Key Points
D816V KIT allele frequency in BM, higher sβ2M levels, and A/R/D pathogenic mutations identify ISM cases at very high risk for progression.
Pathogenic mutations in the A/R/D genes identify ISM cases with multilineal KIT and poorer prognosis.

Abstract
Indolent systemic mastocytosis (ISM) patients have a normal life expectancy, except in the 5% to 10% of cases that progress to more advanced SM (advSM), which has a significantly poorer outcome. Mutations in genes other than KIT frequently found in myeloid neoplasms have been associated with a poorer outcome among advSM, whereas limited information exists about their frequency and prognostic impact in ISM. We investigated the frequency and prognostic impact of variants in 18 genes, found to be altered in advSM, in 322 ISM patients (median follow-up, 5.7 years) divided into discovery (n = 200) and validation (n = 122) cohorts. Overall, 71 genetic variants were detected in 55 of 322 (17%) patients. Mutated ISM cases, particularly those carrying ASXL1, RUNX1, and/or DNMT3A (A/R/D) pathogenic variant allele frequencies (VAFs) ≥ 30%, exhibited significantly shortened (P < .001) progression-free survival (PFS) and overall survival (OS). Multivariate analysis showed that serum β2-microglobulin (sβ2M) levels > 2.5 µg/mL (hazard ratio [HR], 9.8; P = .001), together with a KIT D816V VAF ≥ 1% in bone marrow (BM) (HR, 10.1; P = .02) and pathogenic variants of A/R/D VAFs ≥ 30% (HR, 4.2; P = .02), were the best combination of independent predictors for PFS. In turn, A/R/D gene pathogenic VAF ≥ 30% was the only independent predictor for OS (HR, 51.8; P < .001). Based on these variables, 2 scoring systems were constructed for risk stratification of ISM at diagnosis with significantly different 10-year PFS (100%, 91%, 0% for scores of 0, 1, ≥2, respectively) and OS (100% and 50% for scores of 0 and 1) rates.
Introduction
Indolent systemic mastocytosis (ISM) is the most prevalent subtype of systemic mastocytosis (SM) in adults.1-3 Overall, ISM patients display a good prognosis with a similar outcome to that of the general population1 ; however, in 5% to 10% of cases, the disease will (eventually) progress to more advanced forms of SM (ie, smoldering SM [SSM], aggressive SM [ASM], mast cell leukemia [MCL], and SM associated with another hematologic neoplasm [SM-AHN]) with a significantly reduced life expectancy.4-6
So far, few studies have investigated those variables that might contribute to identify ISM patients at risk for disease progression and death. In these studies, age at diagnosis >60 years and increased serum alkaline phosphatase (SAP), serum/plasma interleukin-6, and serum β2-microglobulin (sβ2M) levels, as well as multilineal involvement of bone marrow (BM) hematopoiesis by the KIT D816V mutation (multilineal KIT), presence of KIT D816V-mutated BM mesenchymal stem cells (MSCs), splenomegaly, and/or a more immature immunophenotype of mast cells (MCs) by flow cytometry, have all been associated with progression to advanced SM (advSM).4,7-9 Of note, among these variables, only increased sβ2M levels and multilineal KIT have proven to be of independent prognostic value for progression-free survival (PFS),4 whereas age and SAP were the only independent predictors for overall survival (OS) described so far for ISM,4 as was confirmed recently in a large retrospective study.10 Altogether, these findings suggest that an increased percentage of KIT-mutated BM hematopoietic cells, together with a chronic proinflammatory immune response, might provide the basis for the progression of ISM to advSM by, for example, potentially providing the required microenvironment and genetic instability for the acquisition of additional mutations in KIT-mutated and nonmutated hematopoietic cells that might ultimately trigger disease progression with the potential for a shortened OS.
In the last decade, numerous mutations in multiple genes, particularly in genes involved in other non-MC myeloid neoplasias (eg, ASXL1, CBL, DNMT3A, EZH2, IDH1/2, U2AF1, RUNX1, SF3B1, SRSF2, and TET2), have been reported in SM patients, in addition to KIT D816V or other rare KIT mutations.11-15 Interestingly, a higher number of mutated genes, together with the presence of SRSF2, ASXL1, RUNX1, and/or EZH2 gene mutations, have been recurrently reported as independent adverse prognostic factors for OS in SM.12,15 However, the frequency of mutations in these and other genes has only been investigated in small series of ISM cases (10 patients,13 15 patients,14 44 patients,16 20 patients,17 and 12 patients15 ) with a rather limited follow-up (median of 8 months,13 24 months,14 25 months,16 40 months,17 and 78 months,15 respectively). Therefore, the frequency and prognostic impact of mutations in genes other than KIT on the outcome of ISM remain largely unknown.
Here, we investigated the frequency and prognostic impact of genetic variants in 18 genes previously found to be recurrently altered in SM (ASXL1, CBL, CDH11, DNMT3A, EPHA7, EZH2, ICK, IKZF1, ITGA10, JAK2, KAT6B, KRAS, PIK3CD, ROS1, RUNX1, SF3B1, SRSF2, TET2),12-16 in parallel with the pattern of involvement of BM hematopoiesis by the KIT D816V, D816H, and D816Y mutations (ie, multilineal KIT and presence of KIT-mutated MSCs and uncommitted hematopoietic precursor cells), as well as the KIT variant allele frequency (VAF) in BM and in peripheral blood (PB) and other previously defined clinical and laboratory prognostic factors, in a large series of 322 ISM patients with a long follow-up grouped into discovery (n = 200) and validation (n = 122) cohorts.
Materials and methods
Patients and samples
Overall, 322 patients (170 males and 152 females), who were divided into a discovery cohort (n = 200) and a validation cohort (n = 122) and who were diagnosed with ISM (median age at diagnosis of 44 years and 50 years; range, 0-76 years and 10-78 years, respectively) at the reference centers of the Spanish Network on Mastocytosis-Cancer Research Center (Salamanca, Spain) and the Virgen del Valle Hospital (Toledo, Spain), were studied. Diagnosis and classification of ISM were (retrospectively) established according to the World Health Organization (WHO) 2016 criteria.18 To rule out an AHN, conventional WHO cytomorphologic and immunophenotypic criteria18 (based on the EuroFlow acute leukemia orientation tube and acute myeloid leukemia/myelodysplastic syndrome antibody panels19 ) were used at diagnosis. All patients who progressed to SSM or advSM showed BM MC aggregates upon histology, with CD25+KIT D816V-mutated and cytologically altered BM MCs. These patients were thoroughly investigated to rule out SSM or advSM, and none showed ≥2 B-findings and/or any C-finding18 at diagnosis. Only those ISM patients with available genomic DNA (gDNA) extracted from whole-BM samples obtained at diagnosis were included in the study. Prior to entering the study, each individual gave his/her written informed consent to participate according to the Declaration of Helsinki, and the study was approved by the local institutional Ethics Committees.
The clinical and biological features of patients within the validation cohort were similar to those of the discovery series, with the exception of older age at diagnosis (50 years vs 44 years, P = .04), lower serum baseline tryptase (sBT) levels (26 µg/L vs 32.9 µg/L, P = .02), and higher SAP (74 U/L vs 66 U/L, P = .03) and immunoglobulin E (IgE; 26.5 KU/L vs 20.7 KU/L, P = .04) levels (supplemental Table 1, available on the Blood Web site).
KIT mutational analyses
Analysis of KIT genetic variants at codon 816 was performed on gDNA obtained from fluorescence-activated cell sorted and purified (ie, ≥98% purity) BM MCs, maturing neutrophils and/or monocytes, T cells, and, whenever available (n = 45), MSCs, using a previously described peptide nucleic acid–mediated polymerase chain reaction (PCR) method.20,21 According to the pattern of involvement of BM hematopoiesis by the KIT mutation, patients were classified as having MC-restricted ISM (ISMMC) or multilineal ISM (ISMMML) when the KIT mutation was detected (only) in BM MCs or in BM MCs and neutrophils/monocytes and/or T cells, respectively.4,20 In those ISM cases who carried the KIT D816V mutation (n = 313/322; 97%), a real-time allele-specific oligonucleotide PCR (ASO-qPCR) method was subsequently used to determine the KIT D816 VAF in BM (n = 313) and, whenever available, in PB (n = 96) using whole (BM and/or PB) gDNA, as described elsewhere.3,22
Targeted BM gDNA sequencing
A customized gDNA library was designed for targeted sequencing of a total of 15 genes (ASXL1, CDH11, DNMT3A, EPHA7, EZH2, ICK, IKZF1, ITGA10, KAT6B, PIK3CD, ROS1, RUNX1, SF3B1, SRSF2, and TET2) previously found to be recurrently mutated in SM patients based on a broader panel of up to 410 genes,15 with a global exon coverage >99% (ie, 219-bp gap distance for a total coverage of 58 305 bp). Briefly, 200 ng of whole BM gDNA per patient was used for the preparation of individual libraries with a TruSeq Custom Amplicon Low Input Kit (Illumina, San Diego, CA), and gDNA was sequenced at a 2 × 150-bp read length on a HiSeq 2500 genome sequencer (Illumina), following the manufacturer’s instructions. In addition, the CBL, KRAS, and JAK2 genes were sequenced in a subset of 107 of 200 (53%) patients in the discovery cohort and, for the CBL gene only, in all 122 patients in the validation cohort.
Sequencing resulted in an average of 645 407 paired-end filter reads per sample, with a mean coverage of 1660×. To align reads to the hg19 human reference sequence, the Smith-Waterman algorithm was used.23 For variant calling, an Illumina Somatic Variant Caller/Genome Analysis Toolkit (GATK) was used, and filtering was manually conducted as previously described15 : (1) only those nonsynonymous coding genetic variants identified with ≥1% allele coverage were initially selected, (2) the sequences obtained were subsequently compared with the 5000 exomes,24 the Iberian Populations in Spain 1000 Genomes Project,25 and the ExAC26 population databases to discriminate between somatic variants and single nucleotide polymorphisms, (3) from the remaining genetic variants, only those that had not been previously reported in Spanish, African, or European (non-Finnish) populations and/or those with a minor allele frequency < 0.1% were considered, and (4) potentially deleterious variants, as defined by the SIFT27 and PolyPhen28 algorithms, were finally selected. All genetic variants identified were subsequently confirmed by Sanger sequencing on gDNA extracted from BM and/or PB from the same patient (kindly provided by the Spanish National DNA Bank Carlos III, University of Salamanca, Salamanca, Spain; http://www.bancoadn.org). For a more stringent approach to pathogenicity, variants were classified as variants of unknown significance (VUS) or pathogenic variants, as described elsewhere.29,30
Statistical analyses
To assess the statistical significance (set at P < .05) of differences observed among groups, the Mann-Whitney U test was used. Receiver operating characteristic curve analysis was used to select the most sensitive and specific KIT-mutated BM and PB allele frequency threshold for the discrimination between ISMMC and ISMMML patients and to identify the most accurate cutoffs to predict for patient outcome for other variables (ie, age at diagnosis, hemoglobin levels, absolute neutrophil counts [ANCs], platelet counts, and VAF of the ASXL1/RUNX1/DNMT3A [A/R/D] genes). Pearson’s correlation coefficient (r) was used to measure the relationship between variables. PFS and OS curves were plotted from the time of diagnosis to disease progression (PFS), death (OS), or the last follow-up visit (PFS and OS) using the Kaplan-Meier method; the log-rank test was used to establish the statistical significance of differences between survival curves. For multivariate analyses, the covariate adjustment model31 was applied to those variables that showed a prognostic impact on PFS and OS in the univariate analysis and then the Cox proportional hazard regression model was used. All statistical analyses were performed with SPSS software (SPSS version 20.0; IBM Corporation, Armonk, NY).
Results
Gene mutational profile of ISM patients
Targeted next-generation sequencing revealed a total of 71 genetic variants in 55 of 322 ISM patients: 35 of 200 (18%) in the discovery cohort (range, 0-2 mutated genes per case) and 20 of 122 (16%) in the validation cohort (range, 0-4 gene variants per case) (Table 1; supplemental Table 1). These variants involved 15 of 18 genes tested (ASXL1, CBL, CDH11, DNMT3A, EPHA7, ITGA10, JAK2, KAT6B, KRAS, PIK3CD, ROS1, RUNX1, SF3B1, SRSF2, and TET2) (supplemental Table 2). With the exception of the KAT6B and CDH11 genes, which were mutated in a single ISM case, the other 13 genes were recurrently mutated in between 2 and 18 patients (median, 2 mutated cases per gene). Most variants identified (50/71; 70%) had been previously reported in the COSMIC database, whereas the remaining 21 of 71 variants (30%) are described here for the first time (supplemental Table 2). Of note, only 30 of 71 (42%) genetic variants other than KIT, involving 8 of 18 genes tested (ie, ASXL1, CBL, DNMT3A, ITGA10, JAK2, RUNX1, SRSF2, and TET2), were considered pathogenic mutations, affecting 23 of 55 (42%) patients (supplemental Table 2). With the exception of SRSF2 (0% vs 2%, P = .04) and ITGA10 (1% vs 5%, P = .03), similar mutational profiles were found in the discovery and validation cohorts (supplemental Table 1).
Of note, the 35 of 200 ISM patients (ie, from the discovery cohort) carrying ≥1 gene variant, in addition to mutated KIT (ie, multimutated ISM cases), showed more advanced age at diagnosis (median, 50 years vs 43 years among ISM patients with isolated KIT mutation; P = .003), together with a higher proportion of cases with increased (≥2.5 µg/mL) sβ2M levels (22% vs 6.5% cases, respectively; P = .006), disease progression (18% vs 4.8% cases, respectively; P = .008), and death (12% vs 0% cases, respectively; P < .001) (Table 2). This translated into shorter PFS (P < .001) and OS (P = .002) among multimutated ISM patients compared with patients with an isolated KIT mutation (n = 166) (Table 3). Interestingly, among the 35 multimutated patients, VAF analyses revealed that, in most cases (32/35, 91%), the KIT mutation was a secondary event to the other gene variant, whereas it could be considered to appear simultaneously or to be an earlier event in only 3 of 35 cases (patients #45, #134, and #193) (supplemental Table 2).
Pattern of involvement of BM hematopoiesis by the KIT mutation
Most ISM patients analyzed in the discovery cohort (191/200; 95.5%) carried the KIT D816V mutation in their BM MCs, whereas a few presented other variants at codon 816 of KIT: KIT D816Y in 6 cases (3%) and KIT D816H in another 3 cases (1.5%). In 94 cases (47%), involvement of BM hematopoietic cells other than MCs was confirmed in highly purified fluorescence activated cell sorting–sorted T cells, monocytes, and/or neutrophils, including 93 of 191 cases (49%) that had the KIT D816V mutation and 1 of 6 cases with the KIT D816Y mutation (Table 1). In the remaining 106 ISM patients (53%), KIT mutation was restricted to BM MCs (ISMMC), with or without involvement of CD34+ precursors (Table 1). Receiver operating characteristic curve analysis showed that, among ISM cases carrying the KIT D816V mutation (n = 191), KIT VAF ≥ 1% in BM and ≥6% in PB were the most discriminating cutoffs to define BM and PB multilineal KIT, with overall accuracies of 87% (sensitivity, 79%; specificity, 95%) and 71% (sensitivity, 32%; specificity, 98%), respectively. Interestingly, a BM KIT VAF ≥ 1% correlated significantly with a PB KIT VAF ≥ 6% (r = 0.5, P < .001), sβ2M ≥ 2.5 µg/mL (r = 0.4, P < .001), and, to a lesser extent, also sBT ≥ 200 µg/L (r = 0.3, P < .001) and the BM MC tumor burden evaluated by flow cytometry (r = 0.31, P < .001).
Of note, although the frequency of multimutated ISM cases was similar (P > .05) in ISMMC and ISMMML patients (16/35 [46%] vs 19/35 [54%]), with an overall similar frequency of mutated and nonmutated genes in both patient groups, a significant difference (P = .01) was observed between the groups for the presence of ≥1 pathogenic variant in genes other than KIT (3/14 [21%] vs 11/14 [79%]) (Table 1). In addition, the number of cases that had KIT mutations other than KIT D816V at codon 816 (ie, KIT D816H/Y) was significantly higher among ISMMC patients than ISMMML patients (7.5% vs 1%, respectively; P = .03) (Table 1).
From a clinical point of view, ISM patients with a BM KIT VAF ≥ 1% (vs <1% KIT BM VAF) showed a greater frequency of skin lesions (92% vs 68%, P < .001), hepatomegaly (12% vs 3.6%, P = .03), and splenomegaly (9.1% vs 1.8%, P = .02); elevated sβ2M (≥2.5 µg/mL in 28% vs 0% cases; P < .001), SAP (≥140 U/L in 8.6% vs 0% patients; P = .002), and sBT (≥200 µg/L in 17% vs 0.9% cases; P < .001) (Table 2); and a greater proportion of cases with KIT D816V+ MSCs (62% vs 0%, P < .001) and lower hemoglobin (145 g/L vs 138 g/L; P < .001) and IgE (29 KU/L vs 11.2 KU/L; P = .001) (Table 2). Similar (P > .05) ANCs and platelet counts were observed in the 2 ISM patient groups defined by the BM KIT D816V VAF cutoff ≥1% (Table 2).
Impact of the gene mutational profile on patient outcome
After a median follow-up of 9 years (range, 2-35), 14 of 200 ISM patients (7%) had progressed to more advanced forms of SM. This included progression from ISM to SSM in 5 cases (2.5%), to ASM in 7 cases (3.5%), and to SM-AHN in 2 patients (1%). Of note, all 14 cases that progressed from ISM to more advanced SM had a BM KIT VAF ≥ 1%, with the presence of the KIT D816V mutation in BM MSCs in all 9 cases tested (supplemental Table 3). During follow-up, 4 of 200 patients (2%) died of SM-related causes, after progression of ISM to ASM (n = 3) or SM-AHN (n = 1) (supplemental Table 3).
Univariate analysis of prognostic factors for PFS and OS revealed that pathogenic variants involving the A/R/D genes (particularly for VAF ≥ 30%), together with the presence of ≥1 pathogenic variant in any of the 8 of 18 genes other than KIT found to be mutated in our series and the presence of multilineal KIT, including BM KIT D816V VAF ≥ 1% and the presence of KIT D816V+ MSCs and PB KIT VAF ≥ 6%, were all associated with shorter PFS and OS (Table 3). Interestingly, the only patient (#149) harboring a pathogenic variant in the CBL gene also carried an ASXL1 pathogenic mutation (supplemental Table 3). Other clinical and biological parameters that showed a significant adverse impact on patient PFS in univariate analyses included age at diagnosis ≥55 years (P = .008), sβ2M ≥ 2.5 µg/mL (P < .001), SAP > 140 U/L (P = .001), and lactate dehydrogenase < 230 IU/L (P = .002); in turn, only sβ2M and SAP levels showed a significant impact (P < .001) on patient OS (Table 3). Of note, among 14 multimutated ISM patients who carried pathogenic variants in genes other than KIT (Table 3), cases with mutations in the A/R/D genes at VAF ≥ 30% (n = 8) displayed significantly higher sBT levels (131 vs 26.2 µg/L, P = .01) and disease progression (to SSM or advSM) rates (75% vs 0%, P = .001) compared with the other mutated cases (n = 6).
Multivariate analysis of prognostic factors (Table 4), based on those parameters that showed a significant impact on patient outcome in the univariate analysis, revealed that a BM KIT D816V allele frequency ≥ 1% (hazard ratio [HR], 10.1; 95% confidence interval [CI], 1.1-90; P = .02), together with increased (≥2.5 µg/mL) sβ2M levels (HR, 9.8; 95% CI, 2.6-36; P < .001) and the presence of pathogenic variants within A/R/D genes with VAF ≥ 30% (HR, 4.2; 95% CI, 1.2-10; P = .02), was the best combination of independent predictors for PFS (P < .001) (Table 4; Figure 1A-B). In turn, the presence of A/R/D pathogenic variants at VAF ≥ 30% (HR: 51.8; 95% CI: 4.1-647) was the only independent predictor for OS (P = .002) (Table 4; Figure 1B-C).
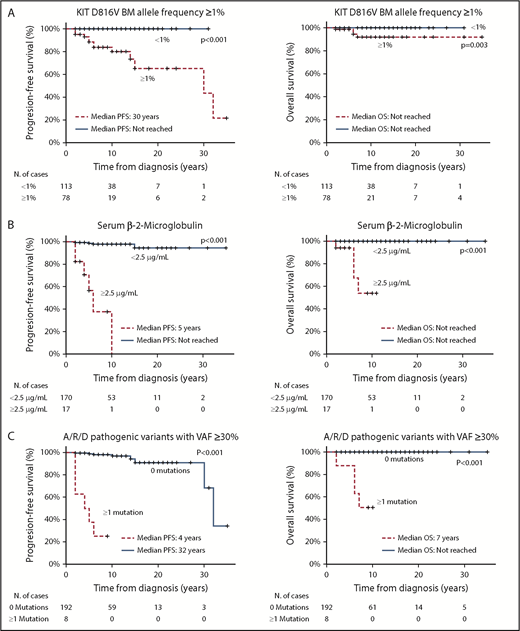
PFS and OS curves of the variables with independent prognostic value in the multivariate analysis. PFS (left panels) and OS (right panels) curves of ISM patients grouped according to the KIT D816V allele frequency in the BM (A), sβ2M levels (B), and the presence vs absence of pathogenic variants in the A/R/D gene panel with VAFs ≥ 30% (C).
PFS and OS curves of the variables with independent prognostic value in the multivariate analysis. PFS (left panels) and OS (right panels) curves of ISM patients grouped according to the KIT D816V allele frequency in the BM (A), sβ2M levels (B), and the presence vs absence of pathogenic variants in the A/R/D gene panel with VAFs ≥ 30% (C).
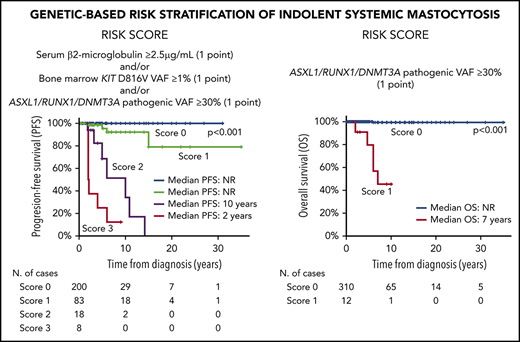
According to the above results, a scoring system was constructed to predict PFS for ISM patients based on the presence of a BM KIT D816V VAF ≥ 1% (1 point), and/or increased (≥2.5 µg/mL) sβ2M levels (1 point), and/or the presence of pathogenic variants in A/R/D genes at VAF ≥ 30% (1 point): score 0, 119 patients (64%); score 1, 51 cases (27%); score 2, 11 cases (6%); and, score 3, 6 cases (3%). As expected, the 17 patients who showed coexistence of ≥2 adverse features (scores 2 and 3) had a significantly (P < .001, for scores 2 and 3) poorer PFS (median of 10 and 2 years, respectively) than did those with lower (≤1) scores (median PFS not reached) (Figure 2A). Similarly, a scoring system based on the presence of pathogenic variants in the ASXL1, RUNX1, and/or DNMT3A genes (A/R/D gene panel) at VAF ≥ 30% (1 point) also proved to be a strong predictor (P < .001) for OS among ISM patients, with median OS rates of 7 years vs not reached for cases with a score 1 vs a score 0, respectively (Figure 2D). When these 2 scoring systems were applied to the validation cohort of ISM patients (Figure 2B-E) and the whole ISM patient series (n = 322), similar results (Figure 2C-F) were observed with regard to PFS (P < .001 and P < .001, respectively) and OS (P = .04 and P < .001, respectively).
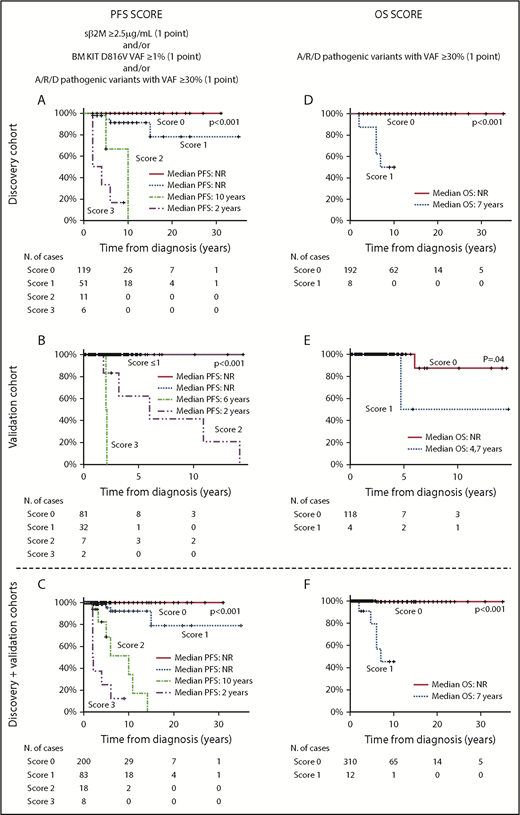
PFS and OS curves of ISM patients, grouped according to the proposed scoring systems, for the discovery and/or the validation patient cohorts. Scoring systems for PFS (A,C,E) and OS (B,D,F) are based on (1) the combination of sβ2M levels at diagnosis (score 1 when ≥ 2.5 µg/mL), and/or the KIT D816V VAF in BM (score 1 when ≥1%), and/or pathogenic variants in the A/R/D gene panel with VAF ≥ 30% (score 1 when ≥1 mutation with VAF ≥ 30% in any of these 3 genes) (A,C,E) and (2) pathogenic variants in the A/R/D gene panel with VAF ≥ 30% (score 1 if there was a mutation in any of these 3 genes with VAF ≥ 30%) (B,D,F). Please note that all patients who scored 1 in (A) had multilineage involvement of BM hematopoiesis by the KIT mutation with sβ2M levels < 2.5 µg/mL. NR, not reached.
PFS and OS curves of ISM patients, grouped according to the proposed scoring systems, for the discovery and/or the validation patient cohorts. Scoring systems for PFS (A,C,E) and OS (B,D,F) are based on (1) the combination of sβ2M levels at diagnosis (score 1 when ≥ 2.5 µg/mL), and/or the KIT D816V VAF in BM (score 1 when ≥1%), and/or pathogenic variants in the A/R/D gene panel with VAF ≥ 30% (score 1 when ≥1 mutation with VAF ≥ 30% in any of these 3 genes) (A,C,E) and (2) pathogenic variants in the A/R/D gene panel with VAF ≥ 30% (score 1 if there was a mutation in any of these 3 genes with VAF ≥ 30%) (B,D,F). Please note that all patients who scored 1 in (A) had multilineage involvement of BM hematopoiesis by the KIT mutation with sβ2M levels < 2.5 µg/mL. NR, not reached.
Discussion
The activating KIT D816V mutation is a hallmark of SM20,32 ; it is present in ≥90% of patients when sensitive molecular approaches are used.3,22 Despite the high frequency of KIT D816V in nonadvanced SM and advSM,20 on its own, this mutation cannot explain the progression of ISM to SSM and advSM or a distinct patient survival. However, acquisition of the KIT D816V mutation in MSCs and uncommitted hematopoietic precursor cells,33 in association with multilineal involvement of BM hematopoiesis by KIT, has been recurrently associated with a poorer outcome in ISM patients.4 At the same time, it represents a hallmark of ASM and ASM-AHN cases, where it is typically observed in association with (additional) mutations in genes other than KIT; in this regard, some specific mutational profiles have been recurrently shown to confer an adverse outcome in SM.12-16,34 Altogether, these findings suggest that multilineal KIT, together with mutations in genes other than KIT, might provide the genetic background required for disease progression and death. However, there are still limited data about the frequency and potential prognostic impact on ISM of mutations in genes other than KIT that are associated with advSM, and its potential relationship with multilineal KIT also remains to be investigated. Thus, of the 18 genes reported to be recurrently mutated in advSM and analyzed here, only 11 (ASXL1, CBL, DNMT3A, EZH2, IKZF1, JAK2, KRAS, SF3B1, SRSF2, RUNX1, TET2) have been investigated in >5 ISM cases/series, for a total of 81 ISM patients analyzed in 4 distinct series13-16 ; no data are available about the relationship between the frequency and prognostic impact of mutations within these genes and its association with multilineal KIT.
We investigated, for the first time, the frequency and prognostic impact of different genetic variants in 18 genes, other than KIT, in a large series of 200 ISM patients studied at diagnosis and followed for a median of 9 years; results were subsequently validated in an additional cohort of 122 ISM patients. Remarkably, all of the ISM patients who progressed to SSM or advSM (and eventually died) showed “high-risk” ISM15,33 features at diagnosis (ie, ISM with BM KIT VAF ≥ 1%, D816V+ MSCs, and high serum baseline tryptase levels), but they did not meet the WHO diagnostic criteria for SSM or advSM (ie, <2 B-findings and no C-findings).18
In line with previous data reported in the literature in smaller series, showing that 16% (mean) of cases are mutated (range, 11-40),13-17 17% of our ISM patients had multiple mutations (ie, ≥1 mutation in addition to KIT D816). Of note, this frequency is significantly lower than that reported previously for SSM patients (3/6 cases; 50%) and advSM patients, including ASM (22/39 cases; 56%), SM-AHN (131/184 cases; 71%), and MCL (23/36 cases; 64%), investigated for a variable number of genes (range, 7-410),12-16,34 including the 18 genes analyzed here.
Similarly to ISM patients with a BM KIT VAF ≥ 1%, multimutated ISM cases showed more adverse prognostic features than ISM patients with an isolated KIT mutation (eg, elevated sβ2M and SAP levels), in association with significantly shorter PFS and OS rates. In line with these findings, a significantly different overall frequency of multimutated cases, as well as of patients carrying pathogenic variants in genes other than KIT, was found among ISM patients with (vs without) BM KIT VAF ≥ 1%. Altogether, these results confirm and extend previous observations from our group and from other investigators with regard to the impact of multilineal KIT on PFS of ISM cases and of pathogenic variants in genes other than KIT on OS of advSM patients.4,12,13,15,16,35 In line with this, as well as with previous findings in advSM,35 the comparison between VAF of KIT and other mutated genes suggests that the KIT mutation might be a secondary event in the great majority of multimutated patients.
Of the 18 genes investigated here, the most frequently mutated genes were ASXL1 and DNMT3A. ASXL1 has frequently been reported to be mutated in advSM, in association with a shorter OS12,17 ; in contrast, despite the fact that the DNMT3A gene has frequently been found to be mutated in SM14-16 and other non-MC myeloid neoplasias,36,37 controversial results have been reported in the literature with regard to the impact of DNMT3A genetic variants on OS of patients with advSM and other non-MC myeloid neoplasias.14-16,36,37 Here, the presence of pathogenic variants in ≥1 of these 2 genes, and/or RUNX1 gene mutations (A/R/D gene panel), had a strong adverse prognostic impact on our ISM patients. These results confirm the adverse prognostic impact of the ASXL1 and RUNX1 genes on advSM,12,15,16 as well as among ISM cases. In contrast, none of our patients from the discovery cohort had SRSF2 and EZH2 genetic variants, suggesting that these adverse genetic markers might emerge at later stages of disease, after advSM is already established (or just prior to disease progression). Interestingly, SRSF2 was found to be mutated in only 2 patients from the validation cohort who progressed to ASM and SM-AHN (supplemental Table 4) and in whom the SRSF2 mutations appeared to be a secondary event to other deleterious variants in genes other than KIT (TET2 and DNMT3A), with VAF of 34% vs 48% and 20% vs 53%, respectively (supplemental Table 2). In line with this hypothesis, SRSF2 and EZH2 gene mutations have been reported in SM-AHN patients12,13,15,16 and, to a lesser extent, in MCL13 and ASM patients,13,16 whereas none of the 81 ISM cases investigated previously in the literature carried an SRSF2 and/or EZH2 gene mutation.
Interestingly, when BM VAF ≥ 30% was established as a cutoff for the A/R/D pathogenic variants, a slight increase in their discriminatory prognostic power for PFS and OS was observed, because all ISM patients that progressed and died had pathogenic variants in A/R/D genes, with high (≥30%) VAF for the mutated genes (supplemental Tables 1 and 2).
Despite all of the above data, multivariate analysis of prognostic factors for PFS and OS showed that increased sβ2M levels, together with KIT VAF ≥ 1% in BM and the presence of pathogenic A/R/D gene VAF ≥ 30%, were the only variables that retained an independent prognostic value for PFS, with the pathogenic A/R/D mutations with VAF ≥ 30% also being the only independent predictor for OS in ISM patients. Based on these 3 variables, 2 scoring systems were constructed that resulted in patient groups with significantly different outcomes in terms of PFS and OS. Thus, these included a large group of patients with high PFS and OS rates (>90%) at 10 years, together with a small subgroup of ISM patients with significantly higher risk of undergoing disease progression and death at 10 years (100% and 50%, respectively). Altogether, these results are fully in line with previous observations from our group in an independent cohort of 145 ISM patients in whom the mutational status for genes other than KIT had not been investigated4 ; in addition, they also show for the first time that detection of multilineal KIT based on laborious analysis of the KIT mutation in (multiple) purified BM cell populations might be replaced by a much simpler, faster, and easy-to-standardize quantitative ASO-qPCR approach to evaluate the KIT VAF in whole-BM samples.22 This change in KIT detection procedure is also probably supported by the fact that assessment of the KIT VAF in BM provides information about the degree of involvement of BM hematopoiesis by the KIT mutation and the overall MC tumor burden, as supported also by the significant, but marginal, correlation observed here and in previous studies between BM KIT VAF ≥ 1% and sBT levels and BM MC tumor burden evaluated by flow cytometry and histopathology.38
Interestingly, we confirm the independent prognostic impact of sβ2M levels in ISM.4 High sβ2M has also been associated with a poorer outcome in many hematological malignancies other than ISM, such as multiple myeloma, mantle cell lymphoma, diffuse large B-cell lymphoma, and non-Hodgkin lymphoma, in general,39-43 whereas it remains to be specifically investigated in advSM. The precise source of sβ2M in ISM (as well in other hematological tumors) still remains to be fully identified. This is primarily due to the fact that β2M is constitutively produced and exposed on the surface membrane of nearly all nucleated cells in association with the HLA class Iα molecule, from which it is released to the serum in a soluble form.44 Previous studies have shown that sβ2M levels increase in parallel with a greater cell turnover40 and/or during an immune response (eg, following native antigen interactions with the α1 and α2 HLA type I domains) and subsequent disruption of HLA-Iα binding to the β2-microglobulin molecule.45
Unlike in previous studies,4,12 we could not confirm the independent prognostic value of high SAP levels for OS, which was replaced by the A/R/D pathogenic variants with VAF ≥ 30%. In this regard, it should be noted that only a small number of all ISM patients showed high sβ2M levels at diagnosis and A/R/D pathogenic variants with VAF ≥ 30%, with all but 1 dying in the first 6 years following diagnosis of ISM, after they had already undergone disease progression. Interestingly, all such patients also had multilineal KIT with BM VAF ≥ 1%, thereby representing a very small subgroup of ISM patients with a particularly adverse outcome. However, because of the low number of deaths observed within our ISM cohort, our results regarding OS (despite confirmation in a validation cohort) should be taken with caution; thus, further studies in other independent and larger ISM patient cohorts, with longer follow-up, are required to confirm these still-preliminary findings. Altogether, our results indicate that BM KIT VAF and sβ2M levels must be evaluated in every newly diagnosed ISM patient, for early risk stratification and identification of the few ISM cases who are at high risk for disease progression and who might benefit from a closer follow-up and, eventually, also (early) cytoreductive therapy. In addition, testing for the A/R/D pathogenic mutations and their VAF should be mandatory in ISM patients, either at diagnosis or just after they progress to SSM or advSM, to identify those cases at higher risk for death.
Data will be published in COSMIC database.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the Instituto de Salud Carlos III (ISCIII) and Fondos Europeos de Desarrollo Regional (FEDER) (PI16/00642, CIBERONC, CB16/12/00400); Consejería de Educación, Junta de Castilla y León (SA013U16, FEDER); Asociación Española de Mastocitosis y enfermedades relacionadas (AEDM 2017); and Fondos de Investigación para Enfermedades Raras del Ministerio de Sanidad, Servicios Sociales e Igualdad. M.J.-A. was supported by Ministerio de Economia y Competitividad (PTA-2016)-Universidad de Salamanca. C.P. was supported by a Personal Técnico de Apoyo (PTA) fellowship (PTA2015-10483-I) from the Spanish Ministry of Economy, Industry and Competitiveness. A.M was supported by CIBERONC (ISCIII). The Spanish National DNA Bank Carlos III was supported by ISCIII and fondos FEDER (PT13/0001/0037 and PT17/0015/0044).
Authorship
Contribution: J.I.M.-G. performed experiments, analyzed the data, and wrote the manuscript; M.J.-A. and I.A.-T. supported the statistical analyses, collected data, and reviewed the manuscript; E.V., C.C., C.P., and A. Mayado analyzed data and reviewed the manuscript; A.H. provided patients’ clinical data and reviewed the manuscript; L.S.-M., A. Matito, and N.D.-F. reviewed the manuscript; and A.O. and A.C.G.-M. designed the study, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alberto Orfao, Centro de Investigación del Cáncer, Campus Miguel de Unamuno, Paseo de la Universidad de Coimbra, 37007 Salamanca, Spain; e-mail: orfao@usal.es; and Andrés C. García-Montero, Edificio Multiusos I+D+i, C/Espejos, 37007 Salamanca, Spain; e-mail: angarmon@usal.es.