenograft model.11 Fibrocytes are spindle-shaped fibroblast-like blood cells derived from a subpopulation of CD14+ monocytes.12-14 In diseases with progressive tissue fibrosis, fibrocytes are associated with the development of fibrosis in pulmonary fibrosis,15 cardiovascular diseases,16 liver fibrosis,17 renal fibrosis,18 autoimmune diseases,19 and PMF.11 To investigate the characteristics and functions of fibrocytes, we established murine fibrocyte cell lines9 from transgenic mice that harbored the temperature-sensitive large T-antigen gene of simian virus 40 (SV40T mice; FACT, Sendai, Japan).20
Using a Rom-induced murine MF model and murine fibrocyte cell lines, we showed that fibrocyte differentiation could be directly induced by the activation of MPL (TPO-R) and could lead to MF progression; furthermore, the elimination of macrophages significantly reduced MF and splenomegaly.9 On the basis of a DNA microarray analysis, human fibrocytes were shown to express a higher level of signaling lymphocytic activation molecule-F7 (SLAMF7, also known as CD319, CS1, 19A24, novel Ly9, and CRACC) compared with macrophages.21 Murine fibrocyte cell lines and cultured human fibrocytes both exhibited a strong SLAMF7 expression on their surfaces.9 We also found that monocytes with high SLAMF7 expression and negative CD16 expression were significantly elevated in the peripheral blood (PB) of MF patients compared with that of healthy controls (HCs). We hypothesized that this monocyte subset reflects fibrocyte activation and could be a noninvasive surrogate marker of MF onset. At present, fibrocytes are a therapeutic target for MF; thus, SLAMF7 could be a potential therapeutic target. Elotuzumab (Elo) is an anti-SLAMF7 antibody recently used for treating relapsed/refractory multiple myeloma, and it has encouraging results and promising safety.22 Thus, Elo could become a potential therapeutic agent for MF. Elo also has a synergistic effect with natural killer (NK) cells by binding with SLAMF7 receptor on the surface of NK cells.22
In this study, we evaluated the SLAMF7highCD16− monocyte percentage in PB of HCs and MPN patients with or without MF in a cross-sectional manner and analyzed the correlation with MF onset, genetic mutation, primary disease, medication, splenomegaly, and various cytokines. Subsequently, we investigated the efficacy of anti-SLAMF7 antibody Elo on human fibrocyte differentiation that promotes MF in vitro and in vivo for this clinical study of Elo.
Methods
Human participants
This study was performed in accordance with the guidelines of the National Defense Medical College Ethics Committee. Between 2017 and 2018, 58 patients who were diagnosed with MPN according to the 2016 World Health Organization classification and diagnostic criteria23 for MPN were enrolled after providing written informed consent. The degree of BM fibrosis was assessed by pathologists according to the European consensus criteria.24 In this study, we defined patients with MPN without MF as MF-0 and those with MPN with MF as MF-1, -2, or -3. The MPN patients were classified as follows: MPN patients with MF (n = 37) and MPN patients without MF (n = 21). The primary diseases were polycythemia vera (n = 23), essential thrombocythemia (n = 23), and PMF (n = 12). For hematologic evaluation, PB samples were collected from all patients and from 21 HCs. In addition, 15 patients were treated with hydroxyurea, 1 was treated with anagrelide, and 2 were treated with ruxolitinib (Rux) when the sample was collected (Table 1). The mutations of JAK2V671F, MPLW515K/L, and CALR were also assessed using genomic DNA isolated from PB and cultured fibrocytes of all patients as previously described.9,25,26 The JAK2V617F allele burden was first determined via alternately binding probe competitive polymerase chain reaction (ABC-PCR).27 In brief, 10 to 100 ng of genomic DNA was subjected to 50 cycles of PCR using Titanium Taq PCR kit (639210; Takara Bio, Kusatsu, Japan), with primers and a fluorescence-conjugated alternately binding probe in a CFX-96 real-time PCR system (184-5096J1; Bio-Rad, Hercules, CA). The allele burden was determined from the detected fluorescence intensity based on a standard curve plotted for a control with a known JAK2V617F allele burden. ABC-PCR accurately determines JAK2V617F allele burdens above 10%; however, allele-specific PCR is more accurate for the measurement of allele burdens below 10%. Therefore, allele-specific PCR was performed when the allele burden determined using ABC-PCR was below 10%. For these assays, genomic DNA (1 µL) was mixed with Universal PCR Master Mix (4304437; Thermo Fisher Scientific, Waltham, MA), primers, and a TaqMan probe, and a total of 50 PCR cycles were performed, as previously reported.28
Human fibrocyte culture assay
As reported, human fibrocytes are easily induced from peripheral blood mononuclear cells (PBMCs) without any cytokine.21 From the PB of 3 HCs, 5 × 106 PBMCs were purified and cultured, as previously described.9 Simultaneously, 6 × 106 purified NK cells were sorted from the PB of HCs using RossetteSep human NK cell enrichment cocktail (15025; STEMCELL Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. After 4 days of incubation, the supernatant fluid was removed and the cultured PBMCs were divided into 4 groups: control group (Dulbecco’s modified Eagle medium [DMEM] alone) (n = 6), NK cell group (n = 6), Elo (kindly provided by Bristol-Myers Squibb) group (n = 6), and Elo plus NK cells group (n = 6). DMEM was added again with or without 1500 μg/mL Elo and/or 6 × 105 NK cells according to the grouping. We performed the same procedure again on day 8, and the frequency of differentiated fibrocytes among all cells was calculated on days 8, 9, and 10, as previously described.29 In detail, differentiated fibrocytes were counted in 5 randomly selected different low-power fields of view for each well using the following criteria: the presence of an adherent cell with an elongated spindle-shaped morphology and oval nucleus.29 We calculated the ratio of spindle-shaped cells among all adherent cells. Then, the same culture assay and analysis using the PBMCs of MPN patients who harbor a JAK2V617F mutation (n = 6) or CALR mutation (n = 6) were conducted with 100 μg/mL Elo. In the culture assay of PBMCs of MPN patients, coculture with NK cells was excluded.
Next, 5 × 106 human PBMCs that were purified as described above were cultured with DMEM with or without Elo for a long period of time. At the start of this experiment, PBMCs were divided into 3 groups: control group (DMEM alone) (n = 6), Elo 50 μg/mL group (n = 6), and Elo 100 μg/mL group (n = 6). PBMCs were resuspended in DMEM with or without 50 or 100 μg/mL Elo according to the grouping. The concentration of Elo was established according to the blood concentration of patients with multiple myeloma. Specifically, the concentration of 100 μg/mL is equivalent to the blood concentration of patients administered Elo 10 mg/kg once per week, and 50 μg/mL is equivalent to the blood concentration of patients administered Elo once every 2 weeks.30 The supernatant fluid was removed after 4 days of incubation, and DMEM was added with or without Elo. We counted fibrocytes (as described above) every 4 days from days 8 to 31.
Furthermore, 5 × 106 human PBMCs were cultured with DMEM with or without Elo and with other agents, such as Rom (TPO-R agonist), Rux (JAK inhibitor) (SC-364729; Santa Cruz Biotechnology, Dallas, TX), and interferon-α2 (IFN-α2) (130-093-873; Miltenyi Biotec, Bergish Gladbach, Germany). At the start of this experiment, PBMCs were divided into 4 groups: Rom group (n = 6), Rom plus Elo group (n = 6), Rom plus Elo plus Rux group (n = 6), and Rom plus IFN-α2 group (n = 6). PBMCs were resuspended in DMEM with 500 ng/mL Rom (which induced human fibrocyte differentiation)9 and 100 μg/mL Elo or 500 nM Rux (which suppressed not only JAK1 and JAK2 but also JAK3)31 or 10 ng/mL IFN-α2 (which was based on the maximal observed concentration after a single subcutaneous dose of 180 μg peg-IFN-α2)32 according to the grouping and incubation. The supernatant fluid was removed, and DMEM was re-added with the same concentration of Rom, Elo, Rux, and IFN-α2 on days 4 and 8. The frequency of spindle-shaped cells was calculated on days 8, 9, and 10.
In addition, 5 × 106 human PBMCs derived from MPN patients with MF who harbored JAK2V617F mutation or CALR mutation were cultured with DMEM with or without Elo. At the beginning of this experiment, PBMCs were divided into the following 4 groups: JAK2V617F control group (n = 6), JAK2V617F plus Elo group (n = 6), CALR control group (n = 6), and CALR plus Elo group (n = 6). PBMCs were resuspended in DMEM with or without 100 μg/mL Elo according to the grouping. The supernatant fluid was removed after 4 days of incubation, and DMEM was added with or without Elo. The frequency of spindle-shaped cells was calculated on days 8, 9, and 10.
Every human fibrocyte cultured assay was performed using 6 PBMC samples (n = 6) derived from HCs of MPN patients, and the data were obtained from 3 replicates. Statistical significance among the results of each day was calculated using 1-way analysis of variance (ANOVA) with post hoc Tukey’s honestly significant difference test.
Murine xenograft model
Because Elo has no reactivity against murine SLAMF7, we had to develop a murine xenograft model transplanted with human hematopoietic cells to verify the effect of Elo in the Rom-induced MF mouse model. Thus, in this study, we used humanized NOG (hNOG) mice (In-Vivo Science, Tokyo, Japan). The hNOG mouse was created by transplanting human cord blood (Allcells, Alameda, CA) into the NOG mouse established by backcrossing an interleukin-2R (IL-2R) γ-chain knockout mouse to a NOD/SCID mouse.33 In our study, the hNOG mice were created by transplanting 1 × 105 human cord blood cells to the NOG mice 24 hours after irradiation at 2.5 Gy and maintained until 20 weeks after transplantation when monocyte engraftment was established.34 We confirmed the engraftment of monocytes in the spleen and PB using flow cytometry (supplemental Figure 1, available on the Blood Web site). Ten hNOG mice were administered saline via intravenous injection on days −7, 1, and 8; they were also administered 1 mg/kg Rom via subcutaneous injection in the neck on day 1 (Rom group, n = 10). Twelve hNOG mice were administered 100 mg/kg Elo on days −7, 1, and 8; they were also administered 1 mg/kg Rom on day 1 (Rom plus Elo group, n = 12). After the mice were euthanized, PB was drawn via cardiac puncture, and the spleens and femurs were extracted on day 15. Five hNOG mice who were given no treatment were euthanized on day 15 as the controls (control group, n = 5). The protocol is presented in Figure 3A. The spleens were weighed, and the spleen cells were isolated as previously described.35 The PB and spleen cells were hemolyzed using BD Pharm Lyse (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions, and flow cytometry analysis was performed. The morphologic analysis of the femur BM of the mice was performed as previously described.9 The degree of BM fibrosis was assessed in a blinded manner according to European consensus criteria24 by 2 hematology experts. All work was conducted using approved protocols from the Center for Laboratory Animal Science of the National Defense Medical College.
Flow cytometry and sorting
Human PBMCs were purified from whole blood samples by density centrifugation using Pancoll (PAN-Biotech, Aidenbach, Germany). Human PBMCs were resuspended in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline with 2% fetal bovine serum and 0.05% NaN3) containing 5 μL human BD Fc block (564220; BD Biosciences) and were incubated for 10 minutes on ice for Fc receptor blocking. PBMCs were resuspended in FACS buffer containing fluorescein isothiocyanate–conjugated anti-human CD14 antibody (IM0645U; Beckman Coulter, Tokyo, Japan), phycoerythrin (PE)-conjugated anti-human CD66b antibody (130-104-414; Miltenyi Biotec), allophycocyanin (APC)-conjugated anti-SLAMF7 antibody (130-099-569; Miltenyi Biotec) or APC-conjugated human REA isotype control (130-104-614; Miltenyi Biotec), and V450-conjugated anti-human CD16 antibody (561310; BD Biosciences) or V450-conjugated mouse immunoglobulin G-1κ isotype control antibody (560373; BD Biosciences). After incubating for 30 minutes on ice, the cells were washed twice and analyzed using BD FACS Aria III (BD Biosciences). We gated the CD14+CD66b− monocytes, and the SLAMF7highCD16− subset was then defined (Figure 1A-B). In addition, we sorted the SLAMF7highCD16− monocytes and the SLAMF7lowCD16− monocytes using the SH800 cell sorter (Sony Biotechnology, San Jose, CA) from 7 patients who harbored a JAK2V617F mutation, according to the manufacturer’s instructions. The JAK2V617F allele burdens of these monocyte subsets were measured, and these subsets were cultured for fibrocyte assay as previously described.
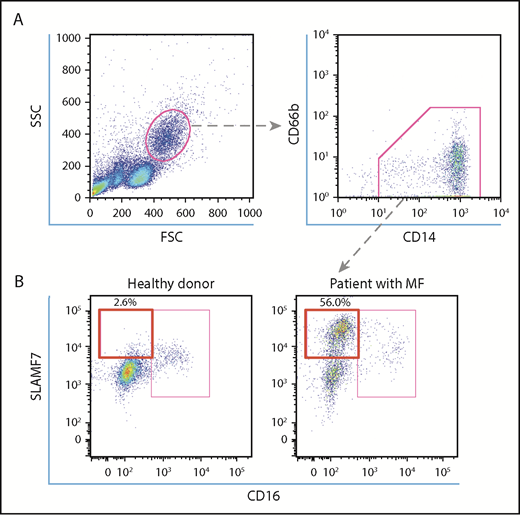
Analysis of SLAMF7high CD16− monocytes in the PB of HCs and MPN patients with MF. (A) To define SLAMF7highCD16− monocytes, we began by gating lymphocyte subsets in a forward scatter/side scatter dot plot. Subsequently, we gated CD14+CD66b− monocytes and divided them into SLAMF7highCD16−, SLAMF7lowCD16−, and CD16+ subsets. (B) The representative flow cytometry dot plot shows that the percentage of SLAMF7highCD16− monocytes increased in MPN patients with MF who harbored JAK2V617F compared with HCs. The ratios of the SLAMF7highCD16− subset to CD14+CD66b− monocytes are presented as percentages.
Analysis of SLAMF7high CD16− monocytes in the PB of HCs and MPN patients with MF. (A) To define SLAMF7highCD16− monocytes, we began by gating lymphocyte subsets in a forward scatter/side scatter dot plot. Subsequently, we gated CD14+CD66b− monocytes and divided them into SLAMF7highCD16−, SLAMF7lowCD16−, and CD16+ subsets. (B) The representative flow cytometry dot plot shows that the percentage of SLAMF7highCD16− monocytes increased in MPN patients with MF who harbored JAK2V617F compared with HCs. The ratios of the SLAMF7highCD16− subset to CD14+CD66b− monocytes are presented as percentages.
The cells in the PB and spleen of the hNOG mice were resuspended in FACS buffer, and we performed Fc receptor blocking as described above. Next, the cells were resuspended in FACS buffer containing Alexa Fluor 488–conjugated anti-mouse CD45 antibody (103121; BioLegend; San Diego, CA), PB-conjugated anti-human CD45 antibody (304029; BioLegend), PE-conjugated anti-human CD14 antibody (A07764; Beckman Coulter) or PE-conjugated anti-human CD19 antibody (R0808; Beckman Coulter), and the isotype control (130-104-614; Miltenyi Biotec) or APC-conjugated anti-SLAMF7 antibody (130-099-569; Miltenyi Biotec). After incubation and washing, we analyzed the cells. First, we gated the human CD45+ mouse CD45− mononuclear cells, followed by the human CD14+ monocytes or human CD19+ B lymphocytes (supplemental Figure 2). Subsequently, we evaluated the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) of the monocytes.
Immunoassays
Serum samples of MPN patients who harbored the JAK2V617F mutation (n = 24) were stored at −20°C until analysis. Serum levels of IL-1ra were determined by using Human Luminex Assays (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.36 Human Luminex Assays are immunoassays formatted on magnetic beads that use principles similar to those of sandwich enzyme-linked immunosorbent assays.33
Statistical analysis
The results were reported as the mean ± standard deviation of the data obtained from 3 individual experiments unless otherwise specified. The sample size was obtained using Power and Sample Size Calculation version 3.1.2, 2014 (Department of Biostatistics, Vanderbilt University, Nashville, TN). Statistical significance was calculated by using the unpaired Student t test unless otherwise specified. All P values were two-sided, and values < .05 were considered statistically significant. Data were plotted and statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA).
Results
The percentage of SLAMF7highCD16− subset in the CD14+CD66b− monocytes significantly increased in patients with MF compared with HCs
Patients were classified into 2 groups (MPN without MF and MPN with MF). The median percentage of SLAMF7highCD16− monocytes in the PB of HCs (n = 21), MPN patients without MF (n = 21), and MPN patients with MF (n = 37) were 0.94%, 3.23%, and 16.90%, respectively. Compared with HCs and MPN patients without MF, MPN patients with MF had significantly increased (1-way ANOVA with post hoc Steel-Dwass test P < .01) SLAMF7highCD16− monocyte percentage (Figure 2A). As the result of subgroup analysis by genetic mutation (JAK2V617F, CALR, and triple-negative mutations), SLAMF7highCD16− monocytes were significantly increased in the MPN patients who harbored a JAK2V617F mutation (n = 28) compared with MPN patients who harbored a CALR mutation (n = 9) or with MPN patients with triple-negative mutations (n = 20) (median: JAK2V617F 28.75% vs CALR 3.26% vs triple-negative mutations 1.73%; 1-way ANOVA with post hoc Steel-Dwass test P < .01) (Figure 2B). In addition, MPN patients with MF who harbored JAK2V617F (n = 19) had a significantly higher SLAMF7highCD16− monocyte percentage than those without MF (n = 9) (median: 43.70% vs 7.74%; Mann-Whitney U test P < .01). In the same manner, MPN patients with MF who did not harbor JAK2V617F (n = 18) had a significantly higher SLAMF7highCD16− monocyte percentage than those without MF (n = 12) (median: 1.24% vs 4.32%; Mann-Whitney U test P < .05); the contrast between patients with and without MF was significant for those with JAK2V617F (Figure 2C). We constructed receiver operating characteristic (ROC) curves to obtain the cutoff of the percentage of SLAMF7highCD16− monocytes in PB for separating MPN patients with MF who harbored a JAK2V617F mutation from those without MF. The resulting area under the curve was 0.901, and by setting the cutoff of the percentage of SLAMF7highCD16− monocytes to >25%, the sensitivity and specificity for classifying MF onset were 79.0% and 77.8%, respectively (Figure 2D).
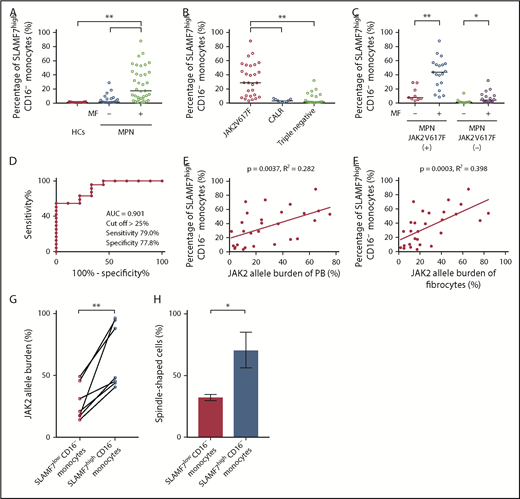
Increase in the percentage of SLAMF7high CD16− monocytes in PB of MPN patients with MF who harbored JAK2V617F and the correlations between SLAMF7high CD16− monocytes and JAK2 allele burden in PB and fibrocytes. (A) Comparison of SLAMF7highCD16− monocytes in PB among HCs and MPN patients with or without MF. The median percentage of SLAMF7highCD16− monocytes in PB of HCs (n = 21), MPN patients without MF (n = 21), and MPN patients with MF (n = 37) were 0.94%, 3.23%, and 19.60%, respectively. The SLAMF7highCD16− monocyte percentage of MPN patients with MF was significantly increased compared with HCs and MPN patients without MF. The results are reported as medians. Statistical significance was calculated using 1-way ANOVA with post hoc Steel-Dwass test. (B) Subgroup analyses by genetic mutation (JAK2V617F, CALR, and triple-negative mutations). In this analysis, we excluded the MPL group because it contained only 1 patient and could not be subjected to statistical analysis. SLAMF7highCD16− monocytes were significantly increased in the MPN patients who harbored a JAK2V617F mutation (n = 28) compared with the MPN patients who harbored a CALR mutation (n = 9) or the MPN patients with triple-negative mutations (n = 20) (median: JAK2V617F 28.75% vs CALR 3.26% vs triple-negative mutations 1.73%; P < .01). The results are reported as medians. Statistical significance was calculated using 1-way ANOVA with post hoc Steel-Dwass test, (C) Subgroup analysis of SLAMF7highCD16− monocytes in PB by the presence or absence of JAK2V617F mutation. MPN patients with MF who harbored JAK2V617F (n = 19) had a significantly higher SLAMF7highCD16− monocyte percentage than those without MF (n = 9) (median: 43.70% vs 7.74%; P < .01). In a similar manner, MPN patients with MF who did not harbor JAK2V617F (n = 18) had a significantly higher SLAMF7highCD16− monocyte percentage than those without MF (n = 12) (median: 1.24% vs 4.32%; P < .05). The contrast between patients with and without MF was clearer in MPN patients who harbored JAK2V617F than in those who did not harbor JAK2V617F. Results are reported as medians. Statistical significance was calculated using the Mann-Whitney U test. (D) ROC curve showing the cutoff value of the percentage of SLAMF7highCD16− monocytes in PB for separating MPN patients with MF who harbored a JAK2V617F mutation from those without MF. The AUC was 0.901, and by setting the cutoff of the percentage of SLAMF7highCD16− monocytes to >25%, the sensitivity and specificity for classifying MF onset were 79.0% and 77.8%, respectively. (E) Correlation between SLAMF7highCD16− monocytes in PB and JAK2 allele burden of PB (n = 28). There was a positive correlation between the SLAMF7highCD16− monocyte percentage in PB and the JAK2V617F allele burden of PB (r = 0.531; R2 = 0.282; P = .0037). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (F) Correlation between SLAMF7highCD16− monocytes in PB and the JAK2 allele burden of cultured fibrocytes (n = 28). There was a positive correlation between SLAMF7highCD16− monocyte percentage in PB and JAK2V617F allele burden of fibrocytes (r = 0.631; R2 = 0.398; P = .0003). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (G) The JAK2V617F allele burden of isolated SLAMF7lowCD16− monocytes and isolated SLAMF7highCD16− monocytes from 7 MPN patients who harbored JAK2V617F. The JAK2V617F allele burden of isolated SLAMF7highCD16− monocytes was significantly higher than the JAK2V617F allele burden of isolated SLAMF7lowCD16− monocytes (paired Student t test P < .01). (H) Culture assay of isolated SLAMF7lowCD16− monocytes and isolated SLAMF7highCD16− monocytes collected from 2 MPN patients who harbored a JAK2V617F mutation. The frequency of fibrocytes derived from SLAMF7highCD16− monocytes was significantly increased compared with that from SLAMF7lowCD16− monocytes. Results are reported as the mean ± standard deviation of data obtained from 3 individual experiments. Statistical significance was calculated by unpaired Student t test. *P < .05; **P < .01.
Increase in the percentage of SLAMF7high CD16− monocytes in PB of MPN patients with MF who harbored JAK2V617F and the correlations between SLAMF7high CD16− monocytes and JAK2 allele burden in PB and fibrocytes. (A) Comparison of SLAMF7highCD16− monocytes in PB among HCs and MPN patients with or without MF. The median percentage of SLAMF7highCD16− monocytes in PB of HCs (n = 21), MPN patients without MF (n = 21), and MPN patients with MF (n = 37) were 0.94%, 3.23%, and 19.60%, respectively. The SLAMF7highCD16− monocyte percentage of MPN patients with MF was significantly increased compared with HCs and MPN patients without MF. The results are reported as medians. Statistical significance was calculated using 1-way ANOVA with post hoc Steel-Dwass test. (B) Subgroup analyses by genetic mutation (JAK2V617F, CALR, and triple-negative mutations). In this analysis, we excluded the MPL group because it contained only 1 patient and could not be subjected to statistical analysis. SLAMF7highCD16− monocytes were significantly increased in the MPN patients who harbored a JAK2V617F mutation (n = 28) compared with the MPN patients who harbored a CALR mutation (n = 9) or the MPN patients with triple-negative mutations (n = 20) (median: JAK2V617F 28.75% vs CALR 3.26% vs triple-negative mutations 1.73%; P < .01). The results are reported as medians. Statistical significance was calculated using 1-way ANOVA with post hoc Steel-Dwass test, (C) Subgroup analysis of SLAMF7highCD16− monocytes in PB by the presence or absence of JAK2V617F mutation. MPN patients with MF who harbored JAK2V617F (n = 19) had a significantly higher SLAMF7highCD16− monocyte percentage than those without MF (n = 9) (median: 43.70% vs 7.74%; P < .01). In a similar manner, MPN patients with MF who did not harbor JAK2V617F (n = 18) had a significantly higher SLAMF7highCD16− monocyte percentage than those without MF (n = 12) (median: 1.24% vs 4.32%; P < .05). The contrast between patients with and without MF was clearer in MPN patients who harbored JAK2V617F than in those who did not harbor JAK2V617F. Results are reported as medians. Statistical significance was calculated using the Mann-Whitney U test. (D) ROC curve showing the cutoff value of the percentage of SLAMF7highCD16− monocytes in PB for separating MPN patients with MF who harbored a JAK2V617F mutation from those without MF. The AUC was 0.901, and by setting the cutoff of the percentage of SLAMF7highCD16− monocytes to >25%, the sensitivity and specificity for classifying MF onset were 79.0% and 77.8%, respectively. (E) Correlation between SLAMF7highCD16− monocytes in PB and JAK2 allele burden of PB (n = 28). There was a positive correlation between the SLAMF7highCD16− monocyte percentage in PB and the JAK2V617F allele burden of PB (r = 0.531; R2 = 0.282; P = .0037). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (F) Correlation between SLAMF7highCD16− monocytes in PB and the JAK2 allele burden of cultured fibrocytes (n = 28). There was a positive correlation between SLAMF7highCD16− monocyte percentage in PB and JAK2V617F allele burden of fibrocytes (r = 0.631; R2 = 0.398; P = .0003). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (G) The JAK2V617F allele burden of isolated SLAMF7lowCD16− monocytes and isolated SLAMF7highCD16− monocytes from 7 MPN patients who harbored JAK2V617F. The JAK2V617F allele burden of isolated SLAMF7highCD16− monocytes was significantly higher than the JAK2V617F allele burden of isolated SLAMF7lowCD16− monocytes (paired Student t test P < .01). (H) Culture assay of isolated SLAMF7lowCD16− monocytes and isolated SLAMF7highCD16− monocytes collected from 2 MPN patients who harbored a JAK2V617F mutation. The frequency of fibrocytes derived from SLAMF7highCD16− monocytes was significantly increased compared with that from SLAMF7lowCD16− monocytes. Results are reported as the mean ± standard deviation of data obtained from 3 individual experiments. Statistical significance was calculated by unpaired Student t test. *P < .05; **P < .01.
Furthermore, the JAK2V617F allele burden of PB (n = 28) was positively correlated with the SLAMF7highCD16− monocyte percentage in PB (Pearson’s correlation coefficient r = 0.531; R2 = 0.282; linear regression analysis P = .0037) (Figure 2E). The JAK2V617F allele burden of fibrocytes (n = 28) was strongly correlated with the SLAMF7highCD16− monocyte percentage in PB (Pearson’s correlation coefficient r = 0.631; R2 = 0.398; linear regression analysis P = .0003) (Figure 2F). Moreover, the JAK2V617F allele burden of isolated SLAMF7highCD16− monocytes was significantly elevated compared with the JAK2V617F allele burden of isolated SLAMF7lowCD16− monocytes in 7 MPN patients who harbored JAK2V617F (paired Student t test, P < .01) (Figure 2G). The culture assay of isolated SLAMF7lowCD16− monocytes and isolated SLAMF7highCD16− monocytes from 2 MPN patients who harbored a JAK2V617F mutation revealed that the frequency of fibrocytes derived from SLAMF7highCD16− monocytes was significantly increased compared with SLAMF7lowCD16− monocytes (unpaired Student t test P < .05) (Figure 2H).
Genomic DNA isolated from PB and cultured fibrocytes was found to harbor the same genetic mutation in all patients. The SLAMF7highCD16− monocyte percentage in PB was not correlated with primary disease or medication (data not shown).
IL-1ra is strongly correlated with the JAK2V617F allele burden and the percentage of SLAMF7highCD16− monocytes in PB from MPN patients
We examined the concentration of IL-1ra in the serum of 24 MPN patients harboring a JAK2V617F mutation. Thereafter, we assessed the correlations between IL-1ra and the JAK2V617F allele burden of PB and fibrocytes and the percentage of SLAMF7highCD16− monocytes in PB. IL-1ra showed positive correlations with the JAK2V617F allele burden of PB, the JAK2V617F allele burden of fibrocytes, and the percentage of SLAMF7highCD16− monocytes in PB (n = 24) (Pearson’s correlation coefficient was 0.784, 0.661, and 0.715, respectively) (Figure 3A-C). Furthermore, MPN patients with MF who harbored JAK2V617F (n = 17) had significantly higher IL-1ra than patients without MF (n = 7) (median: 1550.8 pg/mL vs 411.3 pg/mL; Mann-Whitney U test P < .01) (Figure 3D). For the ROC curve, the sensitivity and specificity for classifying MF onset were 82.4% and 85.7%, respectively, with the cutoff of IL-1ra set to >600 pg/mL (Figure 3E). MPN patients with splenomegaly who harbored JAK2V617F (n = 10) also had significantly higher IL-1ra than those without splenomegaly (n = 14) (median: 1570.1 pg/mL vs 476.1 pg/mL; Mann-Whitney U test P < .01) (Figure 3F). For the ROC curve, the sensitivity and specificity for classifying splenomegaly onset were 90.0% and 78.6%, respectively, with the cutoff for IL-1ra set to >800 pg/mL (Figure 3G).
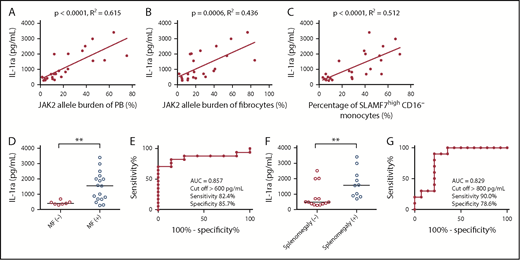
Correlations between IL-1ra and JAK2V617F allele burden of PB and fibrocytes and the percentage of SLAMF7high CD16− monocytes in PB, MF onset, and splenomegaly. (A) The positive correlation between IL-1ra and the JAK2V617F allele burden of PB (n = 24) (r = 0.784; R2 = 0.615; P < .0001). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (B) The positive correlation between IL-1ra and the JAK2V617F allele burden of fibrocytes (n = 24) (r = 0.661; R2 = 0.436; P = .0006). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (C) The positive correlation between IL-1ra and the percentage of SLAMF7highCD16− monocytes in PB (n = 24) (r = 0.715; R2 = 0.512; P < .0001). Pearson’s correlation coefficient was calculated, and the statistical significance was determined using linear regression analysis. (D) Comparison of IL-1ra between MPN patients with MF who harbor JAK2V617F and those without MF. The results are reported as medians. MPN patients with MF who harbor JAK2V617F (n = 17) had significantly higher IL-1ra than those without MF (n = 7) (median: 1550.8 pg/mL vs 411.3 pg/mL, Mann-Whitney U test P < .007). The results are reported as medians. (E) ROC curve showing the cutoff value of IL-1ra for separating MPN patients with MF who harbor a JAK2V617F mutation from those without MF. The AUC was 0.857, and the sensitivity and specificity for classifying MF onset were 82.4% and 85.7%, respectively, with the cutoff value of IL-1ra set to >600 pg/mL. (F) Comparison of IL-1ra between MPN patients with splenomegaly who harbor JAK2V617F and those without splenomegaly. The results are reported as medians. MPN patients with splenomegaly who harbor JAK2V617F (n = 10) also had a significantly higher IL-1ra than those without splenomegaly (n = 14) (median: 1570.1 pg/mL vs 476.1 pg/mL, Mann-Whitney U test P < .007). (G) ROC curve showing the cutoff value of IL-1ra for separating MPN patients with splenomegaly who harbor a JAK2V617F mutation from those without splenomegaly. The AUC was 0.829, and the sensitivity and specificity for classifying MF onset were 90.0% and 78.6%, respectively, with the cutoff of IL-1ra set to >800 pg/mL. The results are reported as medians. Statistical significance was assessed using Mann-Whitney U test. **P < .01.
Correlations between IL-1ra and JAK2V617F allele burden of PB and fibrocytes and the percentage of SLAMF7high CD16− monocytes in PB, MF onset, and splenomegaly. (A) The positive correlation between IL-1ra and the JAK2V617F allele burden of PB (n = 24) (r = 0.784; R2 = 0.615; P < .0001). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (B) The positive correlation between IL-1ra and the JAK2V617F allele burden of fibrocytes (n = 24) (r = 0.661; R2 = 0.436; P = .0006). Pearson’s correlation coefficient was calculated, and the statistical significance was calculated using linear regression analysis. (C) The positive correlation between IL-1ra and the percentage of SLAMF7highCD16− monocytes in PB (n = 24) (r = 0.715; R2 = 0.512; P < .0001). Pearson’s correlation coefficient was calculated, and the statistical significance was determined using linear regression analysis. (D) Comparison of IL-1ra between MPN patients with MF who harbor JAK2V617F and those without MF. The results are reported as medians. MPN patients with MF who harbor JAK2V617F (n = 17) had significantly higher IL-1ra than those without MF (n = 7) (median: 1550.8 pg/mL vs 411.3 pg/mL, Mann-Whitney U test P < .007). The results are reported as medians. (E) ROC curve showing the cutoff value of IL-1ra for separating MPN patients with MF who harbor a JAK2V617F mutation from those without MF. The AUC was 0.857, and the sensitivity and specificity for classifying MF onset were 82.4% and 85.7%, respectively, with the cutoff value of IL-1ra set to >600 pg/mL. (F) Comparison of IL-1ra between MPN patients with splenomegaly who harbor JAK2V617F and those without splenomegaly. The results are reported as medians. MPN patients with splenomegaly who harbor JAK2V617F (n = 10) also had a significantly higher IL-1ra than those without splenomegaly (n = 14) (median: 1570.1 pg/mL vs 476.1 pg/mL, Mann-Whitney U test P < .007). (G) ROC curve showing the cutoff value of IL-1ra for separating MPN patients with splenomegaly who harbor a JAK2V617F mutation from those without splenomegaly. The AUC was 0.829, and the sensitivity and specificity for classifying MF onset were 90.0% and 78.6%, respectively, with the cutoff of IL-1ra set to >800 pg/mL. The results are reported as medians. Statistical significance was assessed using Mann-Whitney U test. **P < .01.
Elo suppressed fibrocyte differentiation in vitro, and NK cells enhanced the inhibitory effect of Elo
To confirm the inhibitory effect on the fibrocyte differentiation of Elo in vitro, we performed a culture assay. On the basis of our results, the frequency of fibrocytes significantly decreased in the Elo group compared with the control group (1-way ANOVA with post hoc Tukey’s honestly significant difference test P < .01 or P < .05) (Figure 4A). Moreover, the frequency of fibrocytes significantly decreased in the Elo plus NK cell group compared with the Elo group on days 8 and 9. In contrast, the frequency of fibrocytes significantly increased in the NK cell group on days 8, 9, and 10 compared with the control group. These results indicate that Elo had an inhibitory effect on fibrocyte differentiation in vitro, and NK cells augmented the inhibitory effect of Elo, although the NK cells alone improved fibrocyte differentiation.
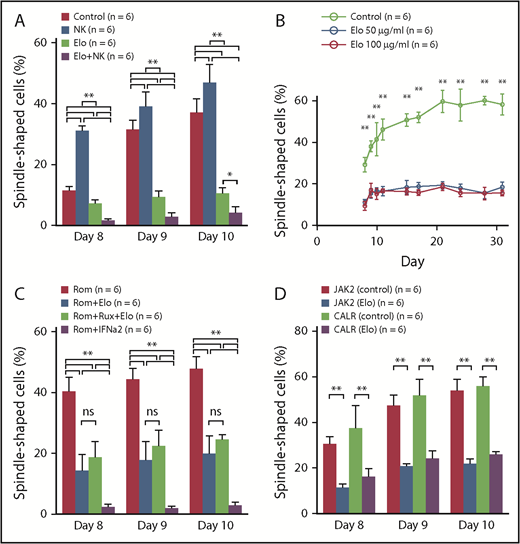
Culture assay of the human PBMCs in vitro. (A) Culture assay of human PBMCs exposed to Elo and NK cells. The frequency of fibrocytes significantly increased in the NK cell group (n = 6) on days 8, 9, and 10 compared with the control group (n = 6). In contrast, the frequency of fibrocytes significantly decreased in the Elo group (n = 6) and Elo plus NK cell group (n = 6). In addition, the frequency of fibrocytes significantly decreased in the Elo plus NK cell group compared with the Elo group on days 8, 9 and 10. (B) The long-term culture assay of human PBMCs exposed to Elo. The frequency of fibrocytes significantly decreased in the Elo 50 μg/mL group (n = 6) and Elo 100 μg/mL group (n = 6) compared with the control group (n = 6) throughout the observation period (from days 8 to 31). No significant difference was observed in the frequency of fibrocytes between the Elo 50 μg/mL and Elo 100 μg/mL groups. (C) Culture assay of human PBMCs exposed to Elo and other agents. Compared with the Rom group (n = 6), the frequency of fibrocytes significantly decreased in the Rom plus Elo group (n = 6), Rom plus Elo plus Rux group (n = 6), and Rom plus IFN-α2 group (n = 6) throughout the observation period (from days 8 to 10). In contrast, there was no significant difference between the Rom plus Elo and Rom plus Elo plus Rux groups in terms of fibrocyte differentiation. Rom plus IFN-α2 had the strongest suppressive effect on fibrocyte differentiation; however, approximately 80% to 90% of the cultured cells were dead and floated in the Rom plus IFN-α2 group, and we considered that IFN-α2 might be too cytotoxic in this setting. (D) Culture assay of PBMCs of from MF patients. Elo significantly decreased the frequency of fibrocytes derived from MPN patients harboring a CALR mutation, similar to fibrocytes derived from MPN patients with a JAK2V617F mutation. Results were reported as the mean ± standard deviation values for data obtained from 3 individual experiments with 6 HCs or 6 patients in each group. Statistical significance of the data for each day was calculated by 1-way ANOVA with post hoc Tukey’s honestly significant difference test. ns: not significant. *P < .05; **P < .01.
Culture assay of the human PBMCs in vitro. (A) Culture assay of human PBMCs exposed to Elo and NK cells. The frequency of fibrocytes significantly increased in the NK cell group (n = 6) on days 8, 9, and 10 compared with the control group (n = 6). In contrast, the frequency of fibrocytes significantly decreased in the Elo group (n = 6) and Elo plus NK cell group (n = 6). In addition, the frequency of fibrocytes significantly decreased in the Elo plus NK cell group compared with the Elo group on days 8, 9 and 10. (B) The long-term culture assay of human PBMCs exposed to Elo. The frequency of fibrocytes significantly decreased in the Elo 50 μg/mL group (n = 6) and Elo 100 μg/mL group (n = 6) compared with the control group (n = 6) throughout the observation period (from days 8 to 31). No significant difference was observed in the frequency of fibrocytes between the Elo 50 μg/mL and Elo 100 μg/mL groups. (C) Culture assay of human PBMCs exposed to Elo and other agents. Compared with the Rom group (n = 6), the frequency of fibrocytes significantly decreased in the Rom plus Elo group (n = 6), Rom plus Elo plus Rux group (n = 6), and Rom plus IFN-α2 group (n = 6) throughout the observation period (from days 8 to 10). In contrast, there was no significant difference between the Rom plus Elo and Rom plus Elo plus Rux groups in terms of fibrocyte differentiation. Rom plus IFN-α2 had the strongest suppressive effect on fibrocyte differentiation; however, approximately 80% to 90% of the cultured cells were dead and floated in the Rom plus IFN-α2 group, and we considered that IFN-α2 might be too cytotoxic in this setting. (D) Culture assay of PBMCs of from MF patients. Elo significantly decreased the frequency of fibrocytes derived from MPN patients harboring a CALR mutation, similar to fibrocytes derived from MPN patients with a JAK2V617F mutation. Results were reported as the mean ± standard deviation values for data obtained from 3 individual experiments with 6 HCs or 6 patients in each group. Statistical significance of the data for each day was calculated by 1-way ANOVA with post hoc Tukey’s honestly significant difference test. ns: not significant. *P < .05; **P < .01.
The results of the long-term culture assay showed that the frequency of fibrocytes significantly decreased in the Elo 50 μg/mL and Elo 100 μg/mL groups compared with the control group throughout the observation period (1-way ANOVA with post hoc Tukey’s honestly significant difference test P < .01) (Figure 4B). In contrast, no significant difference was observed in the frequency of fibrocytes between the Elo 50 μg/mL and Elo 100 μg/mL groups. These observations implied that the blood concentration of Elo administered by using a standard dose in a clinical setting may be enough to suppress fibrocyte differentiation for a long period of time.
To evaluate the efficacy of Elo and other agents used for treating MF in a clinical setting with Rux and IFN-α2 against the activated fibrocytes induced by Rom (one of the stimulators of fibrocyte differentiation9 ), we combined Elo, Rux, IFN-α2, and Rom. Compared with the Rom group, the frequency of fibrocytes significantly decreased in the Rom plus Elo group, Rom plus Elo plus Rux group, and Rom plus IFN-α2 group throughout the observation period (from days 8 to 10) (1-way ANOVA with post hoc Tukey’s honestly significant difference test P < .01) (Figure 4C). Rux did not show a synergistic inhibitory effect with Elo on fibrocyte differentiation. The Rom plus IFN-α2 group had the strongest suppressive effect on fibrocyte differentiation; however, ∼80% to 90% of the cultured cells were dead and floated in the Rom plus IFN-α2 group, and we considered that IFN-α2 might be too cytotoxic in this setting (data not shown). Whereas Rom induced fibrocyte differentiation,9 Elo exhibited an inhibitory effect on fibrocyte differentiation, even after Rom administration. In contrast, although we demonstrated that Rux suppressed fibrocyte differentiation,9 the combination of Elo and Rux did not show a synergistic inhibitory effect with Elo on fibrocyte differentiation.
Even in the culture assay of PBMCs derived from MPN patients with MF who harbored JAK2V617F, the frequency of fibrocytes significantly decreased in the Elo group compared with the control group. Moreover, Elo significantly decreased the frequency of fibrocytes derived from MPN patients harboring CALR mutation, similar to the fibrocytes derived from MPN patients with the JAK2V617F mutation (1-way ANOVA with post hoc Tukey’s honestly significant difference test P < .01) (Figure 4D). This result suggested that Elo inhibited fibrocyte differentiation in patients with MF who presented with JAK2V617F or CALR mutations.
Elo improved the Rom-induced MF-like phenotype in hNOG mice and attenuated the SLAMF7 expression of monocytes in the PB and spleen
By using the mouse xenograft model with Rom-induced MF, we examined the effects of Elo in vivo (Figure 5A). An analysis of the silver staining and immunohistochemical staining with α-smooth muscle actin of the femur BM sections showed that the mice treated with Rom alone exhibited diffused BM fibrosis and those treated with Rom plus Elo exhibited partial BM fibrosis (Figure 5B). In this study, almost all the mice treated with Rom alone presented with MF-3 in the degree of BM fibrosis. On the contrary, mice treated with Rom plus Elo showed no more than MF-2 with one exception (Table 2). These results indicated that Elo suppressed Rom-induced MF in vivo.
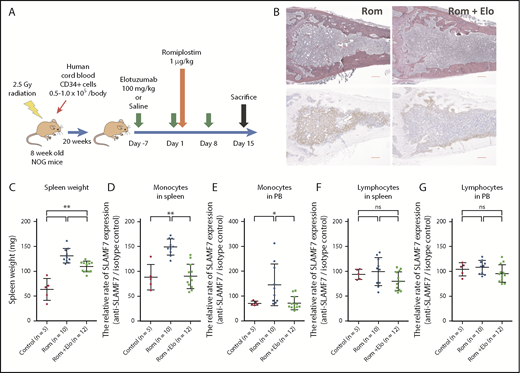
Analyses of femur BM sections, spleen weight, and SLAMF7 expression of monocytes in the spleen and PB of hNOG mice treated with Rom and Elo. (A) The protocol for the transplantation of human cord blood into NOG mice and the administration of Rom and Elo to transplanted hNOG mice in this study. (B) The silver staining and immunohistochemical staining with α-smooth muscle actin (α-SMA) of a femur BM section from hNOG mice treated with Rom and Elo. Both the silver-stained and α-SMA-stained BM sections showed increased fibrosis. (Left) The BM section of mice treated with Rom alone. The BM stroma with silver staining exhibited dense reticulin fibers intermingled with bundles of collagen. In addition, the BM stroma was diffusely α-SMA positive and stained brown in α-SMA staining. MF grade was evaluated as MF-3. The image is representative of 10 mice treated with Rom alone. (Right) The BM section of mice treated with Rom and Elo. The BM stroma exhibited focally dense reticulin fibers forming extensive intersections and was partially α-SMA positive. MF grade was evaluated as MF-2. Elo suppressed Rom-induced MF. The image is representative of 12 mice treated with Rom plus Elo. Pictures were taken and digitized by a BZ-X700 microscope and with BZ-H3A software. Original magnification ×20 for panel B. Scale bar, 300 μm. (C) The spleen weight of the treated hNOG mice. Both mice treated with Rom alone (n = 10) and mice treated with Rom plus Elo (n = 12) exhibited higher spleen weight than control mice (n = 5). The spleen weight significantly decreased in mice treated with Rom plus Elo compared with mice treated with Rom alone. (D) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) of CD45+CD14+ monocytes in the spleen of the treated hNOG mice. The relative rate of SLAMF7 expression of monocytes in the spleen significantly increased in mice treated with Rom alone (n = 10) compared with control mice (n = 5) and mice treated with Rom plus Elo (n = 12). (E) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) of CD45+CD14+ monocytes in the PB of the treated hNOG mice. The relative rate of SLAMF7 expression in monocytes in PB significantly increased in mice treated with Rom alone (n = 10) compared with control mice (n = 5) and mice treated with Rom plus Elo (n = 12). (F) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) in lymphocytes in the spleen of treated hNOG mice. No significant changes were observed in the relative rate of SLAMF7 expression of lymphocytes in the spleen among control mice (n = 5), mice treated with Rom alone (n = 10), and mice treated with Rom plus Elo (n = 12). (G) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) in lymphocytes in PB of treated hNOG mice. No significant changes were observed in the relative rate of SLAMF7 expression of lymphocytes in PB among control mice (n = 5), mice treated with Rom alone (n = 10), and mice treated with Rom plus Elo (n = 12), which is the same as the lymphocytes in the spleen. Results are reported as mean ± standard deviation values. Statistical significance was calculated using 1-way ANOVA with post hoc Tukey’s honestly significant difference test. *P < .05; **P < .01.
Analyses of femur BM sections, spleen weight, and SLAMF7 expression of monocytes in the spleen and PB of hNOG mice treated with Rom and Elo. (A) The protocol for the transplantation of human cord blood into NOG mice and the administration of Rom and Elo to transplanted hNOG mice in this study. (B) The silver staining and immunohistochemical staining with α-smooth muscle actin (α-SMA) of a femur BM section from hNOG mice treated with Rom and Elo. Both the silver-stained and α-SMA-stained BM sections showed increased fibrosis. (Left) The BM section of mice treated with Rom alone. The BM stroma with silver staining exhibited dense reticulin fibers intermingled with bundles of collagen. In addition, the BM stroma was diffusely α-SMA positive and stained brown in α-SMA staining. MF grade was evaluated as MF-3. The image is representative of 10 mice treated with Rom alone. (Right) The BM section of mice treated with Rom and Elo. The BM stroma exhibited focally dense reticulin fibers forming extensive intersections and was partially α-SMA positive. MF grade was evaluated as MF-2. Elo suppressed Rom-induced MF. The image is representative of 12 mice treated with Rom plus Elo. Pictures were taken and digitized by a BZ-X700 microscope and with BZ-H3A software. Original magnification ×20 for panel B. Scale bar, 300 μm. (C) The spleen weight of the treated hNOG mice. Both mice treated with Rom alone (n = 10) and mice treated with Rom plus Elo (n = 12) exhibited higher spleen weight than control mice (n = 5). The spleen weight significantly decreased in mice treated with Rom plus Elo compared with mice treated with Rom alone. (D) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) of CD45+CD14+ monocytes in the spleen of the treated hNOG mice. The relative rate of SLAMF7 expression of monocytes in the spleen significantly increased in mice treated with Rom alone (n = 10) compared with control mice (n = 5) and mice treated with Rom plus Elo (n = 12). (E) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) of CD45+CD14+ monocytes in the PB of the treated hNOG mice. The relative rate of SLAMF7 expression in monocytes in PB significantly increased in mice treated with Rom alone (n = 10) compared with control mice (n = 5) and mice treated with Rom plus Elo (n = 12). (F) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) in lymphocytes in the spleen of treated hNOG mice. No significant changes were observed in the relative rate of SLAMF7 expression of lymphocytes in the spleen among control mice (n = 5), mice treated with Rom alone (n = 10), and mice treated with Rom plus Elo (n = 12). (G) Analysis of the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) in lymphocytes in PB of treated hNOG mice. No significant changes were observed in the relative rate of SLAMF7 expression of lymphocytes in PB among control mice (n = 5), mice treated with Rom alone (n = 10), and mice treated with Rom plus Elo (n = 12), which is the same as the lymphocytes in the spleen. Results are reported as mean ± standard deviation values. Statistical significance was calculated using 1-way ANOVA with post hoc Tukey’s honestly significant difference test. *P < .05; **P < .01.
We also investigated the effects of Elo on splenomegaly and the spleen weight of the treated hNOG mice. Although both mice treated with Rom alone (n = 10) and mice treated with Rom plus Elo (n = 12) exhibited higher spleen weight than control mice (n = 5), the spleen weight significantly decreased in mice treated with Rom plus Elo compared with mice treated with Rom alone (1-way ANOVA with post hoc Tukey’s honestly significant difference test P < .01). (Figure 5C). Elo ameliorated Rom-induced splenomegaly.
We analyzed the relative rate of SLAMF7 expression (anti-SLAMF7 antibody/isotype control) of human CD45+CD14+ monocytes in the spleen and PB of the treated hNOG mice. The relative rate of SLAMF7 expression of monocytes in the spleen significantly increased in mice treated with Rom alone compared with control mice and mice treated with Rom plus Elo (1-way ANOVA with post hoc Tukey’s honestly significant difference test P < .01) (Figure 5D). The relative rate of SLAMF7 expression of monocytes in the PB also significantly increased in mice treated with Rom alone compared with control mice and mice treated with Rom plus Elo (1-way ANOVA with post hoc Tukey’s honestly significant difference test P < .05) (Figure 5E). In contrast, Elo did not significantly decrease the relative rate of SLAMF7 expression of lymphocytes in the spleen and PB (Figure 5F-G), which demonstrated that Elo did not disturb detection of SLAMF7+ cells.
Discussion
Generally, BM fibrosis in PMF is presumed to be caused by fibrogenic transforming growth factors (TGFs), for example, TGF-β released from clonal megakaryocytes and stimulated mesenchymal stromal cells.37 Recent experimental studies have shown that TGF-β1 is important in the development of MF in animal models.38 However, pirfenidone, an antifibrotic agent that inhibits fibrogenic cytokines (including TGF-β) had no positive effect on the treatment of patients with MF.39 This suggests that the pathogenesis of MF does not depend on TGF-β alone.
Recently, Verstovsek et al11 showed that serum amyloid P, an inhibitor of human-derived fibrocyte differentiation,40 caused the reduction of more than 1 grade of MF in 35% of patients.41 Fibrocytes are possible therapeutic targets for MF, and we have investigated their potential. A microarray analysis showed that fibrocytes expressed the cell surface molecule SLAMF7 at a higher level than macrophages did.21 SLAMF7 is a 66-kDa glycoprotein member of the SLAM superfamily.42 The expression of SLAMF7 is restricted to cells of hematopoietic origin, including CD8+ T cells, plasma cells, plasmacytoid dendritic cells, and resting NK cells, with minimal expression on resting CD4+ T cells, resting B cells, and monocytes.43 In contrast, the upregulation of SLAMF7 expression has been observed after the activation of CD4+ T cells, B cells, monocyte-derived dendritic cells, and monocytes.44 SLAMF7 is also highly expressed in multiple myeloma cells.45 We confirmed that murine and human fibrocytes highly expressed SLAMF7 on their surface.9 We also confirmed that the SLAMF7highCD16− monocytes were significantly increased in the PB of MPN patients with MF, irrespective of the existence of genetic mutation (Figure 2A-B), and showed potential for the frequency of SLAMF7highCD16− monocytes as a surrogate marker of MF onset in MPN patients who harbor a JAK2V617F mutation (Figure 2C).
In contrast, there was no significant difference in the percentage of CD16+ monocyte subsets among HCs and MPN patients with or without MF, and MPN patients with JAK2V617F, CALR, and triple-negative mutations (supplemental Figure 3). Moreover, the SLAMF7highCD16− monocyte subset contains a higher level of JAK2V617F+ neoplastic clones and could be a molecular target of anti-MF agents. Several reports demonstrated that MPN patients with MF exhibited a higher JAK2V617F allele burden in PB than did MPN patients without MF.25,26,46,47 A significantly higher rate of MF transformation in MPN patients who harbor homozygous JAK2V617F than in those who harbor heterozygous JAK2V617F was also reported.46 Our findings indicate that a high JAK2V617F allele burden might promote MF progress by increasing SLAMF7highCD16− monocytes, which tend to differentiate into fibrocytes. However, the accurate process that SLAMF7high monocytes or fibrocytes contribute to the progression of MF remains to be elucidated. The molecular mechanisms of MF pathogenesis by fibrocytes is an important subject that needs to be investigated.
IL-1ra was strongly correlated with JAK2V617F allele burden and SLAMF7highCD16− monocytes. IL-1ra, which is a member of the IL-1 family, binds to IL-1 receptors without inducing any intracellular response.48 IL-1ra acts as an antifibrotic agent against the murine pulmonary fibrosis model,49 murine systemic sclerosis models,50 and murine and human cystic fibrosis.51 In our study, IL-1ra was significantly increased in MF patients, which is believed to be the result of negative feedback against MF. One limitation of our study is that because of its cross-sectional nature, not all patient blood samples were collected at the same time as BM biopsy (the maximum interval was 1 year). However, because both SLAMF7highCD16− monocytes and IL-1ra were markedly correlated with the results of BM biopsy, it would be worth evaluating the efficacy of these 2 factors as noninvasive markers of BM biopsy in a prospective study.
Our results showed that Elo suppressed fibrocyte differentiation from human monocytes for a long period of time at a concentration greater than 50 μg/mL in vivo. Furthermore, the addition of NK cells enhanced the inhibitory effect of Elo. Balasa et al52 reported that Elo enhanced NK cell activation and myeloma cell killing through IL-2 and tumor necrosis factor-α pathways. However, the process by which NK cells activated by Elo suppress fibrocyte differentiation remains to be elucidated. In contrast, Rux did not show a synergistic inhibitory effect with Elo on fibrocyte differentiation, although Rux itself suppressed fibrocyte differentiation.9 Regarding the downstream signal transduction, SLAMF7 functions as either an inhibitory or activating receptor depending on the SLAM-associated protein-related adaptor protein EWS/Fli1-activated transcript 2 (EAT-2),53 which is an intracellular protein containing the Src homology 2 domain devoid of enzymatic activity.42 However, our investigation showed no evidence that supported the relevance between SLAMF7 and the JAK/STAT pathway. Why Rux showed no synergistic inhibitory effect with Elo on fibrocyte differentiation also remains to be elucidated. Furthermore, Elo improved MF and splenomegaly in hNOG mice that harbored a Rom-induced MF phenotype in vivo. It also attenuated the SLAMF7 expression of monocytes in the PB and spleen. These results showed that Elo selectively eliminated fibrocytes that highly expressed SLAMF7 and could ameliorate the symptoms of MF in a murine xenograft model.
With respect to this clinical study of Elo against MF patients, one of the advantages of selecting MPN patients with MF who harbor a JAK2V617F mutation is that the SLAMF7highCD16− monocyte percentage and JAK2V617F allele burden could be surrogate markers for evaluating the efficacy of Elo. In contrast, the inclusion of MPN patients with MF who are receiving Rux or other medications might modify the adverse effects of Elo against MPN patients. We plan to start a clinical study to confirm the safety and efficacy of Elo against MPN patients with MF who harbor JAK2V617F who were not initially receiving any medication. We will then proceed with a clinical study to confirm the efficacy of Elo against not only MPN patients with MF who harbor JAK2V617F but also in MPN patients with MF who do not harbor JAK2V617F and MPN patients with MF who are receiving various medications.
In summary, our findings indicate that SLAMF7highCD16− monocytes and IL-1ra were correlated with MF onset in MPN patients who harbor JAK2V617F, that Elo suppressed fibrocyte differentiation and MF progression in vitro and in vivo, and that Elo could be a potential therapeutic agent for MF.
For original data, please contact fkimura@ndmc.ac.jp.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the members of the Department of Immunology and Microbiology for their support for the experimental installation and scientific discussion.
This work was partly supported by Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research No. 18K08344 (F.K.) and a grant from Bristol-Myers Squibb (M.M.).
Authorship
Contribution: T.M. and S. Kato designed the research, performed experiments, analyzed data, and wrote the manuscript; R.H.-S., K.E.-U., S.U., A.K., S. Kobayashi, K. Sato, M.H., S.S., M.A., M.M., and N.K. supervised the study; K.U. provided patients’ samples and supervised the study; T.K., H.O., and Y. Osawa performed experiments and analyzed data; K. Takada, T.S., K. Saito, T.I., S.N., K. Takano, Y. Okada, N.T., M.T., and T.H. collected and preserved patients’ samples and analyzed the data; S.M. examined mutations; and F.K. supervised and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Fumihiko Kimura, Division of Hematology, Department of Internal Medicine, National Defense Medical College, Namiki 3-2, Tokorozawa 359-8513, Japan; e-mail: fkimura@ndmc.ac.jp