

TO THE EDITOR:
β-thalassemia (BT) is a global health problem affecting millions of patients, and current therapies to treat BT present major clinical challenges.1,2 Luspatercept (ACE-536), an erythroid maturation agent that functions independently of the erythropoietin (EPO) pathway,3 has been shown to improve anemia in BT.4,5 In clinical trials, treatment with ACE-536 increased hemoglobin (Hb) levels and significantly reduced red blood cell (RBC) transfusions in adults with BT, with little to no adverse effects.4,5 ACE-536 is a fusion protein composed of a modified extracellular domain of activin receptor IIB (ACVR2B) and the Fc part of human immunoglobulin G1. This fusion protein competes with ACVR2B to bind members of the transforming growth factor-β (TGF-β) superfamily.6 Studies in murine models of BT using the murine analog of ACE-536 (RAP-536) show that RAP-536 targets the protein growth differentiation factor 11 (GDF11).3,7,8 These studies have proposed that overexpression of GDF11 blocks terminal erythroid differentiation by increasing oxidative stress and that treatment with the ligand trap ACE/RAP-536 sequesters GDF11, unblocking terminal erythroid differentiation and, thereby, ameliorating ineffective erythropoiesis. However, ACE-536 and RAP-536 have been shown to stimulate RBC synthesis in healthy humans and mice, where GDF11 overexpression has not been reported.5,9 Because of the incongruency of the proposed model, our study resorted to genetic tools to reduce GDF11. We investigated whether deletion of the Gdf11 gene improved anemia in a BT mouse model (Hbbth3/+).10 We also examined whether RAP-536 was efficacious in the absence of Gdf11.
Gdf11 was functionally inactivated in the entire hematopoietic compartment of mice (Gdf11Δ2-3/Δ2-3) by crossing VavCre-transgenic animals11 with Hbb+/+ and Hbbth3/+ LoxP flanked Gdf11 (Gdf11flox/flox) mice12 to produce Hbb+/+VavCreGdf11Δ2-3/Δ2-3 and Hbbth3/+VavCreGdf11Δ2-3/Δ2-3 mice (supplemental Figure 1, available on the Blood Web site). The resulting progeny were viable, and complete blood count (CBC) results did not show changes in RBC, Hb, hematocrit (Hct), or reticulocyte levels (Figure 1A-H). Gdf11 recombination in the spleens of Hbbth3/+VavCreGdf11Δ2-3/Δ2-3 mice was confirmed by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) (Figure 1Q). Similarly, crossing EpoRCre-transgenic animals13 with Gdf11flox/flox mice, in which Gdf11 was reduced in the early erythroid progenitor, did not result in changes to the hematological profile (supplemental Figure 2).

Conditional deletion of Gdf11 in the entire hematopoietic compartment or pancellularly in thalassemic (Hbbth3/+) mice does not improve hematological parameters. (A-D) Conditional deletion of Gdf11 in the entire hematopoietic compartment of thalassemic mice (Hbbth3/+VavCreGdf11Δ2-3/Δ2-3) (n = 9) does not result in any differences in RBC count, Hb, Hct, or reticulocytes compared with Hbbth3/+ controls (n = 15). (E-H) Conditional deletion of Gdf11 in erythroid cells of nonthalassemic wild-type animals (Hbb+/+) does not result in altered hematopoietic parameters in Hbb+/+VavCreGdf11Δ2-3/Δ2-3 mice (n = 23) compared with Hbb+/+ controls (n = 22). CBCs were analyzed at 2 months of age. Ubiquitous deletion of Gdf11 in Hbbth3/+RosaCreGdf1flox/flox mice treated with TAM (Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3) does not improve hematological parameters. (I-L) Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 9) did not show increases in RBC, Hb, or Hct or lower reticulocyte counts compared with Hbbth3/+Gdf11flox/flox control mice (n = 7). (M-P) No hematological differences were detectable in Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 17) compared with Hbb+/+Gdf11flox/flox controls (n = 15). RosaCreGdf11Δ2-3/Δ2-3 mice and Gdf11flox/flox controls were analyzed between 3 and 6 months of age. CBCs were analyzed 2 weeks post-TAM administration. Females and males were included in the analysis. (Q) Messenger RNA analysis of Gdf11Δ2-3/Δ2-3 mice confirms reduction of Gdf11 in spleens. Messenger RNA from Hbbth3/+VavCreGdf11Δ2-3/Δ2-3 (n = 3) and Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 (n = 3) age-matched males (5 months old) was assessed for Gdf11 reduction in the spleen, by qRT-PCR, using a Gdf11 probe specific for exon 2. Both show significant reductions normalized by Hprt (left panel). Gdf8 was undetectable in all samples tested. No statistical differences were found in the Gypa positive control (right panel). Data are mean ± standard deviation. *P ≤ .05, Student t test.
Conditional deletion of Gdf11 in the entire hematopoietic compartment or pancellularly in thalassemic (Hbbth3/+) mice does not improve hematological parameters. (A-D) Conditional deletion of Gdf11 in the entire hematopoietic compartment of thalassemic mice (Hbbth3/+VavCreGdf11Δ2-3/Δ2-3) (n = 9) does not result in any differences in RBC count, Hb, Hct, or reticulocytes compared with Hbbth3/+ controls (n = 15). (E-H) Conditional deletion of Gdf11 in erythroid cells of nonthalassemic wild-type animals (Hbb+/+) does not result in altered hematopoietic parameters in Hbb+/+VavCreGdf11Δ2-3/Δ2-3 mice (n = 23) compared with Hbb+/+ controls (n = 22). CBCs were analyzed at 2 months of age. Ubiquitous deletion of Gdf11 in Hbbth3/+RosaCreGdf1flox/flox mice treated with TAM (Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3) does not improve hematological parameters. (I-L) Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 9) did not show increases in RBC, Hb, or Hct or lower reticulocyte counts compared with Hbbth3/+Gdf11flox/flox control mice (n = 7). (M-P) No hematological differences were detectable in Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 17) compared with Hbb+/+Gdf11flox/flox controls (n = 15). RosaCreGdf11Δ2-3/Δ2-3 mice and Gdf11flox/flox controls were analyzed between 3 and 6 months of age. CBCs were analyzed 2 weeks post-TAM administration. Females and males were included in the analysis. (Q) Messenger RNA analysis of Gdf11Δ2-3/Δ2-3 mice confirms reduction of Gdf11 in spleens. Messenger RNA from Hbbth3/+VavCreGdf11Δ2-3/Δ2-3 (n = 3) and Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 (n = 3) age-matched males (5 months old) was assessed for Gdf11 reduction in the spleen, by qRT-PCR, using a Gdf11 probe specific for exon 2. Both show significant reductions normalized by Hprt (left panel). Gdf8 was undetectable in all samples tested. No statistical differences were found in the Gypa positive control (right panel). Data are mean ± standard deviation. *P ≤ .05, Student t test.
We then tested whether GDF11 produced by nonhematopoietic tissues indirectly influenced erythropoiesis. Because Gdf11-null mice (Gdf11−/−) are embryonically lethal,12 we generated mice with a pancellular deletion of Gdf11 using the tamoxifen (TAM)-inducible RosaCre strain14 and generating Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 and Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice (supplemental Figure 3). Neither Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 mice nor Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice showed any alterations in RBC, Hb, Hct, or reticulocyte levels 2 weeks (Figure 1I-P), 5 weeks (supplemental Figure 5A-H), or 5 to 6 months (supplemental Figure 5I-P) post-TAM treatment. We confirmed Gdf11 recombination by qRT-PCR in the spleens of Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice (Figure 1Q) and by polymerase chain reaction in the spleen, liver, heart, duodenum, kidney, and bone marrow of Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 and Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 animals (supplemental Figure 4). These findings were consistent in females (supplemental Figure 6) and males (supplemental Figure 7). No differences were detected in the spleen-to-body weight ratios of VavCreGdf11Δ2-3/Δ2-3 or RosaCreGdf11Δ2-3/Δ2-3 mice compared with controls (supplemental Figure 8). GDF11 has also been proposed to exert a negative effect on late-stage erythropoiesis in myelodysplastic syndromes, a heterogeneous group of clonal hematopoietic disorders.3,15 To test this, we crossed NUP98-HOXD1316 (NHD13), a mouse model that recapitulates all key features of myelodysplastic syndromes, with VavCreGdf11Δ2-3/Δ2-3 mice. Results show that genetic removal of Gdf11 from all hematopoietic lineages in NHD13 mice did not confer a therapeutic benefit (supplemental Figure 9).
We then evaluated whether the effect of RAP-536 is mediated by the synthesis of Gdf11 from erythroid or nonerythroid cells. We administered 12 doses of RAP-536 to Hbb+/+ and Hbbth3/+ mice of the VavCreGdf11Δ2-3/Δ2-3- and RosaCreGdf11Δ2-3/Δ2-3-derived lines. In all mice treated, RAP-536 significantly increased RBC, Hb, and Hct parameters; additionally, reticulocyte counts were normalized in Hbbth3/+ mice (Figure 2A-P). Thus, lack of Gdf11 did not prevent responsiveness to RAP-536. Because activin receptor ligand traps have been shown to stimulate RBC synthesis and Hb increases in healthy humans,5,17 we investigated whether CD34+ cells respond to RAP-536 treatment in vitro. CD34+ were differentiated and expanded in a 3-phase liquid erythroid-specific differentiation medium containing RAP-536.18 Cell counts were conducted at the end of an 8-day differentiation assay to determine whether cells cultured in RAP-536 produced more mature cells. No differences were observed in the number of hemoglobinized cells after treatment with 5 µg/mL or 150 µg/mL RAP-536 (Figure 2Q).
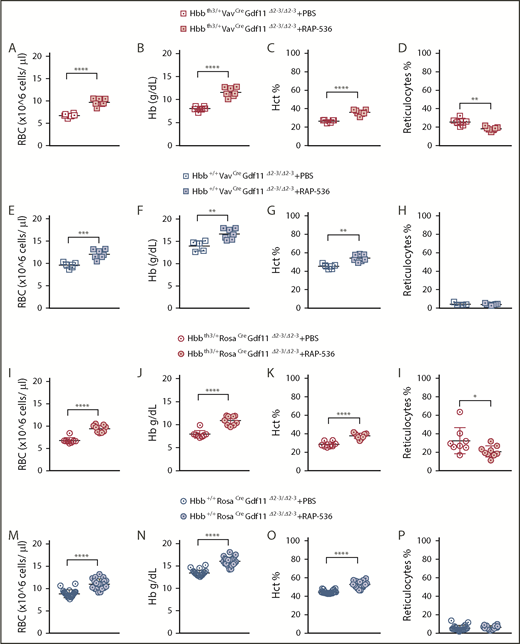

Gdf11 deletion from the hematopoietic compartment or pancellularly from all tissues did not suspend RAP-536 action.Hbbth3/+VavCreGdf11Δ2-3/Δ2-3 mice treated with RAP-536 (n = 7) showed increased RBCs (A), Hb (B), and Hct (C), as well as reduced reticulocytes (D), compared with phosphate-buffered saline (PBS)-treated controls (n = 6). Hbb+/+VavCreGdf11Δ2-3/Δ2-3 mice also exhibited increased RBCs (E), Hb (F), and Hct (G). No statistically significant difference was found in reticulocyte number (H). VavCreGdf11Δ2-3/Δ2-3 mice were treated with RAP-536 between 3 and 4 months. Similarly, Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 9) and Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 20) treated with RAP-536 exhibited increased RBC (I,M), Hb (J,N), and Hct (K,O) levels compared with Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 (n = 8) and Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 (n = 20) PBS-treated animals. (L) Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice showed a significant reduction in reticulocytes. (P) No statistical differences were observed in reticulocytes from Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 animals. RosaCreGdf11Δ2-3/Δ2-3 animals were treated with RAP-536 between 4 and 7 months. CBCs were assessed 2 days after last dose of RAP-536 treatment. Females and males were included in the analysis of all groups. CD34+ cells did not respond to RAP-536 in vitro. (Q) Treatment of CD34+-derived cells isolated from healthy donors with 5 µg/mL or 150 µg/mL RAP-536 (n = 3) did not result in increased cell number at the end of erythroid differentiation assay day 8 (DD8). mRNA levels of GDF11 and ACVR2B were low in human thalassemia and healthy erythroid progenitor cells. Quantification of GDF11 and ACVR2B mRNA in thalassemia (R) and healthy (S) donor-derived erythroblasts showed low GDF11 and ACVR2B expression relative to HPRT after day 1 (DD1) and day 4 (DD4) of differentiation in an erythroid differentiation assay, as determined by qRT-PCR (n = 3). BT CD34+ cells showed higher relative levels of GDF11 compared with healthy CD34+ cells. RHO was used as a negative control (data not shown), and TFRC and GYPA were used as positive controls. Gdf11 and Acvr2b mRNA was expressed at low levels in splenic Ter119+ cells isolated from PBS- and RAP-536–treated Hbbth3/+ mice. (T) Erythroid cells, isolated from the spleens of PBS-treated Hbbth3/+ mice and RAP-536–treated mice, were analyzed for Cd71 and Ter119 marker expression before mRNA extraction. (U) mRNA from Ter119+ cells isolated from the spleens of Hbbth3/+ PBS- and RAP-536–treated mice was analyzed for Gdf11 and Acvr2b by qRT-PCR; results show low expression relative to Hprt compared with Tfrc and Gypa in either group. Rho was used as a negative control (data not shown). Data are mean ± standard deviation. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, Student t test.
Gdf11 deletion from the hematopoietic compartment or pancellularly from all tissues did not suspend RAP-536 action.Hbbth3/+VavCreGdf11Δ2-3/Δ2-3 mice treated with RAP-536 (n = 7) showed increased RBCs (A), Hb (B), and Hct (C), as well as reduced reticulocytes (D), compared with phosphate-buffered saline (PBS)-treated controls (n = 6). Hbb+/+VavCreGdf11Δ2-3/Δ2-3 mice also exhibited increased RBCs (E), Hb (F), and Hct (G). No statistically significant difference was found in reticulocyte number (H). VavCreGdf11Δ2-3/Δ2-3 mice were treated with RAP-536 between 3 and 4 months. Similarly, Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 9) and Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 mice (n = 20) treated with RAP-536 exhibited increased RBC (I,M), Hb (J,N), and Hct (K,O) levels compared with Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 (n = 8) and Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 (n = 20) PBS-treated animals. (L) Hbbth3/+RosaCreGdf11Δ2-3/Δ2-3 mice showed a significant reduction in reticulocytes. (P) No statistical differences were observed in reticulocytes from Hbb+/+RosaCreGdf11Δ2-3/Δ2-3 animals. RosaCreGdf11Δ2-3/Δ2-3 animals were treated with RAP-536 between 4 and 7 months. CBCs were assessed 2 days after last dose of RAP-536 treatment. Females and males were included in the analysis of all groups. CD34+ cells did not respond to RAP-536 in vitro. (Q) Treatment of CD34+-derived cells isolated from healthy donors with 5 µg/mL or 150 µg/mL RAP-536 (n = 3) did not result in increased cell number at the end of erythroid differentiation assay day 8 (DD8). mRNA levels of GDF11 and ACVR2B were low in human thalassemia and healthy erythroid progenitor cells. Quantification of GDF11 and ACVR2B mRNA in thalassemia (R) and healthy (S) donor-derived erythroblasts showed low GDF11 and ACVR2B expression relative to HPRT after day 1 (DD1) and day 4 (DD4) of differentiation in an erythroid differentiation assay, as determined by qRT-PCR (n = 3). BT CD34+ cells showed higher relative levels of GDF11 compared with healthy CD34+ cells. RHO was used as a negative control (data not shown), and TFRC and GYPA were used as positive controls. Gdf11 and Acvr2b mRNA was expressed at low levels in splenic Ter119+ cells isolated from PBS- and RAP-536–treated Hbbth3/+ mice. (T) Erythroid cells, isolated from the spleens of PBS-treated Hbbth3/+ mice and RAP-536–treated mice, were analyzed for Cd71 and Ter119 marker expression before mRNA extraction. (U) mRNA from Ter119+ cells isolated from the spleens of Hbbth3/+ PBS- and RAP-536–treated mice was analyzed for Gdf11 and Acvr2b by qRT-PCR; results show low expression relative to Hprt compared with Tfrc and Gypa in either group. Rho was used as a negative control (data not shown). Data are mean ± standard deviation. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, Student t test.
Next, we investigated messenger RNA (mRNA) expression levels of GDF11 and ACVR2B in early erythroid progenitors derived from healthy and BT donor CD34+ cells. Because GDF11 has been proposed to inhibit early erythroid progenitors from differentiating, cells were cultured as previously mentioned18 and collected for mRNA early in the differentiation phases: 24 hours later and 4 days later. qRT-PCR results showed low levels of GDF11 and ACVR2B in CD34+ cells from BT (Figure 2R) and healthy (Figure 2S) donors compared with TFRC and GYPA controls (supplemental Figure 10B for probes used). Similarly, Ter119+ erythroid progenitors isolated from Hbbth3/+ spleens (Figure 2T; supplemental Figure 10A) showed low expression of Gdf11 and Acvr2b in phosphate-buffered saline– and RAP-536–treated mice (Figure 2U). However, in RAP-536–treated mice, Tfrc and Gypa levels were significantly different compared with phosphate-buffered saline controls, which is consistent with a reduction in the erythroid progenitor pool and an increase in RBC differentiation.
The EPO-independent ability of RAP-536 to increase RBC number and Hb content offers the potential for understanding an undiscovered pathway in erythropoiesis. Studies have identified GDF11 as the primary target of RAP-536.3,7,8 However, evidence for overexpression of GDF11 was obtained using nonspecific antibodies for GDF1119 in BT erythroid cells7 and does not explain how RAP-536 is also effective in Hbb+/+ mice, in which GDF11 overexpression in erythroid cells has not been observed. RAP-536 has also been reported to bind other members of the TGF-β family, including GDF8 and activin B.3,20 Based on these observations, analyses were performed in Hbb+/+ mice using antibodies against activin B, GDF8, or GDF8/11.20 These studies indicate that the antibodies had modest effects on RBC synthesis, and none could recapitulate the effect of RAP-536.20 Here, we show that lack of Gdf11 does not improve anemia in Hbbth3/+ mice or increase hemoglobin in Hbb+/+ mice. In accordance with these findings, Hbbth3/+ and Hbb+/+ animals, with a deletion in the hematopoietic compartments and a pancellular deletion of Gdf11, respond to RAP-536. In addition, we show that Gdf11 and Acvr2b expression is low in a BT mouse model and in human erythroid cells. Altogether, our studies strongly indicate that decreasing GDF11 in erythroid cells has no effect on anemia. Lastly, GDF11 has been reported to have protective properties on cardiovascular health; patients with decreased levels of GDF11 had increased incidences of heart failure, stroke, and death.21 Animal studies have also implicated GDF11 as important in neuronal vascularization and skeletal muscle development.22-24 Targeting GDF11 as an agent to increase hematological parameters could compromise cardiovascular protection or affect other important pathways in development. Future work will focus on the role of the TGF-β superfamily in erythropoiesis and on identifying the targets responsible for the therapeutic effects produced by RAP-536, because the mechanism for RAP-536 remains to be elucidated.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Acceleron Pharma for providing RAP-536.
This work was supported by National Institutes of Health, T32 Kirschstein National Research Service Award 5T32HL007439-39 for funding and training of A.G., and by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease grants R01 DK90554 and R01 DK095112 (S.R.).
Authorship
Contribution: A.G., P.R.O., L.B., and S.R. designed experiments, analyzed and interpreted data, and wrote the manuscript; A.G., P.R.O., C.R.H., S.S., J.Z., V.L.P., C.C., P.L., and A.C.M. performed experiments and collected data; and S.S., A.C.M., L.B., A.K.S., and M.F. edited the manuscript.
Conflict-of-interest disclosure: S.R. is a member of the scientific advisory board for Ionis Pharmaceuticals and Meira GTx, has acted as a consultant for Disc Medicine, Protagonist, and La Jolla Pharmaceutical Company, and is a coinventor for patents US8058061 B2 C12N 20111115 and US7541179 B2 C12N 20090602. The consulting work and intellectual property of S.R. did not in any way affect the design, conduct, or reporting of this research. The remaining authors declare no competing financial interests.
Correspondence: Stefano Rivella, Department of Pediatrics, Division of Hematology, Children’s Hospital of Philadelphia, 3615 Civic Center Blvd, Room 316B, Philadelphia, PA, 19104; e-mail: rivellas@email.chop.edu.

