

Key Points
High PRMT1 expression maintains MLL-r ALL cell survival and growth by regulating FLT3 methylation at R972/973.
PRMT1 inhibition enhances ablation of MLL-r ALL by tyrosine kinase inhibitor treatment.

Abstract
Relapse remains the main cause of MLL-rearranged (MLL-r) acute lymphoblastic leukemia (ALL) treatment failure resulting from persistence of drug-resistant clones after conventional chemotherapy treatment or targeted therapy. Thus, defining mechanisms underlying MLL-r ALL maintenance is critical for developing effective therapy. PRMT1, which deposits an asymmetric dimethylarginine mark on histone/non-histone proteins, is reportedly overexpressed in various cancers. Here, we demonstrate elevated PRMT1 levels in MLL-r ALL cells and show that inhibition of PRMT1 significantly suppresses leukemic cell growth and survival. Mechanistically, we reveal that PRMT1 methylates Fms-like receptor tyrosine kinase 3 (FLT3) at arginine (R) residues 972 and 973 (R972/973), and its oncogenic function in MLL-r ALL cells is FLT3 methylation dependent. Both biochemistry and computational analysis demonstrate that R972/973 methylation could facilitate recruitment of adaptor proteins to FLT3 in a phospho-tyrosine (Y) residue 969 (Y969) dependent or independent manner. Cells expressing R972/973 methylation-deficient FLT3 exhibited more robust apoptosis and growth inhibition than did Y969 phosphorylation-deficient FLT3-transduced cells. We also show that the capacity of the type I PRMT inhibitor MS023 to inhibit leukemia cell viability parallels baseline FLT3 R972/973 methylation levels. Finally, combining FLT3 tyrosine kinase inhibitor PKC412 with MS023 treatment enhanced elimination of MLL-r ALL cells relative to PKC412 treatment alone in patient-derived mouse xenografts. These results indicate that abolishing FLT3 arginine methylation through PRMT1 inhibition represents a promising strategy to target MLL-r ALL cells.
Introduction
MLL rearrangements are found in 10% of acute leukemias overall.1 A substantial fraction of MLL-rearranged (MLL-r) leukemia cases are acute lymphoblastic leukemias (ALLs) with pro-B phenotype, seen in 70% to 80% of ALL cases in infants.2 Among childhood B-cell ALL (B-ALL) patients, MLL-r ALL patients have a relatively poor prognosis.3 This unsatisfactory treatment outcome is largely a result of the persistence of leukemia clones after treatment, including currently available cell cycle–based or signaling protein–targeted therapies.1,4,5 The gene encoding the Fms-like receptor tyrosine kinase 3 (FLT3) is significantly upregulated in MLL-r ALL6 ; therefore, aberrant FLT3 activation occurs after unchecked binding of the autocrine FLT3 ligand (FL) to highly expressed wild-type (WT) FLT3 protein, initiating downstream survival and growth signals.7 However, administration of FLT3 tyrosine kinase inhibitors (TKIs), such as PKC412 or AC220, which potently inhibit FLT3 autophosphorylation, only partially impairs MLL-r ALL cell survival and has modest efficacy in patients.8,9 Moreover, in addition to tyrosine phosphorylation, FLT3 protein can undergo posttranslational modifications, including glycosylation and ubiquitylation,10,11 and it remains unclear whether other posttranslational modifications can affect FLT3 function.
Protein arginine methyltransferases (PRMTs) 1-9 are responsible for adding 1 or 2 methyl groups to the arginine residue, resulting in epigenetic modification of their substrates. PRMT1 is the predominant type I PRMT, generating about 85% of asymmetric dimethylarginine (ADMA) proteins within mammalian cells.12,13 EGFR is methylated by PRMT1 at R198/200 residues, which facilitates ligand binding and enhances downstream signaling.14 PRMT1 is highly expressed in multiple cancers.15,16 In leukemias, the pathogenic function of PRMT1 was defined only within rare leukemia subtypes, such as acute megakaryoblastic leukemia (AML),17 MLL-GAS7+, or MLL-EEN+ AML.18
Here, we show that PRMT1 is aberrantly overexpressed in MLL-r ALL, and its inhibition reduces MLL-rALL cell survival and growth. We revealed that PRMT1 methylates FLT3 at C-terminal arginine residues and enhances recruitment of signaling adaptor proteins. Ectopic expression of methylation-deficient FLT3 in cells phenocopied effects of PRMT1 inhibition. We also evaluated potential therapeutic effects of the type I PRMT inhibitor MS023 in combination with an FLT3 TKI. Our studies not only demonstrate that PRMT1 plays a pivotal role in maintaining MLL-r ALL cell survival and growth by modulating FLT3 arginine methylation, but also suggest a promising therapeutic opportunity for future leukemia treatment through specifically targeting PRMT1 in the FLT3-activating leukemia.
Materials and methods
Human samples
Detailed patient information is shown in supplemental Table 1, available on the Blood Web site. The patient derived B-ALL cells could recapitulate characteristics of the original disease, including immunophenotype, gene expression profiles, and drug sensitivity.19,,,,-24 Sample acquisition was approved by the City of Hope Institutional Review Board in accordance with the Declaration of Helsinki.
In vivo treatment of MLL-r ALL-engrafted mouse model
For studying inducible short hairpin PRMT1 (shPRMT1), SEM cells stably expressing either doxycycline (DOX)-inducible short hairpin control (shCtrl) or PRMT1 short hairpin RNA (shRNA) (shPRMT1) were transplanted into irradiated immunodeficient NOD-scid IL2Rgnull (NSG) mice (1 × 106 cells per mouse). Each group was administered DOX treatment (10 mg/kg) orally for 3 weeks after engraftment. To assess R972/973 function in leukemogeneis, KOCL45 cells transduced with constructs expressing an FLT3 variant were sorted by using red fluorescent protein and injected into mice (1 × 105 cells per mouse), and mouse survival was monitored daily. To assess MS023 effects in vivo, we transplanted primary MLL-r ALL cells into NSG mice (0.5 × 106 cells per mouse). After engraftment, grouped mice were treated with vehicle, PKC412 (100 mg/kg, intragastrically), MS023 (160 mg/kg, intraperitoneally), or a combination for 4 weeks. After treatment, engrafted human cells were identified. Secondary transplantations of whole bone marrow (BM) cells from treated or control mice were then performed. Animal procedures were performed in accordance with federal and state government guidelines and established institutional guidelines and protocols approved by the Institutional Animal Care and Use Committee at City of Hope.
Cell culture and drugs
Leukemia cells from patients with MLL-r ALL were xenografted into irradiated NSG mice. After passaging, leukemia cells were collected. Human normal hematopoietic CD34+ cells were maintained as previously described.25 Detailed information relevant to in vivo treatment is provided in supplemental Methods.
Statistics
Data obtained from multiple experiments were reported as the mean ± standard error of the mean. Statistical analyses were performed with Student t test or analysis of variance for nonlinear distributions. P < .05 was considered statistically significant.
Results
MLL-r ALL cells show increased PRMT1 expression and sensitivity to PRMT1 knockdown
We first analyzed PRMT1 messenger RNA levels using Gene Expression Omnibus (GEO) data sets (GSE13204; GSE34861) that included large cohorts of ALL patients.26,27 BM mononuclear cells of MLL-r ALL specimens showed significantly increased PRMT1 levels compared with those of normal healthy donors (Figure 1A; supplemental Figure 1A). We further confirmed that PRMT1 messenger RNA was overexpressed in MLL-r ALL samples (n = 8) compared with that seen in normal donors (Figure 1B). Western blot analysis also revealed elevated PRMT1 protein levels in CD19+ blasts from MLL-r ALL samples (n = 8) compared with that of CD34+ (Figure 1C) or CD34−CD19+ cells (supplemental Figure 1B) from normal mobilized peripheral blood stem cell donors. Notably, PRMT1 expression was also increased in blasts from non–MLL-r B-ALL samples (n = 6) (Figure 1C). High PRMT1 protein levels were consistently observed in all the B-ALL cell lines (Figure 1D). Next, to assess PRMT1 function, we knocked down PRMT1 expression in ALL cells using lentiviral vectors expressing PRMT1 shRNAs (shPRMT1-1, shPRMT1-2) (supplemental Figure 1C). Efficient PRMT1 knockdown (PRMT1-KD) promoted apoptosis and growth inhibition of the MLL-r ALL SEM and RS4;11 cell lines (supplemental Figure 1D-E). To confirm shPRMT1 specificity, we overexpressed either PRMT1 complementary DNA lacking the 3′ untranslated region (UTR) or a control vector (MOCK) in SEM cells and then knocked down PRMT1 in both lines using shPRMT1-1 targeting the 3′UTR. We observed increased PRMT1 protein levels and rescued cell viability in PRMT1 complementary DNA and shPRMT1 doubly transduced cells compared with that of cells expressing MOCK plus shPRMT1 (supplemental Figure 1F-G).
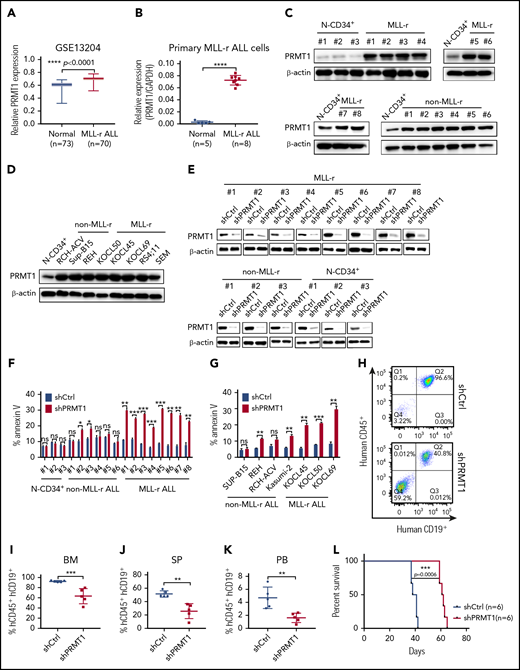
MLL-r ALL cells show increased PRMT1 expression and sensitivity to PRMT1 knockdown. (A) PRMT1 expression (presented on a log scale) in GSE13204 data sets containing specimens from patients with MLL-r ALL vs healthy donors. (B) PRMT1 messenger RNA (mRNA) expression in normal peripheral blood stem cell (PBSC) CD34+ cells (n = 5) and MLL-r ALL primary blasts (n = 8). (C) Western blot showing PRMT1 expression in CD19+ blast cells from primary MLL-r B-ALL (n = 8) and non–MLL-r B-ALL (n = 6) cells compared with normal CD34+ (N-CD34+) cells from PBSC donors (n = 3). (D) Western blot analysis of PRMT1 expression in non–MLL-r (RCH-ACV, Sup-B15, and REH) and MLL-r ALL (KOCL45, KOCL50, KOCL69, RS4;11, and SEM) cell lines compared with that of normal CD34+ cells. (E) Western blot for PRMT1 in primary MLL-r ALL cells, primary non–MLL-r ALL cells, and normal CD34+ cells transduced with a vector expressing shCtrl or shPRMT1. (F) Apoptosis of normal CD34+, primary non–MLL-r, or MLL-r ALL cells, each transduced with shCtrl or shPRMT1, as analyzed by annexin V-Cy5/4′,6-diamidino-2-phenylindole (DAPI) labeling. (G) Apoptosis of shCtrl- or shPRMT1-expressing non–MLL-r ALL or MLL-r ALL cell lines. (H) Shown are representative fluorescence-activated cell sorted profiles for human CD45+/CD19+ cells engrafted in BM from SEM-shCtrl- or SEM-shPRMT1-transplanted mice. Q1: hCD45+CD19−; Q2: hCD45+CD19+; Q3: hCD45−CD19+; Q4: hCD45−CD19−. Percentage of human CD45+/CD19+ (hCD45+/hCD19+) cells engrafted in BM (I), spleen (SP; J), or peripheral blood (PB; K) of recipient NSG mice at 5 weeks after bone marrow transplantation (n = 5 per group). (L) Survival of NSG mice engrafted with SEM cells transduced with either shCtrl or shPRMT1 (n = 6 mice per group). Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ns, not significant.
MLL-r ALL cells show increased PRMT1 expression and sensitivity to PRMT1 knockdown. (A) PRMT1 expression (presented on a log scale) in GSE13204 data sets containing specimens from patients with MLL-r ALL vs healthy donors. (B) PRMT1 messenger RNA (mRNA) expression in normal peripheral blood stem cell (PBSC) CD34+ cells (n = 5) and MLL-r ALL primary blasts (n = 8). (C) Western blot showing PRMT1 expression in CD19+ blast cells from primary MLL-r B-ALL (n = 8) and non–MLL-r B-ALL (n = 6) cells compared with normal CD34+ (N-CD34+) cells from PBSC donors (n = 3). (D) Western blot analysis of PRMT1 expression in non–MLL-r (RCH-ACV, Sup-B15, and REH) and MLL-r ALL (KOCL45, KOCL50, KOCL69, RS4;11, and SEM) cell lines compared with that of normal CD34+ cells. (E) Western blot for PRMT1 in primary MLL-r ALL cells, primary non–MLL-r ALL cells, and normal CD34+ cells transduced with a vector expressing shCtrl or shPRMT1. (F) Apoptosis of normal CD34+, primary non–MLL-r, or MLL-r ALL cells, each transduced with shCtrl or shPRMT1, as analyzed by annexin V-Cy5/4′,6-diamidino-2-phenylindole (DAPI) labeling. (G) Apoptosis of shCtrl- or shPRMT1-expressing non–MLL-r ALL or MLL-r ALL cell lines. (H) Shown are representative fluorescence-activated cell sorted profiles for human CD45+/CD19+ cells engrafted in BM from SEM-shCtrl- or SEM-shPRMT1-transplanted mice. Q1: hCD45+CD19−; Q2: hCD45+CD19+; Q3: hCD45−CD19+; Q4: hCD45−CD19−. Percentage of human CD45+/CD19+ (hCD45+/hCD19+) cells engrafted in BM (I), spleen (SP; J), or peripheral blood (PB; K) of recipient NSG mice at 5 weeks after bone marrow transplantation (n = 5 per group). (L) Survival of NSG mice engrafted with SEM cells transduced with either shCtrl or shPRMT1 (n = 6 mice per group). Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ns, not significant.
More importantly, PRMT1-KD significantly increased apoptosis in primary MLL-r ALL cells but showed no marked effect in normal CD34+ cells; its effect in primary non–MLL-r B-ALL cells was either nonsignificant or moderate (Figure 1E-F; supplemental Figure 1H-I). MLL-r ALL cell lines were also more sensitive to PRMT1-KD compared with non–MLL-r B-ALL cell lines (Figure 1G; supplemental Figure 1J).
Given that PRMT1 levels are comparable in non–MLL-r and MLL-r ALL cells (Figure 1C; supplemental Figure 1K), we reasoned that the robust responses in MLL-r ALL were a result of the overexpressed FLT3 protein. PRMT1 was known to regulate tyrosine kinase receptor-like EGFR.14 Thus, it is possible that PRMT1 promotes MLL-r ALL cell maintenance through regulating FLT3. First, we confirmed that FLT3 expression levels were significantly higher in MLL-r ALL compared with non–MLL-r ALL cases by using GEO data set analysis (supplemental Figure 1L). We also confirmed the overexpression of FLT3 in our own samples and cell lines (supplemental Figure 1M-N). To assess a potential association between FLT3 and PRMT1 function, we transfected murine pro–B-cell line BaF3 cells with MOCK/FLT3 plus shPRMT1/shCtrl. Cells ectopically expressing FLT3 showed a greater number of apoptotic cells upon PRMT1-KD than did shCtrl cells (supplemental Figure 1O-Q), suggesting a functional link between overexpressed FLT3 and PRMT1. Furthermore, we infected cells with inducible shPRMT1 or shCtrl vectors expressing puromycin. After puromycin selection, we then transduced cells with shCtrl or short hairpin FLT3 (shFLT3) and at the same time induced shPRMT1 expression with DOX (supplemental Figure 1R). As a result, FLT3-KD significantly induced apoptosis in KOCL50 cells (supplemental Figure 1S-T). And PRMT1-KD markedly induced apoptosis in FLT3 intact cells but did not further enhance apoptosis in FLT3-KD cells, supporting the notion that PRMT1 effects depend on FLT3 function in MLL-r ALL cells.
Finally, we delivered inducible shPRMT1 to SEM cells and then transplanted red fluorescent protein–sorted cells into NSG mice. When engraftment in peripheral blood (PB) reached 1%, we treated mice with DOX to induce PRMT1-KD or shCtrl (supplemental Figure 1U-Z). After treatment (supplemental Figure 1W), mice transplanted with shPRMT1 cells showed significantly reduced engraftment in BM (Figure 1H-I; supplemental Figure 1X), spleen (Figure 1J; supplemental Figure 1Y), and PB (Figure 1K), as well as reduced splenomegaly compared with mice transplanted with control cells (supplemental Figure 1Z). Importantly, PRMT1-KD significantly extended survival of NSG mice compared with control mice (Figure 1L).
PRMT1 methylates FLT3 at R972/973
We next blocked FLT3 expression in primary MLL-r ALL cells and observed robust apoptosis (n = 6; supplemental Figure 2A-B). By contrast, blocking FLT3 kinase activity by treatment with PKC412, although it effectively blocked tyrosine autophosphorylation, modestly induced apoptosis, consistent with findings of others.8 This result prompted us to ask whether FLT3 promotes leukemia cell growth and survival through kinase-independent mechanisms. Thus, we evaluated whether PRMT1 methylates FLT3. To do this, we first assessed potential PRMT1-FLT3 interaction. We ectopically expressed green fluorescent protein–fused PRMT1 in FLT3-overexpressing 293T cells and then performed co-immunoprecipitation with anti-FLT3 antibody. In parallel, we conducted comparable experiments using green fluorescent protein–fused PRMT3, -4, and -6 but excluded brain-specific PRMT-PRMT8. Co-immunoprecipitation analysis showed that FLT3 binds PRMT1 and PRMT6 but not other PRMTs (Figure 2A). Endogenous PRMT1-FLT3 interaction was validated in SEM cells (Figure 2B). But we did not observe interaction of endogenous PRMT6 with FLT3 in SEM cells (supplemental Figure 2C). We used a proximity ligation assay to assess in situ FLT3-PRMT1 interaction in MLL-r ALL specimens and observed specific red fluorescent foci, indicative of FLT3-PRMT1 protein interaction (Figure 2C). Finally, domain-mapping analysis showed that PRMT1 preferentially interacts with the FLT3 C-terminus (Figure 2D).
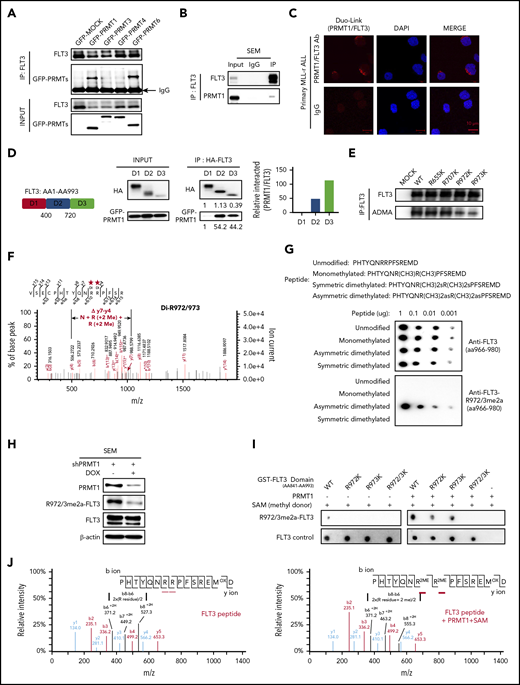
PRMT1 methylates FLT3 at R972 and R973. (A) Green fluorescent protein (GFP)–tagged type I PRMTs were expressed in 293T cells (ectopically expressing FLT3-WT), and then total cell lysates were pulled down with anti-FLT3 antibody, followed by western blotting of FLT3 and GFP. (B) Western blot indicating PRMT1 and FLT3 interaction in SEM cells based on immunoprecipitation (IP) with anti-rabbit immunoglobulin G (IgG) or anti-FLT3 antibodies, followed by western blotting of FLT3 and PRMT1. (C) Representative images from an in situ proximity ligation assay of primary MLL-r ALL cells. Top row (left): red fluorescent spots indicate PRMT1/FLT3 protein interaction, (middle) DAPI-stained nuclei are blue, and (right) merged image. Scale bar, 10 μm. Bottom row: IgG controls. (D) Left: HA-tagged FLT3 fragments and GFP-tagged PRMT1 were co-expressed in 293T cells, and indicated FLT3 protein fragments were pulled down with anti-HA antibody, followed by (middle) western blotting for HA and GFP. Right: quantitative intensity analysis of interaction of FLT3 domains with PRMT1. (E) Western blotting for pan-ADMA levels in MOCK-, FLT3-WT-, FLT3-R655K-, FLT3-R707K-, FLT3-R972K-, or FLT3-R973K-transduced 293T cells. Respective lysates were pulled down with anti-FLT3 antibody. (F) Endogenous FLT3 protein was pulled down from SEM cells followed by MS analysis. R972/973 was identified as dimethylated (Di). The mass-to-charge ratio (m/z) of "y7-y4" indicated the total molecular weight of 3 amino acid residues including N residue and 2 R residues (2Me). (G) Amino acid sequence of peptides corresponding to the FLT3 966-980 region, in which R972/973 was modified as indicated. Different doses of peptides were detected by anti-FLT3 966-980 (control) or anti-FLT3 R972/973me2a antibodies. (H) Western blot of FLT3, R972/973me2a, or PRMT1 in SEM cells transduced with inducible shPRMT1. (I) In vitro methylation assay of indicated GST-tagged FLT3 peptides (aa841-aa993) with or without PRMT1 enzyme plus S-adenosyl methionine (SAM). (J) MS analysis of FLT3 peptide (left) without or (right) with PRMT1 enzyme plus SAM. HA, human influenza hemagglutinin; m/z, represents mass divided by charge number and the horizontal axis in a mass spectrum is expressed in units of m/z.
PRMT1 methylates FLT3 at R972 and R973. (A) Green fluorescent protein (GFP)–tagged type I PRMTs were expressed in 293T cells (ectopically expressing FLT3-WT), and then total cell lysates were pulled down with anti-FLT3 antibody, followed by western blotting of FLT3 and GFP. (B) Western blot indicating PRMT1 and FLT3 interaction in SEM cells based on immunoprecipitation (IP) with anti-rabbit immunoglobulin G (IgG) or anti-FLT3 antibodies, followed by western blotting of FLT3 and PRMT1. (C) Representative images from an in situ proximity ligation assay of primary MLL-r ALL cells. Top row (left): red fluorescent spots indicate PRMT1/FLT3 protein interaction, (middle) DAPI-stained nuclei are blue, and (right) merged image. Scale bar, 10 μm. Bottom row: IgG controls. (D) Left: HA-tagged FLT3 fragments and GFP-tagged PRMT1 were co-expressed in 293T cells, and indicated FLT3 protein fragments were pulled down with anti-HA antibody, followed by (middle) western blotting for HA and GFP. Right: quantitative intensity analysis of interaction of FLT3 domains with PRMT1. (E) Western blotting for pan-ADMA levels in MOCK-, FLT3-WT-, FLT3-R655K-, FLT3-R707K-, FLT3-R972K-, or FLT3-R973K-transduced 293T cells. Respective lysates were pulled down with anti-FLT3 antibody. (F) Endogenous FLT3 protein was pulled down from SEM cells followed by MS analysis. R972/973 was identified as dimethylated (Di). The mass-to-charge ratio (m/z) of "y7-y4" indicated the total molecular weight of 3 amino acid residues including N residue and 2 R residues (2Me). (G) Amino acid sequence of peptides corresponding to the FLT3 966-980 region, in which R972/973 was modified as indicated. Different doses of peptides were detected by anti-FLT3 966-980 (control) or anti-FLT3 R972/973me2a antibodies. (H) Western blot of FLT3, R972/973me2a, or PRMT1 in SEM cells transduced with inducible shPRMT1. (I) In vitro methylation assay of indicated GST-tagged FLT3 peptides (aa841-aa993) with or without PRMT1 enzyme plus S-adenosyl methionine (SAM). (J) MS analysis of FLT3 peptide (left) without or (right) with PRMT1 enzyme plus SAM. HA, human influenza hemagglutinin; m/z, represents mass divided by charge number and the horizontal axis in a mass spectrum is expressed in units of m/z.
Next, endogenous FLT3 was pulled down from SEM cells and subjected to liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis. We identified 6 dimethylated arginine (R) residues (R311, R322, R655, R707, R972, R973). Because PRMT1 specifically interacts with the FLT3 C-terminus, we next focused on C-terminal R residues (R655, R707, R972, R973) and validated whether they were ADMA. Mutating either R972 or R973 residues (R to lysine [K]; R972K, R973K), but not R655 and R707, significantly reduced FLT3 ADMA levels in 293T cells ectopically expressing FLT3 variants (Figure 2E), suggesting that R972 and R973 are the predominant ADMA residues (Figure 2F). Notably, both residues are highly conserved across species (supplemental Figure 2D).
To measure endogenous FLT3 ADMA levels, we generated an anti-FLT3 R972/973 asymmetric dimethylation (FLT3-R972/973me2a) antibody by immunizing rabbits with a synthesized asymmetric dimethylated FLT3 peptide (amino acids 966-980). Dot blot assays confirmed that the antibody specifically recognizes FLT3-R972/973me2a peptide but not unmodified, monomethylated, or symmetrically dimethylated forms of the peptide (Figure 2G). In FLT3-overexpressing 293T cells, mutating both R972 and R973 completely abolished R972/973me2a signals, confirming the specificity of the antibody (supplemental Figure 2E).
Next, we found that knockdown of PRMT1 but not PRMT6 robustly blocked FLT3-R972/973 methylation (Figure 2H; supplemental Figure 2F), indicating that R972/973 methylation of FLT3 is specifically regulated by PRMT1. We also validated that PRMT6 does not methylate FLT3 on other residues, because PRMT6-KD did not alter FLT3 total ADMA levels (supplemental Figure 2G). We then validated that PRMT1 protein directly catalyzes methylation of FLT3-R972/973 in vitro, with S-adenosyl methionine serving as a methyl donor. By using FLT3-R972/973me2a antibody, we found that bacterially purified recombinant glutathione S-transferase (GST)–FLT3 WT protein (aa841-993) but not R972/973K double-mutant protein was methylated at R972/973 after addition of PRMT1 and S-adenosyl methionine (Figure 2I). We further confirmed R972/973 dimethylation by PRMT1 in vitro through LC-MS analysis (Figure 2J; supplemental Table 2) using synthetic FLT3 peptide (aa966-980). Overall, these results reveal that R972 and R973 are the predominant ADMA residues on WT FLT3 protein, and the modifications are catalyzed by PRMT1.
PRMT1-mediated R972/973 methylation regulates leukemia cell maintenance
To assess the function of R972/973 methylation, we first generated an FLT3 R methylation-deficient construct (R972/973K). Interleukin-3 (IL-3)–dependent BaF3 cells were used for functional analysis, because endogenous FLT3 is undetectable in this line. Because FLT3 activity is potentiated by addition of ligand,7 we evaluated BaF3 cells expressing FLT3-WT, FLT3-R972/973K, or the MOCK in the presence of FL. Similar FLT3 levels in the cell lines were confirmed (supplemental Figure 3A). We found that enforced expression of FLT3-WT, but not FLT3-R972/973K, promoted BaF3 cell proliferation (Figure 3A). Moreover, when deprived of IL-3, both MOCK and FLT3-R972/973K cells showed more robust apoptosis than did FLT3-WT cells (Figure 3B), suggesting that R972/973 methylation is required for FLT3 function. Then we compared intracellular tyrosine phosphorylation of FL-stimulated FLT3-WT, FLT3-R972K, FLT3-R973K, or FLT3-R972/973K cells using site-specific antibodies against phospho (p)-tyrosine (Y) 589/591, Y842, or Y969 (Figure 3C).
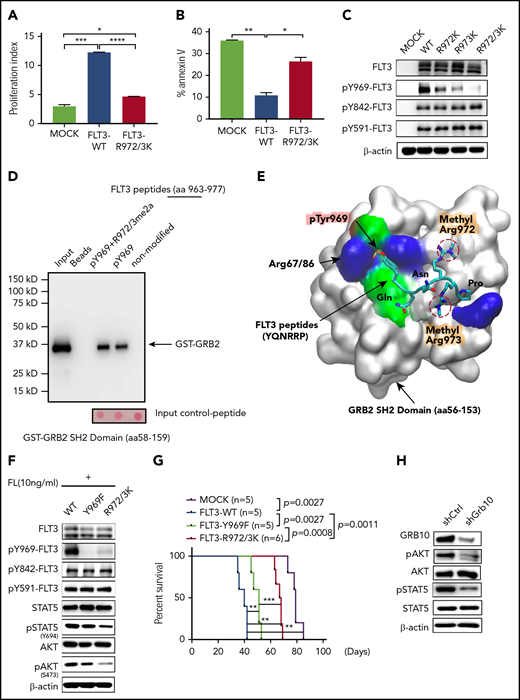
PRMT1-mediated R972/973 methylation regulates leukemia cell maintenance. Analysis of cell proliferation (A) and apoptosis (B) in MOCK-, FLT3-WT-, or FLT3-R972/973K-transduced BaF3 cells with FL stimulation. (C) Western blot analysis of FLT3 and indicated pFLT3 in BaF3-expressing indicated construct upon FL stimulation. (D) After incubation with a purified protein of GST-GRB2 SH2 domain, biotinylated peptides corresponding to FLT3 aa963-977 (non-modified) or containing pY969 or pY969-R972/973me2a were pulled down by streptavidin beads. SH2 domain/FLT3 peptide association, as revealed by immunoblotting with anti-GST antibodies (n = 3). Molecular mass markers (kD) are shown at left. (E) The binding pose of the dimethylated peptide from molecular dynamics simulation is shown on the SH2 domain protein surface. Peptide is shown in licorice mode (aa969-aa974). Hydrogen atoms are omitted for clarity. Protein surface is shown in white with residues within 5 Å of peptide shown in residue type color code: negative surface charge, red; positive surface charge, blue; nonpolar surface, cyan. (F) Western blotting of indicated total and phospho-proteins in FL-stimulated BaF3 cells expressing FLT3-WT, FLT3-Y969F, or FLT3-R972/973K. (G) Survival of NSG mice engrafted with indicated KOCL45 cells. (H) Western blotting of indicated total and phospho-proteins in SEM cells transduced with either shCtrl or shGBR10. Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001.
PRMT1-mediated R972/973 methylation regulates leukemia cell maintenance. Analysis of cell proliferation (A) and apoptosis (B) in MOCK-, FLT3-WT-, or FLT3-R972/973K-transduced BaF3 cells with FL stimulation. (C) Western blot analysis of FLT3 and indicated pFLT3 in BaF3-expressing indicated construct upon FL stimulation. (D) After incubation with a purified protein of GST-GRB2 SH2 domain, biotinylated peptides corresponding to FLT3 aa963-977 (non-modified) or containing pY969 or pY969-R972/973me2a were pulled down by streptavidin beads. SH2 domain/FLT3 peptide association, as revealed by immunoblotting with anti-GST antibodies (n = 3). Molecular mass markers (kD) are shown at left. (E) The binding pose of the dimethylated peptide from molecular dynamics simulation is shown on the SH2 domain protein surface. Peptide is shown in licorice mode (aa969-aa974). Hydrogen atoms are omitted for clarity. Protein surface is shown in white with residues within 5 Å of peptide shown in residue type color code: negative surface charge, red; positive surface charge, blue; nonpolar surface, cyan. (F) Western blotting of indicated total and phospho-proteins in FL-stimulated BaF3 cells expressing FLT3-WT, FLT3-Y969F, or FLT3-R972/973K. (G) Survival of NSG mice engrafted with indicated KOCL45 cells. (H) Western blotting of indicated total and phospho-proteins in SEM cells transduced with either shCtrl or shGBR10. Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Interestingly, in single-mutant R972K or R973K cells, pY969 levels were reduced specifically, and there was no effect at other pY residues. The expression of the double-mutant R972/973K further reduced Y969 phosphorylation, as did PRMT1-KD (supplemental Figure 3B). To further assess whether FLT3 methylation is required for PRMT1-KD effects in an FLT3-activating context, we used RCH-ACV cells, in which endogenous FLT3 levels are very low. Notably, compared with MOCK cells, enforced overexpression of FLT3 promoted receptor autophosphorylation and activated downstream signaling (supplemental Figure 3C). Of note, RCH-ACV cells ectopically expressing FLT3 were more sensitive to PKC412 inhibition (supplemental Figure 3C-D), indicating that FLT3 overexpressed cells were more dependent on FLT3 signaling than MOCK cells. We further transduced RCH-ACV cells with MOCK, FLT3-WT or the corresponding FLT3-R972/973K mutant plus shPRMT1 or shCtrl (supplemental Figure 3E). In contrast to WT FLT3 cells, which showed significantly increased apoptosis, R972/973K-expressing cells did not undergo apoptosis after PRMT1 knockdown (supplemental Figure 3F).
FLT3 pY969 reportedly serves as a docking site for SH2 domains of cytosolic signaling molecules such as GADS and GRB2.28,29 Thus we performed peptide pull-down assays to assess whether R972/973me2a enhanced the association between SH2 domain and FLT3 pY969. We found that purified GST-GRB2 SH2 domain bound pY969 peptide, as expected,29 and greater amounts of GST-GRB2 protein were pulled down by pY969-R972/973me2a (Y phosphorylation plus R methylation) double-modified peptide (Figure 3D). We then conducted computational molecular dynamics stimulations of a peptide pYQNRRP derived from the FLT3 C-terminal (aa 969-974) in complex with GST-GRB2 SH2 domain. To do so, we carried out 100-nanosecond explicit-solvent molecular dynamic simulations of the peptide with or without asymmetric dimethylation of R972/973. The binding energy for dimethylated peptide (–92.30 kcal/mol) is lower than that for nonmethylated peptide (–78.21 kcal/mol). Lower energy for the dimethylated peptide indicates stronger binding of the SH2 domain. Figure 3E shows a snapshot of the binding pose of R972/973me2a on the GRB2 SH2 domain surface, indicating that methylated arginines fit into the SH2 hydrophobic cleft, and addition of hydrophobic methyl groups compensates for unfavorable electrostatic interactions between positively charged arginines and the hydrophobic binding surface. Overall, this analysis demonstrates that R972/973 methylation enhances recruitment of the protein SH2 domain to pY969 of FLT3.
Next, we generated an FLT3-Y969 phosphorylation-deficient construct (Y969F) and compared activation of downstream signals in BaF3 cells expressing Y969F vs R972/973K (Figure 3F). Western blotting analysis indicated that R972/973K cells showed significantly reduced pSTAT5/AKT levels relative to that of Y969F. Moreover, Y969F mutation did not alter R972/973 methylation (supplemental Figure 3G). To validate R972/973 function in leukemogeneis of MLL-r ALL cells, we transduced KOCL45 cells with MOCK, FLT3-WT, FLT3-R972/973K, or FLT3-Y969F constructs and transplanted each into NSG recipients to assess ALL progression. For this analysis, equal FLT3 expression levels were confirmed (supplemental Figure 3H). Cells expressing FLT3-WT exhibited a potent oncogenic role in promoting leukemogenesis as evidenced by shorter overall survival relative to MOCK. However, compared with FLT3-WT, expression of the FLT3-Y969F construct modestly extended leukemic mouse survival, whereas R972/973K expression remarkably prolonged survival (Figure 3G).
Next, to determine whether allosteric changes resulting from FLT3 R972/973 methylation alter protein-protein interactions, we analyzed interactions of adaptor proteins that reportedly bind to FLT3 protein at docking sites other than Y969, including SHC,30 SHIP,31 GAB1,30 GAB2, and Grb10.32 To do this, we immunoprecipitated FLT3 followed by immunoblot with the respective antibody in 293T cells ectopically expressing FLT3-WT, FLT3-R972/973K, or FLT3-Y969F. Among all adaptors tested, only Grb10-FLT3 interaction was disrupted by loss of R972/973 methylation, but not by inhibition of Y969 phosphorylation (supplemental Figure 3I). Grb10-KD also reduced levels of both pSTAT5 and pAKT in SEM cells (Figure 3H), suggesting that Grb10 mediates the effects of R972/973 methylation. We also assessed the FLT3 interactome in 293T cells ectopically expressing FLT3-WT or FLT3-R972/973K by MS analysis of FLT3 immunoprecipitates (supplemental Table 3). Among the high-abundance proteins that are unique to FLT3-WT cells but not FLT3-R972/973K cells, we identified SHP2 and BTK, 2 known FLT3-interacting proteins.33,34 We further confirmed that SHP2 or BTK binding to FLT3 is specifically reduced only in FLT3-R972/973K cells, but not in FLT3-Y969F cells (supplemental Figure 3J).
Taken together, these results suggest that although there is crosstalk between PRMT1-mediated FLT3-R972/973 methylation and Y969 phosphorylation, the effects of methylation on leukemia maintenance do not depend solely on Y969 phosphorylation and that other proteins recruited by allosteric changes after FLT3-R972/973 methylation may mediate the biological effects of methylation.
Pharmacologic PRMT1 inhibition blocks MLL-r ALL cell survival and growth
The type I PRMT inhibitor MS023 significantly blocks the enzymatic activity of PRMT1,35 so we tested the inhibitory effects of MS023 on cell variability in MLL-r ALL primary samples and cell lines (supplemental Table 4). MS023 treatment remarkably reduced FLT3 R972/973 methylation levels in ALL cells (Figure 4A) as well as pSTAT5 and pAKT levels (supplemental Figure 4A). And treatment with TCE-5003, another PRMT1 selective inhibitor,36 also remarkably reduced R972/973 methylation levels, thus increasing MLL-r ALL cell apoptosis (supplemental Figure 4B). We also observed that the extent of MS023-induced growth inhibition paralleled baseline FLT3 R-Me levels in MLL-r ALL cell lines (Figure 4B) and samples (Figure 4C). In addition, primary MLL-r ALL cells transduced with shPRMT1 (Figure 4D), but not shPRMT6 (supplemental Figure 4C), were resistant to MS023-induced apoptosis, confirming that MS023 effects are predominantly PRMT1 dependent in MLL-r ALL cells. We also exposed RCH-ACV cells transduced with FLT3-WT or FLT3-R972/973K to MS023. Notably, MS023-treated R972/973K cells were resistant to apoptosis, unlike FLT3 WT cells (supplemental Figure 4D).
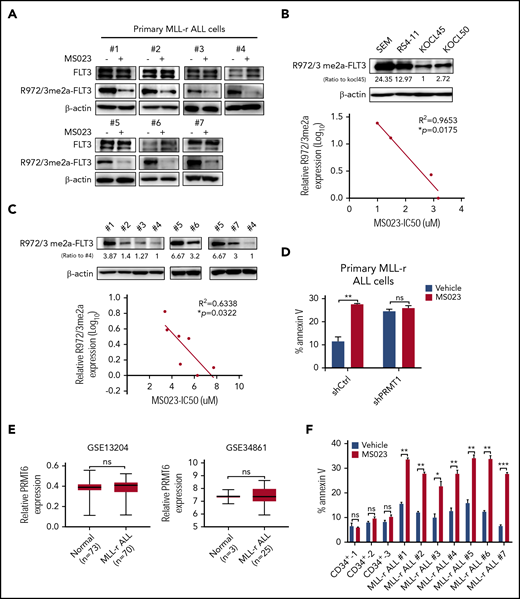
Pharmacologic inhibition of PRMT1 blocks MLL-r ALL cell survival and growth. (A) Western blot for FLT3 and R972/973me2a in MLL-r ALL samples (n = 7) treated with vehicle or MS023 (5 μM). (B-C) Western blot of endogenous R972/3me2a expression in MLL-r ALL cell lines (n = 4) (B, upper panel) and MLL-r ALL samples (n = 7) (C, upper panel). Bottom panels show corresponding correlation analysis of R972/973me2a expression and MS023 IC50 (the half maximal inhibitory concentration). (D) Apoptosis of shCtrl- or shPRMT1-transduced primary MLL-r ALL cells treated with vehicle or MS023. (E) PRMT6 gene expression was analyzed between non–MLL-r and MLL-r ALL specimens in indicated microarray data sets (GSE13204 and GSE34861). (F) Apoptosis of indicated subsets of normal PBSCs (n = 3) in MLL-r ALL samples (n = 7) treated with vehicle or MS023. Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001.
Pharmacologic inhibition of PRMT1 blocks MLL-r ALL cell survival and growth. (A) Western blot for FLT3 and R972/973me2a in MLL-r ALL samples (n = 7) treated with vehicle or MS023 (5 μM). (B-C) Western blot of endogenous R972/3me2a expression in MLL-r ALL cell lines (n = 4) (B, upper panel) and MLL-r ALL samples (n = 7) (C, upper panel). Bottom panels show corresponding correlation analysis of R972/973me2a expression and MS023 IC50 (the half maximal inhibitory concentration). (D) Apoptosis of shCtrl- or shPRMT1-transduced primary MLL-r ALL cells treated with vehicle or MS023. (E) PRMT6 gene expression was analyzed between non–MLL-r and MLL-r ALL specimens in indicated microarray data sets (GSE13204 and GSE34861). (F) Apoptosis of indicated subsets of normal PBSCs (n = 3) in MLL-r ALL samples (n = 7) treated with vehicle or MS023. Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001.
We observed that MS023 treatment could inhibit PRMT6 substrate H3R2me2a in SEM cells after longer exposure (48 hours) (supplemental Figure 4E). In addition, compared with PRMT1 levels, PRMT6 levels are lower in MLL-r ALL cells (supplemental Figure 4F) and are comparable in normal and MLL-r ALL cells (Figure 4E). PRMT6-KD did not alter survival of MLL-r ALL cells (supplemental Figure 4C,G). Taken together, these results indicate that, despite inhibition of PRMT6 activity, the cytotoxicity of MS023 is mediated through PRMT1 targeting.
Importantly, although MS023 treatment significantly inhibited survival in primary MLL-r ALL cells, it barely affected survival of normal hematopoietic CD34+ cells (Figure 4F). We transplanted cord blood CD34+ cells into NSG mice as described previously.25 Eight weeks posttransplantation, when human CD45+ cells showed robust engraftment in PB, we divided mice into 2 groups and treated them with vehicle or MS023 for 4 weeks. MS023 treatment did not alter the long-term engraftment capacity of normal cells in NSG BM (supplemental Figure 4H).
PRMT1 inhibition enhances elimination of MLL-r ALL cells when combined with a TKI
Next, we treated (ex vivo) primary ALL cells with vehicle, MS023, PKC412, or the combination of MS023 and PKC412, and we observed that PKC412 did not alter FLT3-R972/973 methylation levels (Figure 5A). We also found that treatment with the combination resulted in further reduction in pSTAT5/AKT signals as well as in cell survival and growth relative to treatment with MS023 or PKC412 alone (Figure 5A-C). Notably, combined treatment had strong synergistic effects in inhibiting MLL-r ALL cell viability (supplemental Figure 5A; supplemental Table 5). PRMT1-KD combined with PKC412 treatment also further reduced SEM cell survival compared with PKC412 alone (Figure 5D).
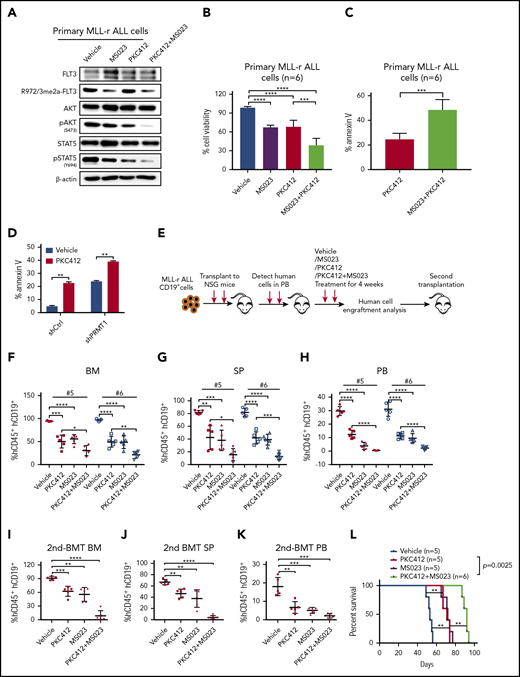
PRMT1 inhibition enhances elimination of MLL-r ALL cells by TKI treatment. (A) Western blotting of indicated total and phospho-proteins in primary MLL-r ALL cells treated with vehicle, MS023, PKC412, or an MS023/PKC412 combination. (B) Cell viability of MLL-r ALL samples (n = 6) treated with vehicle, MS023, PKC412, or MS023/PKC412. (C) Apoptosis of MLL-r ALL samples (n = 6) treated with PKC412 or MS023/PKC412. (D) Apoptosis of shCtrl- or shPRMT1-transduced SEM cells treated with vehicle or PKC412. (E) Experimental design. To assess MS023 effects in vivo, we transplanted primary MLL-r ALL cells into NSG mice. After the engraftment of human cells reached 1%, mice were treated with vehicle, PKC412, MS023, or a combination for 4 weeks. Posttreatment, engrafted human cells were analyzed. Secondary transplantation was then performed. Percentage of human CD45+/CD19+ cells engrafted in BM (F), SP (G), and PB (H) of recipient NSG mice transplanted with #5 or #6 primary MLL-r ALL cells. Mice with patient-derived xenografts were treated as indicated in each panel for 4 weeks. Percentage of human CD45+/CD19+ cells engrafted in BM (I), SP (J), and PB (K) of secondary recipient NSG mice receiving primary donor cells (#5) from mice treated as indicated in each panel. (L) Survival of NSG mice engrafted with primary MLL-r ALL cells treated with vehicle, PKC412, MS023, or PKC412/MS023. Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001.
PRMT1 inhibition enhances elimination of MLL-r ALL cells by TKI treatment. (A) Western blotting of indicated total and phospho-proteins in primary MLL-r ALL cells treated with vehicle, MS023, PKC412, or an MS023/PKC412 combination. (B) Cell viability of MLL-r ALL samples (n = 6) treated with vehicle, MS023, PKC412, or MS023/PKC412. (C) Apoptosis of MLL-r ALL samples (n = 6) treated with PKC412 or MS023/PKC412. (D) Apoptosis of shCtrl- or shPRMT1-transduced SEM cells treated with vehicle or PKC412. (E) Experimental design. To assess MS023 effects in vivo, we transplanted primary MLL-r ALL cells into NSG mice. After the engraftment of human cells reached 1%, mice were treated with vehicle, PKC412, MS023, or a combination for 4 weeks. Posttreatment, engrafted human cells were analyzed. Secondary transplantation was then performed. Percentage of human CD45+/CD19+ cells engrafted in BM (F), SP (G), and PB (H) of recipient NSG mice transplanted with #5 or #6 primary MLL-r ALL cells. Mice with patient-derived xenografts were treated as indicated in each panel for 4 weeks. Percentage of human CD45+/CD19+ cells engrafted in BM (I), SP (J), and PB (K) of secondary recipient NSG mice receiving primary donor cells (#5) from mice treated as indicated in each panel. (L) Survival of NSG mice engrafted with primary MLL-r ALL cells treated with vehicle, PKC412, MS023, or PKC412/MS023. Error bars represent standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001.
We then used MLL-r ALL patient-derived xenografts from 2 different patient specimens to assess BM engraftment of leukemic mice treated with a combination of MS023 and PKC412. Primary MLL-r ALL cells were transplanted into mice, and when human cell engraftment in PB reached 1%, we divided mice into 4 groups and treated them with vehicle, MS023, PKC412, or the combination (Figure 5E). The frequency of hCD45+/CD19+ ALL cells in BM, spleen, and PB significantly decreased in mice treated with single drugs, with a further reduction in the group treated with the combination (Figure 5F-H; supplemental Figure 5B-D). We also assessed the effects of combination treatment on leukemia-initiating activity by transplanting whole BM cells from vehicle-, MS023-, PKC412-, or combination-treated mice to secondary recipients. Although we observed a significant ALL burden in vehicle-, MS023-, or PKC412-treated transplants, minimal residual ALL cells were seen in combination-treated secondary transplants (Figure 5I-K). It is important to note that treatment with the combination extended the survival of leukemic mice compared with treatment with a single agent (Figure 5L). These results suggest that in vivo administration of MS023 in combination with a TKI blocks MLL-r ALL propagation by inhibiting maintenance of functional MLL-r ALL-initiating cells.
Discussion
MLL-r ALL is seen in 70% of infants with ALL.37 Historically, 5-year event-free survival for MLL-r ALL has ranged from 20% to 40% compared with 60% or higher for patients with WT MLL ALL.37,38 Overall survival for adult MLL-r ALL is also lower than that for adult patients with B-ALL.39,40 The poor outcome is a result of relapse in >50% of patients.41 Thus, there is an unmet need for more pertinent treatments to eradicate MLL-r ALL. Herein, we demonstrate that PRMT1-mediated FLT3 arginine methylation promotes MLL-r ALL cell survival and growth. Inhibiting FLT3 methylation blocked MLL-r ALL leukemogenesis. More importantly, MS023 treatment enhanced ablation of MLL-r ALL cells by treatment with PKC412. These studies suggest that PRMT1 inhibition could be developed as a promising therapeutic strategy to antagonize MLL-r ALL.
PRMT1 is important for normal development, because Prmt1−/− knockout mouse embryos die at day 7.5 after implantation.42 By methylating both histone and non-histone proteins (including H4R3, EGFR, and RUNX1), PRMT1 functions in transcription activation and signal transduction.43 Recent studies also show that PRMT1 regulates B-cell lymphoid hematopoietic differentiation.44 Our studies identify FLT3 as a novel PRMT1 methylation substrate and show that MLL-r ALL cell maintenance is regulated by PRMT1. Unlike robust apoptosis induction seen in MLL-r ALL cells, non–MLL-r B-ALL cells show varying degrees of sensitivity to PRMT1-KD. Thus, in addition to the fact that aberrantly high FLT3 levels are positively correlated with robust PRMT1-KD phenotypes, other factors may also regulate the response of B-ALL cells to PRMT1-KD, especially in non–MLL-r B-ALL cells that express relatively low FLT3 levels.
Blocking FLT3 kinase activity as an MLL-r ALL therapeutic strategy has been extensively explored.8 However, administration of FLT3 TKIs that are effective in inhibiting kinase activity (supplemental Figure 2A) only partially impairs MLL-r ALL cell survival (supplemental Table 2B). The recent TACL trial reported that MLL-r ALL patients consistently show modest response to combination treatment with FLT3-TKI AC220 and intensive chemotherapy.45 Herein, we show that compared with TKI treatment, FLT3-KD induces robust apoptosis of MLL-r ALL blasts, suggesting that FLT3 protein might regulate MLL-r ALL cell survival in a kinase-independent manner. FLT3 protein can be modified by phosphorylation or glycosylation, which alter its activity.46 In this study, we found that FLT3-R972/973 methylation positively regulates Y969 phosphorylation (Figure 3C). Like outcomes seen after arginine methylation of histone tails, which promotes docking of other functional factors,47 our results support the idea that methylation of R972/973 enhances the binding of adaptors to FLT3 protein at different docking sites (Figure 3D,H; supplemental Figure 3J). We also observed that phenotypic and signaling effects of R972/973 methylation deficiency are more significant than those after Y969 phosphorylation deficiency alone, strongly suggesting that R972/973 methylation has a function independent of Y969 phosphorylation (Figure 3F). Our study indicates that arginine methylation of the FLT3 C-terminus serves as a novel posttranslational regulation that may explain the lack of efficacy of TKIs in fully eradicating MLL-r ALL. Nevertheless, our work also suggests that combining PRMT1 inhibition with TKI treatment could be appropriate for MLL-r ALL patients whose specimens show FLT3 overexpression. In support of that idea, we sorted cells from 1 FLT3-overexpressing MLL-r ALL sample into 2 subpopulations based on FLT3 expression (CD135high and CD135low). Importantly, FLT3high cells showed more robust apoptosis after treatment with the combination of MS023 and PKC412 than did comparably treated FLT3low cells (supplemental Figure 5E-G). Together with our finding in FLT3-ITD+ AML,48 these results indicate that the effects of PRMT1 on FLT3 signaling could be more pronounced in FLT3high-expressing or FLT3-activating leukemia, even without MLL-r. But the overall dependence of leukemia cells on PRMT1 could also be attributed to factors other than FLT3, which may warrant further investigation.
A common limitation to current ALL chemotherapy is specificity, because most reagents in use are toxic to normal hematopoietic stem and progenitor cells. Here, we found that administering MS023 in vivo had minimal effects on long-term engraftment of human hematopoietic stem and progenitor cells. We also observed no significant toxicity in mice after treatment with MS023 (data not shown). This evidence of safety together with MS023 selectivity toward MLL-r ALL cells provides a strong rationale for further clinical testing of MS023 with a high therapeutic index toward leukemic cells. Although our studies show that the effects of MS023 on MLL-r ALL cells are mainly dependent on PRMT1 (Figure 4), this compound is designated a type I PRMT inhibitor. Thus, further detailed MS023 preclinical evaluation is warranted, particularly regarding its potential toxicity to other organs in which other type I PRMTs are highly expressed.
In conclusion, we demonstrate here how PRMT1 modulates FLT3 activity and contributes to enhanced MLL-r ALL cell maintenance. FLT3 methylation, as a novel mechanism, may facilitate FLT3 downstream survival signaling. Importantly, we identified robust synergy of TKI and MS023 treatment in primary leukemia cells. Our work highlights inhibiting PRMT1-mediated FLT3 methylation as a potential therapeutic approach and encourages further assessment of MS023 or other potent PRMT1 inhibitors for MLL-r ALL or FLT3-activating leukemia treatment.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the City of Hope Comprehensive Cancer Center, as well as patients, donors, and their physicians for providing primary specimens for this study and the University of California at Los Angeles (UCLA) Center for Aids Research Virology core and the UCLA AIDS Institute for cord blood samples.
This work was supported in part by National Institutes of Health, National Cancer Institute grants (5P30 AI028697 and P30CA33572), a National Heart, Lung, and Blood Institute grant (R01 HL141336), an American Cancer Society Research Scholar Grant (RSG-19-036-01-LIB), a St. Baldrick’s Foundation Research Grant (Award #632462), and the Margaret E. Early Medical Research Trust Award.
The work reported here includes work performed in the Analytical Cytometry Core assisted by Lucy Brown, Translational Biomarker Discovery Core, with technical support from Yunan Miao, Pathology (Liquid Tumor) and Animal Resource Center at City of Hope.
The content is solely the responsibility of the authors and does not necessarily represent official views of the National Institutes of Health.
Authorship
Contribution: Y.Z. designed experiments, interpreted results, wrote the manuscript, and performed experiments, including mass spectrometry, GEO database, peptide pull-down, and flow cytometry analysis; Y.Z. and X.H. preformed animal experiments and proximity ligation assay analysis; X.H. performed dot-blot analysis; H.D. prepared samples for in vitro methylation assay; L.Z. searched databases for MS; Y.-C.L., H.Z., W.J., and Y.L. performed structure analysis of SH2 domain binding; Y.S. and J.J. generated MS023 compounds; M.L. performed the statistical analysis; H.W. performed the STR assay; Z.W. and Y.Y. provided technical support for PRMT1 methylation assay; J.C., M.M., C.-W.C., and M.Y.K. provided ALL samples; X.C., L.X.N., and J.S. interpreted results and reviewed the manuscript; J.C., W.S., N.C., and G.M. reviewed the manuscript; and L.L. and Y.Z. prepared the manuscript with input from other authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ling Li, Department of Hematological Malignancies Translational Science, Gehr Family Center for Leukemia Research, Hematologic Malignancies and Stem Cell Transplantation Institute, Beckman Research Institute, City of Hope Medical Center, 1500 E Duarte Rd, Duarte, CA 91010; e-mail: lingli@coh.org.
