Abstract
Sickle cell disease (SCD) leads to significant morbidity and early mortality, and hematopoietic cell transplantation (HCT) is the only widely available cure, with impacts seen on SCD-related organ dysfunction. Outcomes are excellent following matched-related donor (MRD) HCT, leading to significantly expanded application of this treatment over the past decade. The majority of SCD patients lack an MRD, but outcomes following alternative donor HCT continue to improve on clinical trials. Within this framework, we aim to provide our perspective on how to apply research findings to clinical practice, for an individual patient. We also emphasize that the preparation of SCD recipients for HCT and supporting them through HCT have special nuances that require awareness and close attention. Through the use of clinical vignettes, we provide our perpsective on the complex decision-making process in HCT for SCD as well as recommendations for the evaluation and support of these patients through HCT.
Introduction
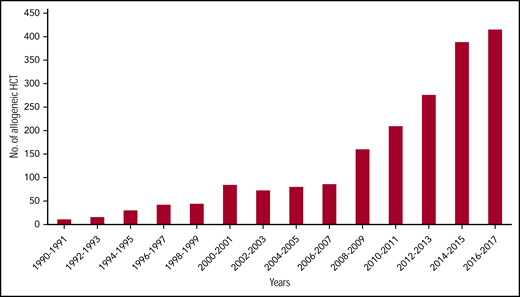
Hematopoietic cell transplantation (HCT), currently the only curative therapy for sickle cell disease (SCD), is associated with excellent outcomes when performed from HLA matched-related donors (MRDs).1-5 Combined with emerging data on the safety and efficacy of HCT from alternate donors6-9 and the use of reduced intensity or nonmyeloablative (NMA) conditioning regimens,7,10-13 there has been a dramatic increase in the acceptability and applicability of HCT for SCD. In fact, >50% of HCT for SCD reported to the Center for International Blood and Marrow Transplant Research were performed after 2013 (Center for International Blood and Marrow Transplant Research, e-mail, 19 November 2019; Figure 1); although the majority have been using an MRD, recent years have included increases in alternative donor HCT, primarily on clinical trials. With >100 000 individuals living in the United States with SCD, millions worldwide, and >300 000 new births each year,14 demand for curative therapies including HCT will continue to increase. As transplant physicians who focus on SCD HCT research, we describe here our perspective on the status and application of >2 decades of research to clinical practice. We believe that performing HCT for SCD requires attention to many details, and as such, is a topic worthy of discussion and education.
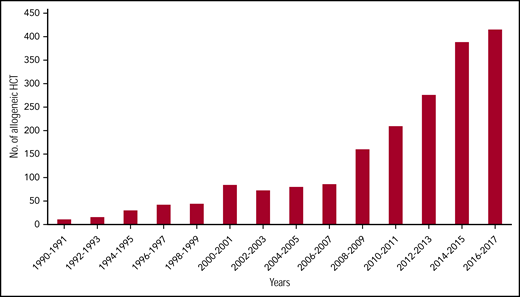
Dramatic increase in number of SCD patients undergoing HCT. In data courtesy of the Center for International Blood and Marrow Transplantation, the annual number of transplants for SCD has quadrupled in the last decade of available data. The data presented here are preliminary and were obtained from the Coordinating Center of the Center for International Blood and Marrow Transplant Research. The analysis has not been reviewed or approved by the Statistical or Scientific Committees of the CIBMTR. No., number.
Dramatic increase in number of SCD patients undergoing HCT. In data courtesy of the Center for International Blood and Marrow Transplantation, the annual number of transplants for SCD has quadrupled in the last decade of available data. The data presented here are preliminary and were obtained from the Coordinating Center of the Center for International Blood and Marrow Transplant Research. The analysis has not been reviewed or approved by the Statistical or Scientific Committees of the CIBMTR. No., number.
Presentation of cases
Case 1 is a 12-year-old boy with hemoglobin SS (HbSS) receiving regular, comprehensive SCD care from a pediatric hematologist. At age 11 years, he presented with an overt stroke. He commenced monthly chronic transfusions to maintain a hemoglobin level of 9 to 11 g/dL and sickle hemoglobin (HbS) level of <30%. His 25-year-old brother, who had sickle cell trait, was HLA matched. One year after the stroke, he underwent HCT using his brother’s bone marrow (BM). On day +20 posttransplant, he developed aphasia and a generalized tonic-clonic seizure. Earlier, he was noted to have relative hypertension (125/70 mm Hg; baseline 110/60). Laboratory investigation revealed magnesium and cyclosporine (CSA) levels of 1.3 mEq/L and 635 μg/L; other laboratory parameters were normal.
Case 2 is an 8-year-old girl with HbSS who commenced hydroxyurea (HU) prophylaxis at age 4. Despite excellent HU adherence and hemoglobin F levels ∼30%, she suffered from 3 episodes of acute chest syndrome (ACS) over 4 years, one of which required intensive care unit admission for exchange transfusion and mechanical ventilation. She also had frequent vasoocclusive crises (VOCs) in the previous year, leading her to miss ∼30 school days. Her hematologist obtained HLA typing for her and her siblings; her 2-year-old brother was HLA identical, and blood grouping revealed a major ABO mismatch.
Case 3 is a 16-year-old girl with hemoglobin SC who despite commencing HU at age 12 due to recurrent VOC, continued to have frequent VOC, requiring 3 admissions per year on average in the preceding 2 years. She has no HLA-matched familial donors. After consulting with her hematologist and the HCT team, she and her parents decided to proceed on a clinical trial of matched unrelated donor (URD) HCT following reduced intensity conditioning (RIC). HCT was successful, with 100% donor cell engraftment, but it was complicated by chronic graft-versus-host disease (GVHD) requiring prolonged systemic immunosuppression. She also continued to have chronic pain post-HCT.
Case 4 is a 25-year-old man with HbSS, complicated by stroke and Moyamoya disease, receiving monthly red blood cell (RBC) transfusions for 10 years with successful oral iron chelation. Due to chronic medical needs that impinge on his quality of life (QoL), he is interested in curative treatment options, having heard about gene therapy and HCT in the news. He does not have an HLA MRD or URD.
Brief review of HCT for SCD
The first successful HCT for SCD was performed in a child with acute myeloid leukemia who also had SCD.15 Since the first international clinical trial,1 HCT for SCD has been increasing in applicability, but until recently, was restricted to children with severe SCD and an available HLA-identical sibling.1,2,4,5,10,16-21 An international, retrospective, registry-based analysis reported an overall survival (OS) of 92.9% and event-free survival (EFS) of 91.4% in 1000 children who underwent HCT.4 On multivariate analysis, survival was better with BM compared with peripheral blood stem cells (PBSCs; with no difference between umbilical cord blood [UCB] and BM) and in those transplanted after 2006. Survival however declined with increasing recipient age (OS and EFS 81% in children >16 years), with GVHD the primary cause of death. The majority of patients underwent HCT using myeloablative conditioning (MAC). More recently, reduced toxicity/intensity2,5,19 as well as fully NMA10,12,20,21 conditioning regimens have been demonstrated to be safe and effective in ameliorating SCD clinical manifestations, with early beneficial results in adults compared with the older MAC regimens.2,5,19 Finally, post-HCT follow-up has revealed normalization of transcranial Doppler velocities, stabilization of IQ, increase in health-related QoL (HRQoL), decrease in chronic opioid use, and improvement in pulmonary and splenic functions, suggesting stabilization or improvement of organ function following donor cell engraftment.3,5,6,22-27 Because a comprehensive review of outcomes is outside the scope of this article, we point interested readers to recently published, detailed reviews on MRD28 and alternative donor29 HCT for SCD.
The challenges
Only a small minority of patients with severe SCD manifestations undergo HCT. In the initial multicenter international trial, only 8% of eligible patients underwent transplant, with lack of an available MRD being the primary barrier.30 Registry data suggest that ∼20% of patients have an HLA-matched URD (8/8), and ∼5% will have a matched (6/6) unrelated UCB product.31,32 Unacceptably high rates of chronic GVHD have limited the acceptability of URD BMT5,6 ; further improvements in GVHD prophylaxis may improve safety. High rates of graft failure following unrelated UCB transplant are a barrier to its wider applicability,33,34 but modifications in conditioning may improve engraftment.7 Low UCB cell dose is a risk factor for rejection, but the ex vivo expansion strategies may overcome this limitation.35 HCT from related haploidentical donors has the potential to significantly expand the applicability of HCT for SCD but has been limited by low rates of stable engraftment.8 Refinements in conditioning regimens have yielded encouraging OS and EFS rates8,9,13,27,36 and are being tested in large clinical trials. Access to care as well as financial and psychosocial hurdles, particularly in low-income countries where the majority of patients reside, add to transplant applicability. In a single-center study, 20% of adults were denied HCT due to insurance denial; 10% lacked an available haploidentical donor; 29% had unacceptably high donor-specific HLA antibodies; and 34% refused consent due to toxicity.9 Addressing these barriers is crucial to making this curative therapy an option for suitable patients.
The decision for HCT
Recipient considerations
Disease-related complications, organ toxicity (including QoL), treatment response, and predictors of potential future complications have been considered in determining the risk justifiable for HCT (Figure 2) and used as eligibility criteria in clinical trials (Table 1). Stroke was the most common indication for HCT in the first multicenter trial,1 although recurrent VOC requiring medical care is currently most common.4 Health care utilization (HCU) for recurrent VOC, however, is an inadequate measure of pain burden because the majority of pain episodes are managed at home.37 There is therefore a need to better estimate pain burden, informing risk-benefit stratification in consideration of HCT. For the first time, the BMT CTN 1503 trial uses high-impact chronic pain with significant disability as an indication for HCT, even in the absence of frequent emergency department visits or hospitalizations for VOC.
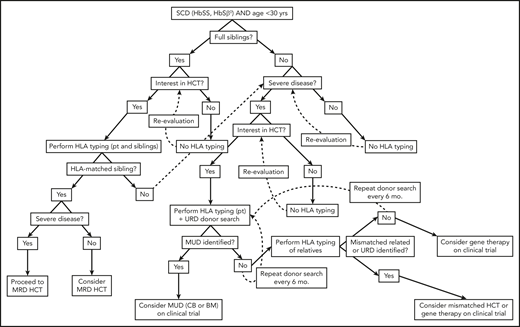
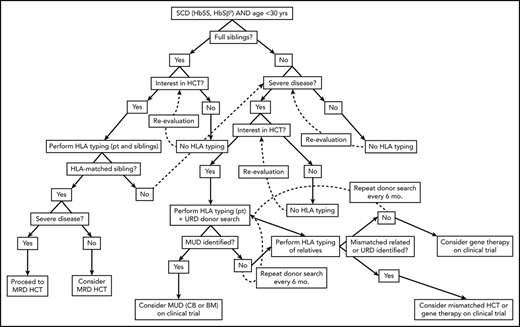
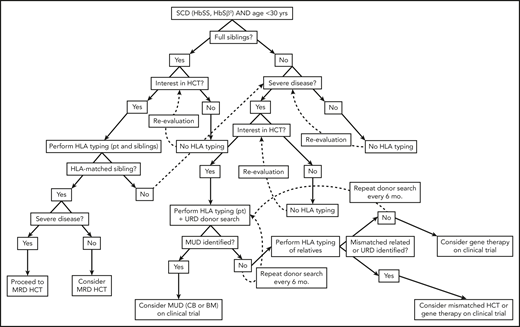
Algorithm for consideration of HCT for SCD. Consideration of HCT and other curative therapies in individuals with SCD must take into consideration severity of disease to date, available HCT donor options, and clinical trials that an individual patient may be eligible for. While this decision-making process is complex, this algorithm provides a framework for how we consider an individual patient. Additionally, this algorithm does not take into account multiple processes that may occur concurrently, such as searching for URD and haploidentical donors, as well as considering gene therapy and mismatched related or unrelated donor HCT on a clinical trial.
Algorithm for consideration of HCT for SCD. Consideration of HCT and other curative therapies in individuals with SCD must take into consideration severity of disease to date, available HCT donor options, and clinical trials that an individual patient may be eligible for. While this decision-making process is complex, this algorithm provides a framework for how we consider an individual patient. Additionally, this algorithm does not take into account multiple processes that may occur concurrently, such as searching for URD and haploidentical donors, as well as considering gene therapy and mismatched related or unrelated donor HCT on a clinical trial.
Age at HCT predicts risk for subsequent OS and EFS, with risk for severe complications including death exceedingly low in patients <14 years.4,6,38 Combined with excellent overall outcomes in MRD HCT, this has led to the consideration of this curative treatment in young children without severe manifestations of disease.39 SCD organ damage is insidious, with glomerular hyperfiltration40 and splenic dysfunction41 found by 1 year and silent stroke in ∼25% <6 years,42 leading to cognitive deficits and decreased school performance.43 We recommend careful consideration of HCT in young children with an available MRD and symptomatic SCD, without severe manifestations. This approach has the potential to yield excellent HCT outcomes but also to avoid the development of irreversible SCD-related organ damage. In addition, excellent outcomes in adult patients following MRD HCT5,10 have led to the consideration of HCT up to 40 to 45 years in clinical trials.
Donor considerations
HLA matching and stem cell source are the primary donor factors that influence outcomes (Table 2). We recommend routinely HLA typing pediatric patients with SCD with available full siblings. Beforehand, we strongly recommend discussing ongoing clinical trials of HCT to accommodate those with biologic randomization (between conservative management and HCT; BMT CTN 1503). In patients without MRD options, HLA typing and URD search should be based on clinical disease severity, eligibility for current clinical trials, and patient and family preference regarding care, with the process of selecting an alternative donor similar to HCT for other diseases. Such informed decision making will necessitate extensive discussion between the patient/family, the primary physician (primary care physician and/or hematologist), and the consulting BMT team.
In MRD HCT, additional considerations include stem cell source and donor age. The majority of sibling donors have sickle cell trait (hemoglobin AS),3 an acceptable donor source. Banked UCB, providing an adequate cell dose, can be the sole source of MRD stem cells. We recommended a threshold of ≥3 × 107 total nucleated cell (TNC) per kilogram recipient weight; no impact of cell dose on engraftment or disease-free survival (DFS) was found with a median dose of 3.9 × 107 TNC per kilogram (range, 1.5 to 14).18 In the case of TNC 1 to 3 × 107 per kilogram, MRD BM can be collected and infused sequentially to UCB.2 We recommend minimum donor size and age of ≥10 kg and ≥1 year, to ensure collection of an adequate cell dose without compromising donor safety. If >1 MRD, we consider donor size/age, ABO typing, and cytomegalovirus (CMV) serostatus, a process not unique to HCT for SCD. Increasing donor age impacts chronic GVHD risk in malignant disease44,45 and may similarly impact SCD HCT recipients, although data are lacking. If significant discrepancy in donor/recipient size, PBSCs yield better cell dose, but outcomes are compromised by high GVHD rates. To offset this, an alternate strategy would be to infuse CD34 selected PBSCs combined with T-cell addback (we use 3 to 5 × 106 CD34+ cells per kilogram plus 1 × 106 CD3 cells per kilogram). In cases of ABO major or minor mismatch, which may add to graft stem cell loss, the donor/recipient size differential must be stringently assessed. Major ABO mismatch should be avoided whenever possible given cell dose impact, delayed RBC engraftment,46 and decreased OS,47 especially with pure RBC aplasia.48 Last, donor-recipient CMV serostatus should be matched (D−/R− or D+/R+) whenever possible to optimize CMV risks.
Family and caregiver decision making
The decision to proceed to HCT often poses a complex decisional dilemma involving risk-benefit tradeoffs. Comprehensive care and disease-modifying therapies have improved outcomes, but there are no data comparing HCT and standard clinical care. HCT offers the possibility of cure but is associated with treatment-related morbidity and mortality risk. Studies of treatment decision making in HCT for SCD found that disease severity and availability of an MRD impact decision making in SCD patients/caregivers49 and their physicians,50 favoring HCT. Family support, resources, and BMT-related education also impact decision to proceed.49 Although approximately one-fourth of SCD patients/caregivers reported an unwillingness to accept any risk of GVHD or mortality,51 no posttransplant patients/caregivers reported decisional regret49 ; this was likely due to education regarding key aspects of the process, including that despite low risk for GVHD and mortality, risk is never totally mitigated. The majority of SCD patients and their caregivers are willing to undertake GVHD and mortality risks commiserate with MRD HCT.51 Ultimately, the decision to proceed with HCT must involve the patient, caregivers/family, primary hematologist, and transplant physician.
Physician perspective
Among SCD experts, there is substantial variability in the readiness to discuss HCT.50 Critical factors impacting decision making include patient/clinical characteristics and how the physician perceives these.50,52 The ethical challenges of this decision making, including in the setting of less than “severe disease,” have been extensively reviewed.52,53 In our practice, we offer MRD HCT to young patients who are symptomatic, after team (BMT and hematology) discussion and consensus regarding patient characteristics and suitability for transplant. Furthermore, family education and opinion are sought in multiple stages over time before proceeding.
To address the complexity of this decision-making process, we recommend the following: (1) physician education of patients/families as a dynamic iterative process, often requiring multiple BMT consultations over time and meeting with different physicians and team members to contribute to various aspects of undertaking the procedure; (2) close collaboration between medical providers caring for SCD patients, including primary care physicians, hematologists, and BMT physicians, to allow multidirectional transfer of knowledge in areas of expertise and presentation of all aspects of care and outcomes with or without transplant; (3) peer education of patients/families, which may occur through SCD “educational symposiums”54 and through connecting with patient/family support groups who have undergone HCT; (4) enhancement of education and decision making through a multidisciplinary approach, which may include social work, psychology, psychiatry, and faith-based personnel, depending on patient/family needs; and (5) use of educational aids, including informational Web sites and online decision aids. Each of these is important to providing a global view of SCD care that includes a transplant option.
Pre-HCT recipient evaluations
Patients proceeding to HCT are evaluated for HCT eligibility, organ function, and SCD-related complications (Table 3). HLA antibody screening must be performed to screen for high-titer donor-directed HLA antibodies, which may predict a high risk for graft rejection. Desensitization strategies have been successful in SCD before haploidentical HCT,36 and the European Society for Blood and Marrow Transplantation recently published general guidelines.55
Assessment for SCD-related organ damage includes evaluation of iron overload and neurologic, splenic, and renal function. All patients must have a baseline brain magnetic resonance imaging/magnetic resonance angiography (MRI/MRA) to evaluate for overt/silent stroke and vasculopathy. We consult neurosurgery in patients with Moyamoya disease, who may be considered for or have already undergone encephaloduroarteriosynangiosis.56 Further neurologic evaluations may include transcranial Doppler velocity (age <16 years) and neurocognitive evaluation, including IQ testing, which may stabilize or improve posttransplant.25 We use a lifetime transfusion burden ≥10 transfusions AND serum ferritin ≥1500 ng/mL as a trigger for workup, including liver iron quantification and liver fibrosis evaluation by imaging and/or liver biopsy in collaboration with hepatology. Because of hyperfiltration seen with SCD,40,57 we recommend obtaining glomerular filtration rate (GFR), urine-specific gravity, and albumin-to-creatinine ratio; notably, normal GFR in adolescence or adulthood may in fact suggest renal impairment in SCD.58 Baseline splenic function may be evaluated with liver/spleen nuclear medicine scan,41 which may be repeated 1 year following successful transplant to document recovery of splenic function.23
We recommend that all patients meet with specialists in social work, psychology, and child life (pediatric patients). We consult transfusion medicine to assist in transfusion burden assessment, in determining alloimmunization status, and to formulate an RBC transfusion plan, including prior to conditioning.59 We refer patients for fertility consultation with endocrinology or obstetrics/gynecology, depending on age. The patient’s primary SCD physician (typically hematology) should be actively engaged in deciding when to discontinue SCD-specific medications (eg, HU).
HCT regimen
The majority of MRD transplants have received MAC,4 initially with busulfan/cyclophosphamide,1 but the use of RIC has increased.2,4,6,7,33,60 The most common RIC approach has been fludarabine ± another agent,4 typically an alkylator.2 Reduced-toxicity strategies61,62 have also been successful in MRD HCT,5,63,64 allowing busulfan deescalation with the addition of fludarabine ± cyclophosphamide63 or substitution of busulfan with treosulfan + thiotepa.5,64 Following initial reports of NMA conditioning (total body irradiation 200 cGy plus fludarabine) in adult and pediatric SCD MRD HCT,65,66 newer NMA strategies have combined total body irradiation (300 cGy) with alemtuzumab successfully in adults10,21 and children12,67 using PBSCs. This approach may be particularly attractive in lower-income countries, due to decreased organ toxicity and length of admission. Overall, certain advantages vs disadvantages are known or can be predicted based on conditioning intensity (Table 4). RIC and NMA transplant strategies and outcomes vary based on agents used. Hence, we recommend that transplants performed using novel regimens be undertaken in a clinical trial setting to determine applicability and determine outcomes.
In vivo T-cell depletion (TCD) has been given to 82.8% of MRD HCT patients,4 using antithymocyte globulin (ATG; 70.6%) or alemtuzumab (11.5%), with rejection decreasing from 22.6% to 3% with ATG in early studies.3 ATG use is uncommon in MRD cord blood (CB).18 Rabbit ATG has been used more commonly than equine19,63,68 but does not appear to impact OS, EFS, or GVHD.69 Alemtuzumab has been administered in both pediatric and adult MRD HCT, especially with NMA or RIC2,10 and URD CB transplant.7,33 Haploidentical HCTs have been performed with a combination of in vivo TCD with posttransplant cyclophosphamide plus ATG or alemtuzumab8,11,13,36,60 or a combination of in vivo (typically ATG) and ex vivo TCD, including CD34+ selection,70 CD3/CD19 depletion,71 or T-cell receptor α/β and CD19 depletion.72
Most patients undergoing MRD HCT have received calcineurin inhibitor (CNI)-based GVHD prophylaxis, most commonly CSA alone (19.9%) or combined with methotrexate (56.5%) or mycophenolate mofetil (7.7%).4 Based on decreased DFS following MRD CB transplant with methotrexate,18 mycophenolate mofetil is substituted in this setting.7,33 Although there are limited SCD-specific data, target cell dose goals can be extrapolated. In a single-center analysis of graft failure risk factors in adult and pediatric patients receiving BM or PBSCs for malignant or nonmalignant indications, graft failure decreased to 5% from 10% when TNC was ≥2.5 × 108/kg.73 In a Center for International Blood and Marrow Transplant Research analysis restricted to NMD and URD, mortality decreased with TNC > 2 × 108/kg and >5 × 108/kg with PBSCs and BM, respectively.74 Analyses of MRD UCB cell dose in SCD did not demonstrate an impact on engraftment, but subjects received a median UCB cell dose of 3.9 × 107 TNC per kilogram.18 In URD UCB, TNC > 5 × 107/kg increased engraftment and DFS.34 For MRD transplant, we recommend a target cell dose of 4 to 5 × 108 TNC per kilogram and 4 to 5 × 107 TNC per kilogram for BM and CB (prethaw) grafts, respectively, unless protocol specified. Given the higher risk of GVHD with PBSCs, we recommend PBSCs only be used in a clinical trial with additional GVHD prophylaxis.
Discussion of cases
HCT should be strongly considered for case 1, because he has clinically severe disease with overt stroke requiring chronic transfusions for prevention of recurrence and an available MRD. Clinicians should discuss available clinical trials of HCT with the patient and his family, or perform HCT as standard of care, involving the patient and his family as well as multidisciplinary medical providers in the decision-making process. Although the most extensive long-term experience is with busulfan/cyclophosphamide,4 substitution of cyclophosphamide with fludarabine has been found to be safe and effective with lower risk of venoocclusive disease. Reduced intensity,2 reduced toxicity,5 and NMA12 regimens have also been demonstrated to be effective in this setting and are our preference to reduce short- and long-term toxicities. His donor’s older age (25) may put him at higher GVHD risk, which should be discussed during the informed consent process. If there were >1 MRD, we would recommend selecting the younger, provided there is no other reason to exclude that donor (medical issues, size differential, etc).
Case 2 does have an HLA-matched sibling but does not meet criteria for severe disease. She has had multiple episodes of ACS, but they were over a period of 4 years. Severity criteria do not account, though, for this patient having had a life-threatening episode of ACS. The pain crises were managed at home; hence, she does not meet the severity threshold. This young patient with an HLA-matched brother is likely to have an excellent outcome following HCT.4 We offer MRD HCT to such young patients whose disease manifestations do not rise to the threshold of severe SCD definitions,39 in conjunction with patient and caregiver preferences and informed decision making. For patients not meeting severity criteria, patients/families undergo BMT consultation at least twice; they also undergo detailed psychosocial evaluations to determine transplant readiness and understanding. In some cases, families may meet with our team multiple times over years before deciding to proceed to HCT. Because of limited data on long-term outcomes following HCT for SCD, particularly compared with other supportive therapies, and the heterogeneity of disease severity without good predictors for such, there is substantial practice variability in the disease severity threshold (or lack thereof) used to proceed to MRD HCT.
In case 2, donor-recipient size discrepancy could compromise harvest of an adequate cell dose, with further loss by RBC depletion due to ABO mismatch. Strategies to achieve an adequate cell dose may become necessary, such as CD34+ selection from PBSCs. The combination of disease severity in the recipient and the nuances of stem cell collection from the young donor will affect the risk/benefit discussion of HCT.
Case 3 is a young adult patient with clinically severe disease due to recurrent VOC whose sibling is not an HLA match. She met eligibility criteria for a clinical trial of matched unrelated donor HCT,6 which she consented for after discussion with BMT and her primary hematology team.
Case 4 is an adult man also with clinically severe disease due to Moyamoya disease, but who unfortunately does not have any HCT donor options. Our BMT coordinators maintain a patient list on whom to perform biannual URD searches, in case a new HLA-matched donor joins the registry. Patients with severe SCD who could benefit from curative therapies should be referred to and evaluated at transplant centers offering such therapies (URD or haploidentical HCT) on clinical trials so the relevant discussions are imparted. Such discussions currently include applicability of transplant or autologous transplantation of gene-modified cells if eligibility criteria are met.75
Engraftment following transplant
Although analyses of risk factors for graft failure specific to SCD have not yet been systematically performed, HLA mismatch, high titers of donor-directed HLA antibodies, lower intensity of conditioning, and active infection at the time of engraftment are generally accepted to predict higher rates of rejection.73
Donor engraftment should be evaluated serially posttransplant ideally using cell-sorted chimerism (lymphoid [CD3] and myeloid [CD15 or CD33]), hemoglobin level, and hemoglobin electrophoresis (for HbS%). Although whole blood chimerism of 11% to 74% may adequately predict stable donor-derived erythropoiesis,17,76,77 lineage-specific chimerism may provide additional information, with research studies demonstrating the advantage of specifically tracking RBC engraftment.19,78 Clinically, myeloid chimerism is likely to be the best predictor of stable engraftment, with a threshold of 20% to 25%.5,76,77 Rising HbS, particularly >50%, suggests impending autologous recovery. In the setting of mixed chimerism, stability is as important as the level. We recommend chimerism testing monthly through day +100, at 6 months, and then yearly for at least 1 to 2 years; if donor chimerism is low (<50%) or declining or HbS is higher than expected or increasing, we recommend more frequent assessments (at least monthly until stabilized or improved). In the event of decreasing chimerism, stem cell boosts with minimal conditioning have been successful. There are no data to support the use of donor lymphocyte infusions to stabilize engraftment in nonmalignant disorders, and the risks may outweigh the benefits.
Rejection with aplasia is uncommon using an MRD, with the majority of patients having autologous reconstitution and few reported as undergoing a second HCT.3,76 Even in URD and alternative donor settings, autologous recovery is usual following rejection.5-8,76 If marrow aplasia or prolonged cytopenias prevail, a second HCT should be undertaken promptly.5 In patients with rejection and autologous reconstitution, following risk/benefit discussions with the patient and caregiver, a second HCT may be considered after ≥6 months, especially in patients who received NMA or RIC conditioning.
Prevention and management of complications during HCT
Attention is required for complications that SCD patients are uniquely susceptible to during HCT. These include neurologic, cardiovascular/pulmonary, hepatobiliary, renal, and infection susceptibility (Table 5). Detailed recommendations regarding transplant and disease-related follow-up and late effects screening have been previously described and serve as a guideline for uniformity in follow-up.79
Neurologic
The high incidence of neurologic complications, including seizures and hemorrhagic stroke, was initially noted in one-third of patients who underwent MRD HCT80 and brought to light several risk factors: (i) higher stroke risk in patients with pretransplant stroke; (ii) susceptibility to hypertension due to neurologic and renal compromise as well as medications (CNI and steroids); and (iii) hemorrhagic stroke with concurrent thrombocytopenia or polycythemia in patients with cerebral vasculopathy, not seen in patients transplanted in France where platelets were maintained ≥50 000/μL.3 Although posterior reversible encephalopathy syndrome (PRES), also known as reversible posterior leukoencephalopathy syndrome, was not specifically described initially, a high incidence (22% to 34%) has been subsequently reported.6,81 SCD patients are susceptible to PRES,82 but the incidence increases significantly with HCT, a phenomenon also noted less frequently in patients with thalassemia.81 SCD is a risk for PRES, in addition to hypertension and acute GVHD.81 Patients who developed PRES following MRD HCT had decreased OS and DFS,81 and although the multicenter URD trial did not find a survival association,6 we believe that PRES impaired early posttransplant HRQoL.
Based on the above, we recommend strict parameters attempting to minimize neurologic complications, including (i) seizure prophylaxis during conditioning (especially when busulfan-based) and CNI administration; (ii) strict control of hypertension, noting lower baseline blood pressures (BPs) in SCD compared with age-matched peers83 ; (iii) prevention of CNI-induced hypomagnesemia84,85 ; and (iv) transfusions to keep platelets >50 000/μL and hemoglobin 9 to 11 g/dL. It is crucial to perform serial brain imaging with MRI/MRA ± magnetic resonance venography (baseline, 12 months, and 24 months posttransplant) to detect HCT-induced neurologic changes.1 Additional time points should be performed based on symptoms.
Pain described as consistent with vasoocclusive pain can occur during and after transplant, which is poorly understood but an area of active research. Subjectively, it is more common in young adults and adults and in those with chronic pain pre-HCT, consistent with a recent report demonstrating an association between high pain burden pretransplant and chronic pain at 1-year posttransplant.86 Although pain persisted at 1 year in 40%,86 it is encouraging that HRQoL returned to baseline, pain interference decreased, and physical function improved.5 Thus, patients with substantial pretransplant pain burden are anticipated to have chronic pain until or even beyond 1 year following successful HCT. We recommend close, multidisciplinary (pain/anesthesia, psychology, psychiatry, and social work) follow-up of such patients, considering complementary strategies, including acupuncture and/or pain rehabilitation, including during the transplant admission.
Cardiovascular/pulmonary
Prevention of hypertension may prevent severe neurologic complications, as detailed above.1,80 Pegelow et al reported that individuals with SCD have lower baseline BPs than expected for age, sex, and race,83 a difference that increases with increasing age. Before HCT admission, we recommend determination of baseline BPs via electronic medical record review, aiming to keep BP within 10% of median for age and sex, according to normative data.83 A threshold for systolic and diastolic BP should be determined, with as needed antihypertensives (typically isradipine or hydralazine) administered to maintain goal BP. To prevent CNI-induced hypertension, we recommend considering daily low-dose amlodipine starting day of CNI.
Although pulmonary hypertension is rare in children, TRJ velocity >3.0 m/s, B-type natriuretic peptide >160 pg/mL, and 6-minute walk test <332 m are the strongest predictors of mortality in adult SCD patients.87,88 TRJ velocity may improve in some patients following HCT.5 In patients with preexisting pulmonary hypertension, we recommend having a low threshold of repeating echocardiogram.
Finally, in patients with acute cardiorespiratory symptoms, we recommend evaluating for pericardial and pleural effusions, initially with a chest radiograph. In patients with effusions, transplant-associated thrombotic microangiopathy should be in the differential diagnosis89,90 ; therefore, we recommend peripheral blood smear review and additional workup, including haptoglobin, reticulocyte count, lactate dehydrogenase level, and sC5-9 level. In patients with transplant-associated thrombotic microangiopathy, we recommend switching from the current CNI and instituting appropriate therapy.
Infectious
Because of acquired splenic dysfunction, with poor spleen function present in most by age 3 years,41,91 SCD patients are at risk for infection with encapsulated bacteria, particularly Streptococcus pneumoniae. In the era of pneumococcal conjugate vaccines, the majority of invasive pneumococcal disease in SCD occur with nonvaccine serotypes, although the majority remain penicillin sensitive.92 Pneumococcal infections are rare following HCT for SCD,23 but they should be considered in patients presenting with fever ± cardiorespiratory compromise even if still on prophylaxis, where we have had 1 case of fatal invasive pneumococcal disease occurring nearly 1 year after successful MRD HCT.
We recommend starting (or resuming) penicillin prophylaxis on the day before HCT, if not adequately covered by alternate antibiotics, and continuing post-HCT. Penicillin prophylaxis should be discontinued only if splenic function posttransplant and response to vaccination are documented, and only after discontinuation of systemic immunosuppressants. We recommend assessment of splenic function with 99mTc sulfur colloid liver-spleen scan initially at 1-year posttransplant. In patients with present or normal function, prophylaxis can be discontinued, providing there are no other risk factors (eg, chronic GVHD).23,93 We recommend revaccination with pneumococcal conjugate vaccines 3 to 6 months posttransplant.94
Hepatobiliary
SCD patients are at risk for acute hepatobiliary dysfunction due to chronic RBC hemolysis and iron overload from chronic transfusions. In patients with baseline iron overload, we recommend serum ferritin and transferrin saturation starting 6 months posttransplant and every 3 to 6 months until normal.79 Following recovery from HCT, we recommend initiation of iron depletion by phlebotomy or iron chelation until iron overload and the associated organ damage is reversed.
Renal
While on CNI or other nephrotoxic medications, renal function must be closely monitored. Posttransplant, we recommend the following: (i) serum blood urea nitrogen/creatinine and GFR or 24-hour creatinine clearance yearly × 2 years; (ii) urinalysis for proteinuria and ultrasound of kidneys at 1 year (or if clinical indication); and (iii) urine for microalbumin yearly × 2 years.79
Discussion of cases
Case 1 developed an acute onset of aphasia 20 days after transplant with a subsequent seizure; this episode was preceded by hypertension (>10% above baseline and ∼90th% for age/sex/race83 ) in the setting of elevated CSA level and hypomagnesemia. This patient developed PRES, which responded to treatment, including the correction of hypomagnesemia, administration of antihypertensives, and switching of CSA to an alternative, such as tacrolimus. Despite the supportive care measures detailed in Table 5, neurologic complications, including PRES, occur in approximately one-third of patients.24 Although PRES typically occurs in the absence of hypomagnesemia or elevated CNI levels, hypertension significantly increases the incidence of PRES. The majority of patients present with seizures and with symmetric hyperintense lesions on T2 MRI, predominantly in frontal, parietal, temporal, and occipital regions. A minority of patients develops respiratory failure requiring mechanical ventilation, and neurological symptoms and MRI findings usually completely resolve. On multivariate analysis, risk factors for PRES include hypertension and grade II to IV acute GVHD.
Case 2 is a young adult patient whose primary indication for matched URD HCT is recurrent VOC; because of her age and chronic pain, she is at higher risk for prolonged pain posttransplant, even if successful. In such patients, we advise multidisciplinary management of chronic pain. This approach should seek the active cooperation of the family in establishing specific treatment goals, based on the anticipated prolonged course, and must include pain medicine, psychiatry, psychology, or alternative medicine for acupuncture and mind-body interventions. This patient subsequently developed chronic GVHD, with risk factors being older recipient age and URD6 ; the severity of her GVHD will require close outpatient follow-up and multidisciplinary approach, which may include therapy, anesthesia, social work, nutrition, psychology, or psychiatry.
Case 4 is an adult with Moyamoya disease who is interested in curative treatment options such as alternative donor HCT or gene therapy. Neurosurgical consultation should be sought for the consideration of surgical revascularization.56 If this patient undergoes encephaloduroarteriosynangiosis, we recommend it occur ≥6 months before curative treatment.
Conclusion
Refinements in conditioning regimen, GVHD prophylaxis, and the use of alternative donors have dramatically improved the acceptability and applicability of HCT for SCD. Preliminary reports of the feasibility and safety of gene therapy suggest the likelihood of an even greater expansion of treatments with curative intent.75 We believe, therefore, that all SCD patients should receive information on curative treatment options. We believe that consideration must be given to MRD HCT, if one is available, in young children with symptomatic SCD. Alternative donor transplantation should be reserved for patients with severe symptoms, causing or likely to cause organ damage, and should be undertaken only in the context of clinical trials. Patients undergoing these therapies require care and counseling regarding psychosocial aspects, including importance of treatment compliance. Increasing long-term outcome data following HCT for SCD95,96 is critical to define benefits and pitfalls of curative therapies. It is crucial to support the development of long-term follow-up registries to ensure and investigate the reversibility of disease-related organ dysfunction (see Table 1 in Shenoy et al) and direct uniformity of data collection.79 Newer curative options, including autologous HCT with gene modification, have advantages over allogeneic HCT, including absence of GVHD and donor dependence, but questions remain regarding the level of control required for cure, durability of gene-modified cell engraftment, and long-term risk of insertional mutagenesis.75,97,98 Although consideration must be given to the availability and cost of these therapies,99 these are expected to decrease as the treatments become more prevalent, streamlined, and easier to use. Attention must also be given to individual and societal costs of chronic illness, HCU, and the loss of educational and work potential. Although transplantation at age <10 vs ≥10 years is associated with decreased HCU and cost,99 a direct comparison of HCT and supportive care is lacking to date. Of note, the changing landscape of supportive care and newer agents should be taken into account in this comparison at the current time. Meanwhile, research support to develop systematic trials building up on previous experience, and ensuring adequate follow-up, will continue to enhance discovery in the treatment of this highly prevalent chronic disease.
Acknowledgment
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grant K23HL133446 (E.O.S.).
Authorship
Contribution: E.O.S., S.S., and L.K. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Elizabeth O. Stenger, 4401 Penn Ave, Pittsburgh PA, 15224; e-mail: stengereo@upmc.edu.