Key Points
211At targeted to CD38 eliminates MM cell clones in murine models of low-burden disease.
211At deposits ≥500 times more energy than β-emitters and provides a mechanism of uniform cell kill unique among MM therapeutics.

Abstract
Minimal residual disease (MRD) has become an increasingly prevalent and important entity in multiple myeloma (MM). Despite deepening responses to frontline therapy, roughly 75% of MM patients never become MRD-negative to ≤10−5, which is concerning because MRD-negative status predicts significantly longer survival. MM is highly heterogeneous, and MRD persistence may reflect survival of isolated single cells and small clusters of treatment-resistant subclones. Virtually all MM clones are exquisitely sensitive to radiation, and the α-emitter astatine-211 (211At) deposits prodigious energy within 3 cell diameters, which is ideal for eliminating MRD if effectively targeted. CD38 is a proven MM target, and we conjugated 211At to an anti-CD38 monoclonal antibody to create an 211At-CD38 therapy. When examined in a bulky xenograft model of MM, single-dose 211At-CD38 at 15 to 45 µCi at least doubled median survival of mice relative to untreated controls (P < .003), but no mice achieved complete remission and all died within 75 days. In contrast, in a disseminated disease model designed to reflect low-burden MRD, 3 studies demonstrated that single-dose 211At-CD38 at 24 to 45 µCi produced sustained remission and long-term survival (>150 days) for 50% to 80% of mice, where all untreated mice died in 20 to 55 days (P < .0001). Treatment toxicities were transient and minimal. These data suggest that 211At-CD38 offers the potential to eliminate residual MM cell clones in low-disease-burden settings, including MRD. We are optimistic that, in a planned clinical trial, addition of 211At-CD38 to an autologous stem cell transplant (ASCT) conditioning regimen may improve ASCT outcomes for MM patients.
Introduction
Potent treatments for multiple myeloma (MM) greatly reduce initial disease burden and result in a complete response for a significant subset of patients.1,2 However, most patients relapse as a consequence of minimal residual disease (MRD) defined by occult foci of treatment-insensitive tumor cells.3,-5 Therapy capable of entirely eliminating malignant cell clones from within low-disease-burden settings, including MRD, has remained elusive. Malignant cell escape following treatment with pathway-specific targeting agents may be explained by heterogeneity of MM within afflicted individuals.5 In contrast, radionuclide therapy is agnostic to disease heterogeneity as virtually all MM cells, including clones with high-risk cytogenetic features, are exquisitely sensitive to radiation.6,,-9 The α-emitting radionuclide astatine-211 (211At) is an optimal candidate for eradicating low-level disease and has been characterized as “the most promising α particle” for therapy based on its decay characteristics.10 By depositing a large dose of radiation (6.8 MeV) concentrated within a few cell diameters (50-90 μm), 211At produces a prodigious linear energy transfer (LET) and then quickly decays (7.2-hour half-life) without producing problematic long-lived daughter nuclides. High LET cytotoxicity results from double-strand DNA breaks that overwhelm cellular repair mechanisms.11,12 Studies of other hematopoietic cell types suggest that this high LET cell-killing mechanism will remain effective irrespective of a potentially quiescent state and the hypoxic bone marrow environments in which MRD MM is thought to survive.12,,-15 We therefore hypothesized that 211At targeted to MM cells would be uniquely capable of eliminating isolated cells and small tumor clusters with minimal damage to surrounding tissues.
We selected an anti-human CD38 monoclonal antibody (mAb) as the targeting agent. The CD38 antigen is expressed on malignant plasma cells regardless of mutational status, and anti-CD38 mAbs constitute a proven targeted therapy for MM, but resistance mechanisms prevent unmodified CD38 mAbs from reliably eradicating disease.16,,-19 Using CD38 mAbs as targeting agents for β-emitter radionuclides can eliminate disease in preclinical MM models,20,21 yet these studies also suggest that current β-therapies using yttrium-90 (90Y) and iodine-131 (131I) are optimally effective for larger tumor clusters than typified by MRD.22 In clinical settings, directly targeted β-therapies have been associated with hematologic toxicity that in some cases may prevent dose escalation to levels that eliminate MRD.23,24 Possible contributors to toxicity include a relatively longer decay half-life (eg, 131I, 8 days)23 or longer path length (eg, 90Y, 5 mm).24 Thus, the favorable physical characteristics of α-emitting radionuclides, and new opportunities to harness their potential,25,26 provide rationale for our approach.
To create a therapeutic agent, we conjugated the anti-CD38 mAb OKT10 to the novel amine-reactive agent B10-NCS,25 which is labeled with 211At on the day of treatment. We hypothesized that the resulting 211At-CD38 therapy would effectively treat MM in preclinical models. We tested the specific predictions that 211At-CD38 would selectively bind and kill CD38+ MM cells in vitro, and, in murine xenograft models of bulky vs low-burden disease designed to recapitulate MRD, would optimally eliminate the latter.
Methods
Cell culture
The human MM cell lines NCI-H929, U266B1, and RPMI 8226 were obtained from the American Type Culture Collection (ATCC; Manassas, VA), and OPM-2 and MOLP-8 from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). Cell lines were authenticated by DNA profiling (Fred Hutchinson Cancer Research Center [FHCRC] Research Cell Bank, Seattle, WA), tested for mycoplasma, and maintained in log-phase growth in RPMI 1640 media supplemented with 10% to 20% fetal bovine serum, 50 U/mL penicillin G, and 50 µg/mL streptomycin sulfate for no more than 6 weeks after thawing. Luciferase (Luc) cell lines were generated by retroviral transduction to stably express firefly luciferase for in vivo bioluminescent imaging (BLI).
Reagents and radiolabeling
The OKT10 hybridoma cell line expressing the murine immunoglobulin G1 (IgG1) anti-human CD38 mAb, and the bovine herpes virus-1 (BHV1) hybridoma (IgG1 isotype control), were purchased from ATCC. OKT10 and BHV1 antibodies (Abs) were produced from the hybridomas using a hollow-fiber bioreactor system in the mAb production facility at FHCRC. The amine-reactive bifunctional labeling reagent B10-NCS was prepared as previously reported and conjugated to the Abs using the method of Wilbur.25 Produced on a Scanditronix MC-50 cyclotron at the University of Washington, 211At was used to label OKT10-B10 and BHV1-B10 as previously described.27,28
Cell binding and cytotoxicity of radiolabeled anti-CD38 OKT10-B10
To assess cell binding of anti-CD38 (OKT10) mAb constructs, 5 × 105 MM cells per well of CD38+ NCI-H929 or CD38− U266B1 were incubated in 50 µL of either OKT10, OKT10-DOTA, or OKT10-B10 at 0.2 mg/mL for 30 minutes at 4°C, washed twice, resuspended in 50 µL of 1:100 anti-mouse IgG (F(ab′)2)-allophycocyanin (APC; Jackson ImmunoResearch, West Grove, PA), incubated 30 minutes at 4°C, washed twice, and analyzed using flow cytometry. To assess cell binding of radiolabeled anti-CD38 mAb, 3.3 × 106 MM cells per well of CD38+ OPM-2 were first incubated in 50 µL of OKT10-B10 (0.2 mg/mL) or media (RPMI 1649, 1% fetal bovine serum, 0.1% sodium azide) for 45 minutes at 4°C. Then, 50 µL of 4 µg/mL 211At-OKT10-B10 or 211At-BHV1-B10 was added and cells were incubated for 60 minutes at 4°C, washed twice, and cell-associated radioactivity measured in a γ counter. To assess cytotoxicity, 2 × 104 MM cells per well of CD38+ NCI-H929 or CD38− U266B1 were incubated at 37°C in 100 µL of cell media (control) or media plus serial dilutions of unlabeled OKT10-B10 (maximum concentration 20 μg/mL), or 211At-OKT10-B10 (maximum concentration 20 μg/mL labeled with 250 µCi 211At). After 60 hours, cell viability was assessed using CellTiter-Glo (Promega, Madison, WI).
Mouse models
Female and male NOD.Cg-Rag1tm1MomIl2rgtm1Wjl/SzJ (NRG) mice (FHCRC colony) were maintained under standard protocols approved by the FHCRC Institutional Animal Care and Use Committee (IACUC). To model bulky disease, MM xenografts were generated by subcutaneous flank injection of 1 × 107 OPM-2Luc or NCI-H929 cells 7 to 9 days prior to treatment to produce tumors of 25 mm3 to 75 mm3 at treatment. A low-disease-burden model was developed through optimization of protocols identifying minimal IV-injected tumor cell doses capable of producing 100% engraftment rates and similar low disease burdens within, and between, cell line models at time of treatment (supplemental Figure 1, available on the Blood Web site). In the final models, mice received 2.75 Gy external beam cesium-137 (137Cs; JL Shepard Mark I irradiator) 6 days prior to treatment, followed 1 day later by IV injection of 1.2 × 105 MOLP-8Luc, 2.5 × 105 OPM-2Luc, or 5 × 105 NCI-H929Luc cells (5 days prior to treatment). In all studies, mice were randomized into groups with equivalent body weights, tumor sizes, and sex ratios.
Biodistribution studies
NRG mice bearing flank MM xenografts received IV injections of 15 µg of mAb radiolabeled with 15 µCi 211At, as either 211At-OKT10-B10 (211At-CD38), or IgG1 (isotype-matched) control 211At-BHV1-B10 (211At-Ab−) (n = 5 per group). Retro-orbital blood (ROB) samples were taken from 3 mice per group at 1, 25, 120, and 250 minutes posttherapy. At 24 hours posttherapy, all mice were euthanized, and blood, tumor, and relevant normal tissues (lung, liver, kidneys, spleen, stomach, small and large intestines, muscle, and tail) were harvested, weighed, and 211At activity was measured. Decay-corrected percentage injected dose (ID) of 211At absorbed per gram of tissue (percent ID per gram), and tumor-to-normal tissue ratios of absorbed radioactivity were calculated.
Therapy studies
Female and male NRG mice bearing either subcutaneous flank xenografts or disseminated disease received IV injections of 15 µg of mAb radiolabeled with 7.5 to 45 µCi as either 211At-CD38 or isotype control 211At-Ab− (n = 5-10 per group). All mice received syngeneic donor bone marrow transplant 3 days posttherapy, potentially required to reverse myelotoxicity at high-dose levels.29 In subcutaneous models, tumor dimensions were monitored 3 times weekly and, in disseminated models, disease progression was monitored weekly via BLI. Body weight was monitored 3 times weekly and mice were euthanized if they experienced >30% weight loss, hind-limb paralysis, or exceeded tumor volume limits per IACUC requirements. In the study using the 2 highest 211At doses (30 and 45 µCi, OPM-2Luc disseminated model), ROB samples were taken at intervals to assess hematopoietic, renal, and hepatic function. ROB sampling was limited to 1 study in consideration of animal safety and prior observations of minimal 211At-therapy–related toxicities.28
Statistical analyses
In biodistribution studies, effects of 211At-CD38 vs 211At-Ab− on tumor and normal tissue 211At absorption were examined by the Student t test, and relative 211At absorptions of tumor vs normal tissues were examined by the paired Student t test. In therapy studies, treatment effects on mouse survival were determined by log-rank (single independent variable) or proportional hazards (multivariate) analyses of Kaplan-Meier survival functions. Analyses were performed using JMP 14 (SAS Institute, Cary, NC) and GraphPad Prism 7.03 (GraphPad Software, La Jolla, CA).
Results
In vitro cell binding and cytotoxicity
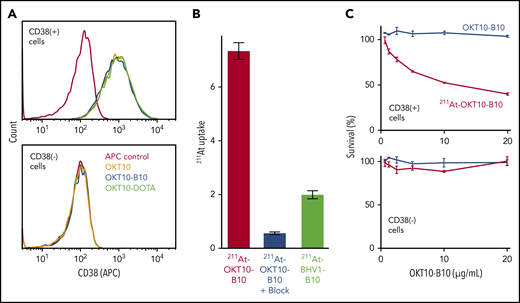
We confirmed the antigen specificity of our anti-CD38 constructs with flow cytometry, showing that CD38+ MM cells bound OKT10, OKT10-B10, and OKT10-DOTA with an equally high affinity, whereas CD38− MM cells bound none of these constructs (Figure 1A). To confirm binding specificity of radiolabeled mAbs, we assayed 211At levels in MM cell lines after incubation with 211At-CD38 mAb (OKT10-B10) or 211At-nontargeting, isotype control mAb (BHV1-B10). The 211At level (percentage uptake) was significantly higher after CD38 vs control mAb, and over 92% of the 211At uptake from 211At-CD38 was blocked by preincubating cells with unlabeled CD38 mAb (Figure 1B). Low-level 211At uptake with the control mAb reflects nonspecific binding as seen in previous 211At-mAb cell-binding studies.28 We assayed selective cytotoxicity of 211At-CD38 mAb by comparing viability of CD38+ and CD38− cell lines following incubation with either 211At-CD38 mAb or unlabeled CD38 mAb (OKT10-B10) and demonstrated that 211At-CD38 selectively eliminates CD38+ cells, but not CD38− cells, and that unlabeled CD38 mAb did not affect cell viability (Figure 1C).
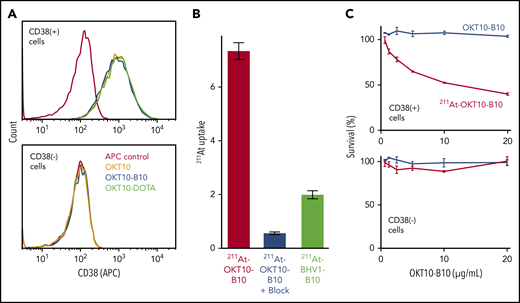
211At-CD38 selectively binds and kills CD38+ MM cells. (A) CD38+ or CD38− cell lines were incubated with either anti-CD38 OKT10 mAb, or OKT10 conjugated with amine-reactive B10, or OKT10 conjugated with yttrium chelator DOTA. Cells were then washed twice, incubated with anti-mouse IgG Fc-APC, washed twice, and analyzed by flow cytometry. (B) CD38+ cells were preincubated with buffer or CD38 mAb, then 211At-OKT10-B10 or isotype control 211At-BHV1-B10 was added and cells incubated, washed twice, cell-associated radioactivity measured in a γ counter, and 211At uptake calculated as a percentage of applied 211At. N = 4 replicates per treatment; error bars = 1 standard error of the mean (SEM). (C) CD38+ or CD38− cells were incubated in media (control) or serial dilutions of OKT10-B10 or 211At-OKT10-B10 at 37°C for 60 hours. Cytotoxicity was then assessed using CellTiter-Glo. N = 3 replicates per treatment; error bars = 1 standard deviation (SD).
211At-CD38 selectively binds and kills CD38+ MM cells. (A) CD38+ or CD38− cell lines were incubated with either anti-CD38 OKT10 mAb, or OKT10 conjugated with amine-reactive B10, or OKT10 conjugated with yttrium chelator DOTA. Cells were then washed twice, incubated with anti-mouse IgG Fc-APC, washed twice, and analyzed by flow cytometry. (B) CD38+ cells were preincubated with buffer or CD38 mAb, then 211At-OKT10-B10 or isotype control 211At-BHV1-B10 was added and cells incubated, washed twice, cell-associated radioactivity measured in a γ counter, and 211At uptake calculated as a percentage of applied 211At. N = 4 replicates per treatment; error bars = 1 standard error of the mean (SEM). (C) CD38+ or CD38− cells were incubated in media (control) or serial dilutions of OKT10-B10 or 211At-OKT10-B10 at 37°C for 60 hours. Cytotoxicity was then assessed using CellTiter-Glo. N = 3 replicates per treatment; error bars = 1 standard deviation (SD).
211At-CD38 biodistribution and pharmacokinetics
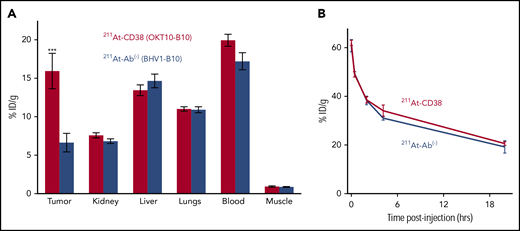
Biodistribution experiments demonstrated the ability of 211At-CD38 to selectively target MM xenografts in NRG mice. Mice bearing subcutaneous tumors were IV injected with 211At-CD38 or 211At-Ab− and tissues harvested 24 hours later, the time of maximum 211At retention.28 Targeted 211At-CD38 delivered more radiation to tumor than did isotype control 211At-Ab− (15.9% vs 6.6%, respectively, of ID per gram; P = .007; Figure 2A), and created favorable tumor:normal organ ratios of absorbed activity for kidney, liver, and lungs. As expected, blood retained a similar amount of activity as targeted tumor, and consistent with previous observations,28 blood-rich nontargeted organs retained similar activity following 211At-CD38 or 211At-Ab−. To examine pharmacokinetics of 211At-mAbs, we sampled ROB at intervals from 1 minute to 24 hours posttherapy injection. Blood activity, which was 60% ID per gram at 1 minute, reduced to 38% at 2 hours and 20% at 24 hours, and this clearance profile was nearly identical for targeted and isotype control treatments (Figure 2B).
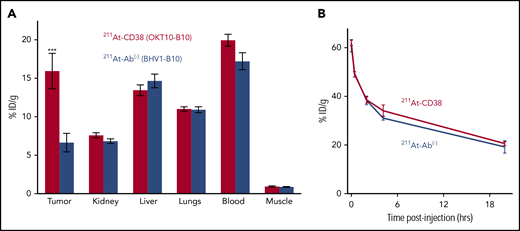
211At-CD38 biodistribution and pharmacokinetics. Mice bearing subcutaneous MM xenografts were IV injected with 211At-CD38 or 211At-Ab−, and ROB samples taken at 1, 25, 120, and 250 minutes posttherapy. At 24 hours posttherapy, mice were euthanized and blood, tumor, and normal tissues harvested, weighed, and γ counted, and then decay-corrected percent-injected dose of 211At absorbed per gram of tissue calculated. (A) Biodistribution of 211At-CD38. Tumors targeted by 211At-CD38 absorbed 2.4 times more activity than tumors exposed to 211At-Ab− (P = .007), and 211At-CD38 created favorable tumor-to-normal tissue ratios of absorbed activity for kidney, liver, and lungs. N = 5 mice per group; error bars = 1 SEM. (B) Blood clearance of 211At-CD38. Blood radioactivity of 211At-CD38 treated mice reduced from 60% ID per gram at 1 minute posttherapy to 38% at 2 hours and 20% at 24 hours. N = 3 mice per time point per treatment; error bars = 1 SEM.
211At-CD38 biodistribution and pharmacokinetics. Mice bearing subcutaneous MM xenografts were IV injected with 211At-CD38 or 211At-Ab−, and ROB samples taken at 1, 25, 120, and 250 minutes posttherapy. At 24 hours posttherapy, mice were euthanized and blood, tumor, and normal tissues harvested, weighed, and γ counted, and then decay-corrected percent-injected dose of 211At absorbed per gram of tissue calculated. (A) Biodistribution of 211At-CD38. Tumors targeted by 211At-CD38 absorbed 2.4 times more activity than tumors exposed to 211At-Ab− (P = .007), and 211At-CD38 created favorable tumor-to-normal tissue ratios of absorbed activity for kidney, liver, and lungs. N = 5 mice per group; error bars = 1 SEM. (B) Blood clearance of 211At-CD38. Blood radioactivity of 211At-CD38 treated mice reduced from 60% ID per gram at 1 minute posttherapy to 38% at 2 hours and 20% at 24 hours. N = 3 mice per time point per treatment; error bars = 1 SEM.
211At-CD38 therapy in subcutaneous MM models
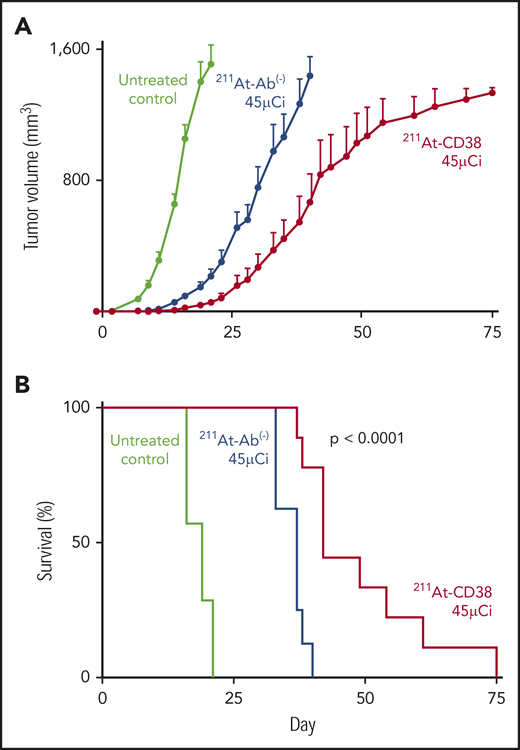
Subcutaneous xenograft studies were designed to recapitulate bulky disease, and, in 2 therapy studies of these models, 211At-CD38 significantly slowed disease progression and doubled median survival of mice. In the first study, mice bearing NCI-H929 flank tumors were treated with 211At-CD38 at 7.5 or 15 µCi. As anticipated,28 in a bulky tumor model, 211At therapy was not capable of eliminating disease, and the median survival of untreated mice (17 days) and 211At-CD38 7.5-µCi mice (21 days) was not different (P > .1, Kaplan-Meier log-rank test). 211At-CD38 15-µCi treatment extended median survival to 34 days (greater than other groups; P < .01), but all mice eventually succumbed to disease (data not shown). In the second study, mice bearing OPM-2Luc flank tumors were treated with 211At-CD38 at 30 and 45 µCi. α-emitter–labeled anti-CD38 mAbs again slowed tumor growth (Figure 3A), and extended median survival beyond untreated mice (Figure 3B; untreated [19 days] vs 211At-CD38 30-µCi [40 days] or vs 211At-CD38 45-µCi [42 days]; P < .0001), and beyond 211At-Ab− 45-µCi–treated mice (median survival, 37 days; P = .0005; Kaplan-Meier log-rank tests). However, no long-term survival was observed (Figure 3B).
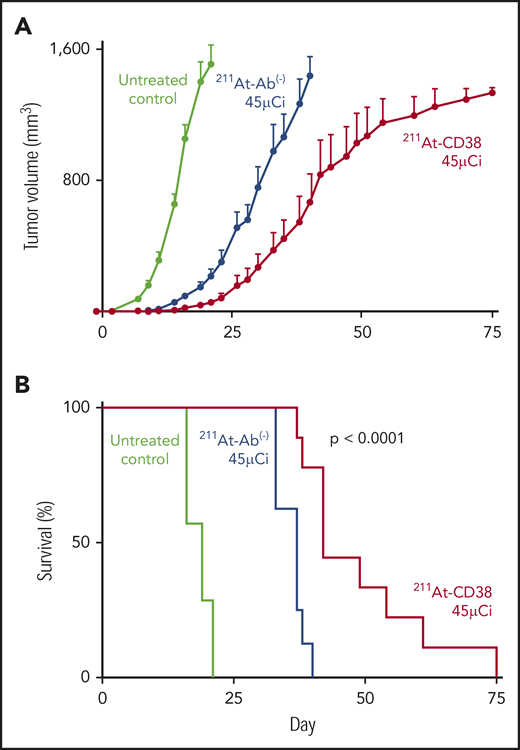
211At-CD38 therapy in subcutaneous MM models. Mice bearing subcutaneous OPM-2Luc xenografts were treated on day 0 with 30 or 45 µCi 211At-CD38 or 211At-Ab− (isotype control); n = 8 to 9 mice per group. Tumor dimensions were monitored 3 times weekly and mice euthanized when tumor volumes met IACUC requirements. (A) Tumor progression for untreated controls and 45-µCi groups; error bars = 1 SEM. (B) Kaplan-Meier survival curves. The 45-µCi 211At-CD38 treatment increased median survival relative to untreated (P < .0001) and to 45-µCi 211At-Ab− treated mice (P = .0005). The 30-µCi groups are not shown for visual clarity; data are described in “Results.”
211At-CD38 therapy in subcutaneous MM models. Mice bearing subcutaneous OPM-2Luc xenografts were treated on day 0 with 30 or 45 µCi 211At-CD38 or 211At-Ab− (isotype control); n = 8 to 9 mice per group. Tumor dimensions were monitored 3 times weekly and mice euthanized when tumor volumes met IACUC requirements. (A) Tumor progression for untreated controls and 45-µCi groups; error bars = 1 SEM. (B) Kaplan-Meier survival curves. The 45-µCi 211At-CD38 treatment increased median survival relative to untreated (P < .0001) and to 45-µCi 211At-Ab− treated mice (P = .0005). The 30-µCi groups are not shown for visual clarity; data are described in “Results.”
211At-CD38 therapy in disseminated MM models
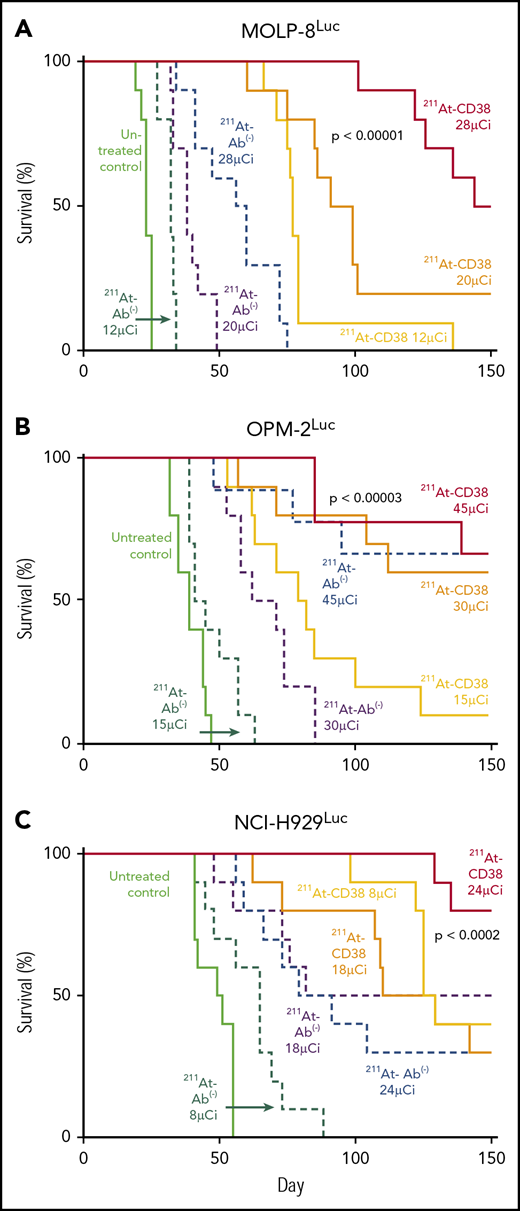
Disseminated xenograft models were designed to recapitulate the isolated tumor cells and small cell clusters associated with MRD and exploit the salient features of α-emitter therapy. In 3 therapy studies of these models, single-dose 211At-CD38 at 24 to 45 µCi produced sustained remissions that resulted in long-term survival (>150 days) for 50% to 80% of mice.
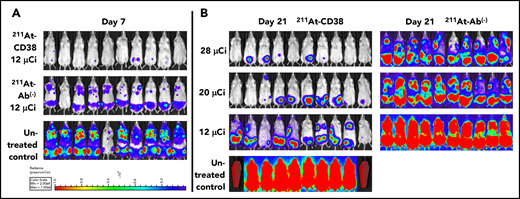
In MOLP-8Luc models, mice bearing disseminated disease were treated with 211At-CD38 or 211At-Ab− at 12, 20, or 28 µCi. At 7 days posttreatment, all mice treated at even the lowest dose level showed significant disease regression when compared with controls (Figure 4A). At 21 days, disease had recurred in all 211At-CD38 12-µCi–treated mice, but 20% of 211At-CD38 20-µCi and 50% of 211At-CD38 28-µCi mice lacked detectable tumors by BLI (Figure 4B), and mice lacking visible tumors at day 21 all survived over 100 days. In contrast, all untreated and 211At-Ab− groups showed extensive disease progression at day 21 (Figure 4B) and untreated mice had a median survival of only 23 days (Figure 5). 211At-CD38 treatments extended life at least threefold, providing a median survival of 77 and 95 days for 211At-CD38 12- and 20-µCi mice, respectively, and 90% survival of 211At-CD38 28-µCi mice through day 120. Accordingly, 211At-CD38 treatment strongly predicted improved survival relative to untreated controls (P < .00001) and to 211At-Ab− treated mice (P < .00001) in a dose-dependent manner (P < .00001; Kaplan-Meier proportional hazards tests; Figure 5).
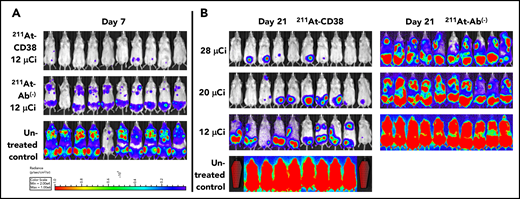
211At-CD38 therapy in a disseminated MOLP-8Luc model. Mice bearing disseminated MOLP-8Luc xenografts were treated on day 0 with 211At-CD38 or 211At-Ab− at 12, 20, or 28 µCi; n = 10 mice per group. Disease progression was monitored via weekly BLI and thrice weekly observations of mouse weight, condition, and mobility. Mice were euthanized when they experienced hind-limb paralysis or met IACUC weight loss or condition requirements. (A) BLI of untreated control and 12-µCi treatment groups at 7 days posttherapy. (B) BLI of all groups at 21 days posttherapy. Red coffins indicate deceased mice. BLI scale identical to panel A.
211At-CD38 therapy in a disseminated MOLP-8Luc model. Mice bearing disseminated MOLP-8Luc xenografts were treated on day 0 with 211At-CD38 or 211At-Ab− at 12, 20, or 28 µCi; n = 10 mice per group. Disease progression was monitored via weekly BLI and thrice weekly observations of mouse weight, condition, and mobility. Mice were euthanized when they experienced hind-limb paralysis or met IACUC weight loss or condition requirements. (A) BLI of untreated control and 12-µCi treatment groups at 7 days posttherapy. (B) BLI of all groups at 21 days posttherapy. Red coffins indicate deceased mice. BLI scale identical to panel A.
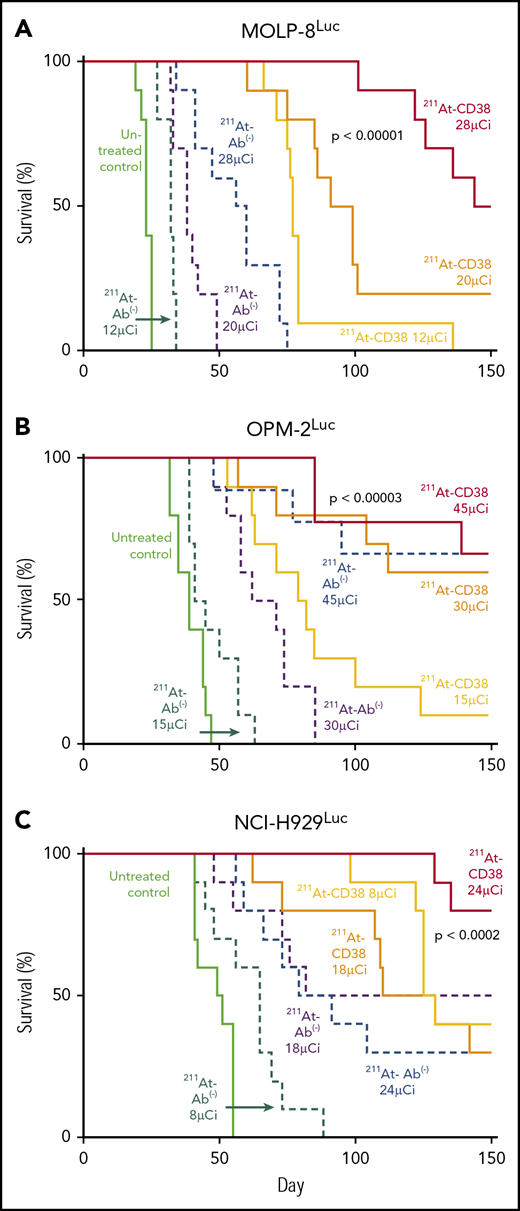
Kaplan-Meier survival curves for disseminated models. Mice bearing disseminated MM xenografts were treated on day 0 with 211At-CD38 or 211At-Ab−; n = 9 to 10 mice per group. Disease progression was monitored via weekly BLI and thrice weekly observations of mouse weight, condition, and mobility. Mice were euthanized when they experienced hind-limb paralysis or met IACUC weight loss or condition requirements. Statistics report Kaplan-Meier proportional hazards tests. (A) MOLP-8Luc model, treated at 12, 20, and 28 µCi. Across treatment levels, 211At-CD38 therapy strongly benefitted survival relative to untreated controls (P < .00001) and to 211At-Ab− groups (P < .00001) in a dose-dependent manner (P < .00001). (B) OPM-2Luc model, treated at 15, 30, and 45 µCi. 211At-CD38 therapy strongly improved survival across groups, relative to untreated controls (P < .00001) and to 211At-Ab− treatments (P = .00002) in a dose-dependent manner (P < .00001). Additional statistics in “Results.” (C) NCI-H929Luc model, treated at 8, 18, or 24 µCi. Mice treated with 211At-CD38 survived significantly longer than untreated control (P < .0001) and 211At-Ab− treated mice (P < .0006). Treatment benefits were dose-dependent with higher 211At dosing improving survival (P < .002).
Kaplan-Meier survival curves for disseminated models. Mice bearing disseminated MM xenografts were treated on day 0 with 211At-CD38 or 211At-Ab−; n = 9 to 10 mice per group. Disease progression was monitored via weekly BLI and thrice weekly observations of mouse weight, condition, and mobility. Mice were euthanized when they experienced hind-limb paralysis or met IACUC weight loss or condition requirements. Statistics report Kaplan-Meier proportional hazards tests. (A) MOLP-8Luc model, treated at 12, 20, and 28 µCi. Across treatment levels, 211At-CD38 therapy strongly benefitted survival relative to untreated controls (P < .00001) and to 211At-Ab− groups (P < .00001) in a dose-dependent manner (P < .00001). (B) OPM-2Luc model, treated at 15, 30, and 45 µCi. 211At-CD38 therapy strongly improved survival across groups, relative to untreated controls (P < .00001) and to 211At-Ab− treatments (P = .00002) in a dose-dependent manner (P < .00001). Additional statistics in “Results.” (C) NCI-H929Luc model, treated at 8, 18, or 24 µCi. Mice treated with 211At-CD38 survived significantly longer than untreated control (P < .0001) and 211At-Ab− treated mice (P < .0006). Treatment benefits were dose-dependent with higher 211At dosing improving survival (P < .002).
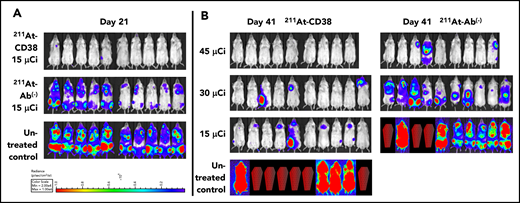
We explored a higher 211At dose in our study of OPM-2Luc, where mice bearing disseminated disease received 211At-CD38 or 211At-Ab− at 15, 30, or 45 µCi. For this cell line, 211At-CD38 15-µCi eliminated detectable disease in 80% of mice at day 21, whereas no untreated mice and no mice receiving 211At-Ab− 15-µCi were disease-free (Figure 6A). At day 41, only 20% of 211At-CD38 15-µCi mice demonstrated an absence of MM, but 70% of 211At-CD38 30-µCi and 100% of 211At-CD38 45-µCi mice had no detectable disease by imaging (Figure 6B). In contrast, all untreated, all 211At-Ab− 15-µCi and 30-µCi mice, and 33% of 211At-Ab− 45-µCi mice displayed measurable disease at this time point (Figure 6B). These patterns persisted through the end of the study: all untreated mice died of progressive disease by day 47 (Figure 5B), whereas as of day 150, disease had recurred in only 1 of 9 211At-CD38 45-µCi mice. Two 211At-CD38 45-µCi mice died spontaneously during the study without detectable cause (no apparent disease or toxicity, see “Toxicity”), and as of day 150, survival did not differ between the CD38-targeted and isotype-matched treatment group receiving 45 µCi (P = .87; Kaplan-Meier log-rank test; Figure 5B). Nonetheless, 211At-CD38 efficacy significantly surpassed 211At-Ab− overall. Among 30-µCi groups, 60% of 211At-CD38 mice remained alive at day 150 whereas all 211At-Ab− mice were dead from disease by day 85 (P = .0007), and, in 15-µCi groups, 211At-CD38 cured 10% of mice whereas all 211At-Ab− mice died by day 57 (P < .0001; Kaplan-Meier log-rank tests; Figure 5B). Analyzed across all 3 211At treatment levels, 211At-CD38 strongly outperformed 211At-Ab− (P = .00002) and extended survival in a dose-dependent manner (P < .00001; Kaplan-Meier proportional hazards test).
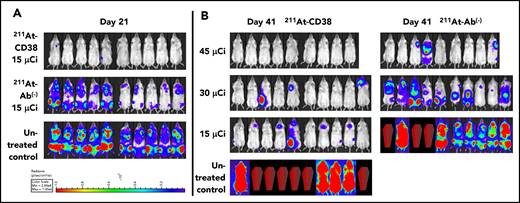
211At-CD38 therapy in a disseminated OPM-2 Luc model. Mice bearing disseminated OPM-2Luc xenografts were treated on day 0 with 211At-CD38 or 211At-Ab− at 15, 30, or 45 µCi; n = 9 to 10 mice per group. Disease progression was monitored via weekly BLI and thrice weekly observations of mouse weight, condition, and mobility. Mice were euthanized when they experienced hind-limb paralysis or met IACUC weight loss or condition requirements. (A) BLI of untreated control and 15 µCi treatment groups at 21 days posttherapy. (B) BLI of all groups at 41 days posttherapy. Red coffins indicate deceased mice. BLI scale identical to panel A.
211At-CD38 therapy in a disseminated OPM-2 Luc model. Mice bearing disseminated OPM-2Luc xenografts were treated on day 0 with 211At-CD38 or 211At-Ab− at 15, 30, or 45 µCi; n = 9 to 10 mice per group. Disease progression was monitored via weekly BLI and thrice weekly observations of mouse weight, condition, and mobility. Mice were euthanized when they experienced hind-limb paralysis or met IACUC weight loss or condition requirements. (A) BLI of untreated control and 15 µCi treatment groups at 21 days posttherapy. (B) BLI of all groups at 41 days posttherapy. Red coffins indicate deceased mice. BLI scale identical to panel A.
In the studies of disseminated disease discussed above, 211At-CD38 doses above 20 µCi appeared sufficient to produce long-term survival for the majority of mice. Thus, with the goal of confirming both therapeutic efficacy and the dose-response relationship, mice in disseminated NCI-H929Luc models received 211At-CD38 or 211At-Ab− at 8, 18, or 24 µCi. Consistent with our other studies, 211At-CD38 treatment strongly benefitted mice across treatment levels, increasing survival relative to untreated controls (P < .0001) and to 211At-Ab− treatments (P < .0006), and these benefits were dose-dependent as higher 211At dosing improved survival (P < .002; Kaplan-Meier proportional hazards tests; Figure 5C). At day 28 posttreatment, imaging showed progressive disease in 100% of untreated mice, 100% of 211At-Ab− 8-µCi mice and 90% of 211At-Ab− 24-µCi mice, compared with 50% of 211At-CD38 8-µCi and 30% of 211At-CD38 24-µCi mice (not shown). At the end of the study (day 150), all untreated, all 211At-Ab− 8-µCi mice, and 70% of 211At-Ab− 24-µCi mice had died due to progressive disease, whereas 30% of 211At-CD38 8-µCi mice and 80% of 211At-CD38 24-µCi mice remained alive (Figure 5C).
Toxicity
The impact of therapy on liver and kidney function (Table 1) and mouse body weight (supplemental Figure 1) was mild, no clinical evidence of toxicity was observed, and all values returned to within normal limits by 32 days following treatment. Hematologic toxicity was limited to platelet counts where mice receiving the highest (45-µCi) dose of 211At-CD38 experienced a 54% reduction relative to untreated control values at day 14, however, no bleeding events were observed and platelet counts recovered to 92% of untreated values by day 32 (Table 1). In the therapy study of disseminated OPM-2Luc, 2 mice in the 211At-CD38 45-µCi group died without detectable MM or toxicity, 1 at 85 days and 1 at 139 days posttreatment (Figure 5B). We have observed similar rates of spontaneous mortality in untreated NRG mice in our colony,30 suggesting that late deaths in study mice are likely not therapy related.
Discussion
MM that is detectable only as MRD has become prevalent due to deeper therapeutic responses and more sensitive methods of MRD detection.31,-33 Fifty percent to 60% of MM patients now achieve a complete response to frontline therapy,31,34,-36 but the majority never achieve MRD-negativity to ≤10−5; for example, 75% of patients treated with bortezomib, lenalidomide, and dexamethasone remained MRD-positive by next-generation sequencing (NGS).31 This finding is concerning as MRD elimination at any point during therapy is a major prognostic indicator for progression-free and overall survival.31,32,37 The persistence of low-level disease likely reflects subclonal heterogeneity that allows small clusters of treatment-resistant MM cells to survive in protective bone marrow microenvironments. Disease eradication requires preventing all mechanisms of tumor cell escape. The potential removal of all clones, thus precluding selective emergence of resistant clones, may distinguish 211At-CD38 therapy from other approaches to MM treatment. We have shown that targeting the CD38 antigen with the very high-energy α-emitter 211At can quickly ablate MM in models designed to recapitulate low-tumor-burden settings or MRD. Single-dose treatments of 211At-CD38 resulted in long-term survival of 50% to 80% of mice in 3 MRD xenograft models (Figures 4,5-6). Efficacy of 211At-CD38 appears to be a function of disease burden and distribution, indicated by the shorter remissions seen in our subcutaneous models. Bulky tumor geometry may limit mAb penetration28 and thus the ability of short-path-length α-emissions to reach every cell, and, indeed, even at our highest treatment dose, 211At-CD38 45-µCi, all mice bearing subcutaneous MM tumors eventually succumbed to disease (Figure 3). Our data support the postulate that isolated cells and small tumor clusters readily accessible to mAbs create optimal conditions for α-emitter cell kill.28,38,,,-42 We therefore hypothesize that 211At-CD38 therapy has the potential to improve patient outcomes by comprehensively removing residual myeloma cell clones.
In an initial test of this hypothesis, we plan to include single-dose 211At-CD38 therapy in an autologous stem cell transplant (ASCT) conditioning regimen for treatment of MM, as part of a phase 1 clinical trial. ASCT is a standard of care for eligible MM patients, yet, for 2 decades, no modification to high-dose chemotherapy conditioning regimens has demonstrably improved ASCT outcomes. We predict that adding 211At-CD38 to conditioning will eliminate MRD in a greater proportion of patients than is currently achieved with high-dose chemotherapy conditioning alone and, if 211At-CD38 is found to be safe and effective in conjunction with stem cell rescue, the approach may ultimately be most useful as a stand-alone therapy in any MRD-positive setting. Furthermore, although MRD-negative status to ≤10−5 predicts significantly extended survival, the majority of these patients eventually relapse,31,32 indicating that residual disease can exist below the limit of detection of even state-of-the-art depth assessments. Our current data for MRD vs subcutaneous xenograft models demonstrate that 211At-CD38 efficacy increases with decreasing disease burden, suggesting that 211At-CD38 might prove extremely effective at eliminating even MM lurking below the limits of MRD detection.
The success and frontline approval by the US Food and Drug Administration (FDA) of the unmodified CD38 mAb daratumumab43,-45 validates CD38 as a desirable target for radionuclide delivery, yet experience with CD38 targeting and the frequency with which daratumumab is being used in clinical settings demands attention when proposing CD38 as a target for 211At therapy. Daratumumab monotherapy for MM ultimately results in relapse. Multiple factors have been reported to contribute to daratumumab resistance, including a protective bone marrow microenvironment,46 loss of cell-adhesion antigen CD56 on MM cells,19 reduction in natural killer cells,47 upregulation of complement-inhibitory proteins on MM,48 and impacts on immune effector cells.49 We posit that the efficacy of 211At-CD38 is not susceptible to these factors, because the cell-killing mechanism of 211At-CD38 is direct, immediate, and independent of host immune function. 211At-CD38 efficacy could, however, be hampered in patients pretreated with daratumumab if CD38 expression is downregulated. Studies of MM patient samples mitigate this concern, reporting that CD38 expression rebounds to original levels after daratumumab is discontinued, and that treatment with certain agents (panobinostat,50 all-trans retinoic acid,48 DNA methyl-transferase inhibitors51 ) can upregulate CD38, even in samples from daratumumab-resistant patients.48 Furthermore, the OKT10 mAb binds a different epitope and is not blocked by daratumumab. Thus, provided CD38 remains expressed, 211At-OKT10 therapy should be effective in patients with circulating daratumumab or selective loss of the daratumumab-binding epitope. To address concerns raised by prior CD38 mAb treatments, the upcoming clinical trial will limit enrollment to patients with CD38+ MM, and will examine the relationship between CD38 antigen density and 211At-CD38 efficacy.
CD38-targeted 211At has notable benefits in the era of precision medicine. A key advantage derives from the spatial and temporal heterogeneity of MM; patients may have different clones at different sites of disease, and these clonal populations evolve over time. Precision medicine often aims to increase therapeutic ratios by targeting patient-specific oncogenetic mutations, yet such therapies may ultimately fail when coexistent disease subclones harbor differing mutations.52,53 In some cases, targeted therapies can even stimulate oncogenetic pathways in nontargeted subclones.53 These problems may be overcome with improved methods of identifying actionable pathways in multiple subclones and treating with targeted drug combinations, though toxicity can limit the extent of such combinations. Thus, therapies agnostic to clonal type while optimizing therapeutic ratios should benefit both the logistics of drug administration and patient outcomes. Virtually all subtypes of MM are exquisitely sensitive to radiation. This includes disease with high-risk cytogenetic features, giving 211At-CD38 the potential to effectively treat MM patients across a wide spectrum of disease presentations.
211At-CD38 has additional properties that favor clinical application. First, 211At lacks toxic daughter radionuclides and has a moderate 7.2-hour half-life. These decay features aid logistical feasibility in the clinic and are better suited to the biological half-life of the targeting Ab than other α-emitters, of which actinium-225 (225Ac) and bismuth-213 (213Bi) are among the best studied. 225Ac produces daughter radionuclides that can reduce ligand stability and increase treatment toxicity54,55 and has a 10-day half-life that can contribute to dose-limiting extended myelosuppression.56 Although 213Bi has a contrastingly short 46-minute half-life, this property can reduce pharmacokinetic and therapeutic efficacy.54,57 Second, in common with other α-emitters, the short path length of 211At minimizes toxicity to healthy tissues and simplifies clinical handling. Third, 211At-CD38 should have a highly favorable dosing schedule. A preclinical study of 211Bi-CD38 therapy for MM reported that a series of 6 treatments extended median survival of xenograft-bearing mice from 60 days (control) to 100 days.58 In contrast, using only a single treatment, we found that optimal dosing of 211At-CD38 at least quadrupled median mouse survival and offered curative potential. Fourth, we previously demonstrated that a pretargeted, β-emitter (90Y)-CD38 therapy was highly effective in preclinical, bulky tumor models of both MM and non-Hodgkin lymphoma,20,21 suggesting that, in low-disease-burden settings, 211At-CD38 might be similarly relevant to a broad patient population. Finally, we anticipate that 211At therapies will be accessible to patients in the United States, as the US Department of Energy has made 211At availability a high priority of the National Isotope Development Center, and is funding the upgrade of existing cyclotrons to provide 211At regionally throughout the country.59 Taken together, these factors suggest that 211At-CD38 is ready for clinical translation and offers the potential for therapeutic efficacy as a single-dose treatment of MM.
We believe our results are robust despite limitations associated with our approach. No mouse model perfectly represents MRD, a state of low-volume disease lacking a uniform definition, and, in our disseminated model, 211At-CD38 did not achieve 100% cure rates (defined as survival to 150 days posttreatment without disease visible to BLI). However, the minimal toxicity and the consistent, dose-dependent therapeutic efficacy of 211At-CD38 show that higher cure rates are clearly achievable. As mouse dosing does not directly translate, we contend that further dose-finding efforts are most usefully implemented in a clinical setting. Paradoxically, the capacity to achieve high cure rates is further suggested by our finding that the highest dose level of nontargeted 211At-Ab− treatments cured some mice in 2 of our MRD experiments. This result is unsurprising given the extreme radiosensitivity of all MM clones, the high LET of 211At, and the existence of low-level nonspecific binding by the 211At-Ab− isotype control (Figure 1B). This result does not support a nontargeted approach: our biodistribution data demonstrate that 211At-CD38 delivers significantly more radiation to tumor than nontargeted 211At-Ab− (Figure 2A), thus reducing potential systemic toxicity at a given tumor dose. Most importantly, in all 5 of our therapy experiments, 211At-CD38 comprehensively outperformed 211At-Ab− (Figures 3,45-6). Another potential limitation derives from the risk of radiation-induced secondary malignancies, particularly myelodysplastic syndrome (MDS). This risk may have limited the clinical use of β-emitter therapies in lymphoma, although large trials using β-emitters have reported no increase in MDS incidence.60,,-63 The very short path length and high LET of 211At may reduce risk relative to β-therapies. This potential safety of α-therapies is supported by the low incidence of MDS among patients receiving FDA-approved radium-223 treatment of prostate cancer, despite a much longer period of exposure (11.4-day half-life).64
To our knowledge this is the first study of an 211At treatment of MM. Our approach is agnostic to high-risk cytogenetic features and offers the potential to eliminate all residual MM cell clones in low-disease-burden settings. We are optimistic that the addition of 211At-CD38 to an ASCT-conditioning regimen in our upcoming clinical trial may improve ASCT outcomes for MM patients.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Nancy Ottman Press and Katie Ahlgren for comprehensive support.
This work was supported by grants from the National Institutes of Health, National Cancer Institute R01CA205248 (D.J.G.), R01CA076287 (D.J.G. and B.G.T.), K01CA188151 (J.J.O.), and K24CA184039 (A.K.G.); and by The Defeat Myeloma Fund, Multiple Myeloma Opportunities in Research and Education, The Brotherton Family, The Multiple Myeloma Cure Seekers Society, and The Quest for Truth Foundation.
Authorship
Contribution: S.O. designed and performed research, analyzed data, and wrote the paper; M.L.C. and J.C.J. designed and performed research and analyzed data; D.K.H., A.K., M.E.N., Y.L., and B.W.M. performed research; T.A.G., J.J.O., S.A.T., B.G.T., A.K.G., B.M.S., and O.W.P. analyzed data; D.S.W. contributed vital reagents; and D.J.G. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: J.J.O. receives research funding from Actinium Pharmaceuticals. J.C.J. is employed by, and has equity ownership in, Juno Therapeutics. T.A.G. consults for Nohla Therapeutics and REGiMMUNE, and holds independent data monitoring committee membership in Kiadis Pharma and data safety monitoring board membership in Astellas and Pharmacyclics. B.G.T. holds patents with, and receives royalties and research funding from, Mustang Bio. A.K.G. reports grants and nonfinancial support from Teva, Bristol-Myers Squibb, Merck, Takeda, TG Therapeutics, and Effector; grants, personal fees, and nonfinancial support from Seattle Genetics, Pfizer, Janssen, Gilead, Spectrum, Amgen, Bayer, I-Mab Bio, and Incyte; and personal fees from Aptevo, BRIM Bio, Seattle Genetics, and Sanofi. B.M.S. reports consulting for Kiadis Pharma, Actinium Pharmaceuticals, Frazier Healthcare Ventures, and Bristol-Meyers Squibb; equity ownership in AnaptysBio, Oncoresponse, Inipharm, EpiThany, and Blaze Bioscience; employment and equity ownership in Mavupharma, Inc; and research funding from Bellicum. D.J.G. holds patents with, and receives royalties and research funding from, Juno Therapeutics, and consults for Cellectar Bio, Seattle Genetics, Celgene, and GlaxoSmithKline. The remaining authors declare no competing financial interests.
Correspondence: Damian J. Green, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA 98109; e-mail: dgreen@fredhutch.org.