Key Points
Aging-associated inflammation by TNF-α plays an important role in the development of platelet hyperreactivity during aging.
Aging-associated platelet hyperreactivity is associated with megakaryocytic inflammatory, metabolic, and mitochondrial reprogramming.

Abstract
Aging and chronic inflammation are independent risk factors for the development of atherothrombosis and cardiovascular disease. We hypothesized that aging-associated inflammation promotes the development of platelet hyperreactivity and increases thrombotic risk during aging. Functional platelet studies in aged-frail adults and old mice demonstrated that their platelets are hyperreactive and form larger thrombi. We identified tumor necrosis factor α (TNF-α) as the key aging-associated proinflammatory cytokine responsible for platelet hyperreactivity. We further showed that platelet hyperreactivity is neutralized by abrogating signaling through TNF-α receptors in vivo in a mouse model of aging. Analysis of the bone marrow compartments showed significant platelet-biased hematopoiesis in old mice reflected by increased megakaryocyte-committed progenitor cells, megakaryocyte ploidy status, and thrombocytosis. Single-cell RNA-sequencing analysis of native mouse megakaryocytes showed significant reprogramming of inflammatory, metabolic, and mitochondrial gene pathways in old mice that appeared to play a significant role in determining platelet hyperreactivity. Platelets from old mice (where TNF-α was endogenously increased) and from young mice exposed to exogenous TNF-α exhibited significant mitochondrial changes characterized by elevated mitochondrial mass and increased oxygen consumption during activation. These mitochondrial changes were mitigated upon TNF-α blockade. Similar increases in platelet mitochondrial mass were seen in platelets from patients with myeloproliferative neoplasms, where TNF-α levels are also increased. Furthermore, metabolomics studies of platelets from young and old mice demonstrated age-dependent metabolic profiles that may differentially poise platelets for activation. Altogether, we present previously unrecognized evidence that TNF-α critically regulates megakaryocytes resident in the bone marrow niche and aging-associated platelet hyperreactivity and thrombosis.
Introduction
Aging is an independent risk factor for atherothrombosis and cardiovascular disease (CVD), the leading cause of death worldwide.1,2 Platelet activation and the subsequent formation of platelet-rich coronary thrombi are, in part, responsible for the elevated morbidity and mortality associated with CVD. Chronic inflammation due to high levels of tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) is critical in the development of CVD and is a significant component of human and murine aging.3,4 Similarly, diseases in which systemic TNF-α levels are high and contribute to disease pathogenesis, including rheumatoid arthritis (RA), inflammatory bowel disease, and sepsis, are associated with a higher incidence of CVD and atherothrombotic events, suggesting that TNF-α plays a significant role in the pathophysiology of thrombosis in these conditions.5-7 Likewise, genetic variants in the promoter region of TNF have been associated with increased levels of circulating TNF-α, higher incidence of thrombotic events,8-10 and with elevated levels of TNF-α in patients with RA, renal failure, sickle cell disease, and myeloproliferative neoplasms (MPNs).9,11-14 Moreover, older prefrail and frail adults exhibit significantly higher levels of TNF-α when compared with older healthy adults, and have a higher risk of atherothrombosis.15,16 Interestingly, the platelet hyperreactivity observed in patients with RA, an aging disease characterized by high levels of TNF-α, can be significantly downregulated after the initiation of anti–TNF-α therapy, suggesting that the platelet hyperreactivity observed in arthritis may be, at least in part, TNF-α dependent.6,17,18
In the work presented here, we interrogated the interplay of inflammation and thrombosis in the context of aging-associated chronic inflammation and its effect on platelet development and function. Single-cell RNA-sequencing (scRNA-seq) transcriptomic analysis of directly isolated megakaryocytes from old mice demonstrated evidence of substantial reprogramming with numerous pathways affected, including inflammation, mitochondrial dysfunction, and oxidative phosphorylation. Functional platelet studies using murine models of inflammation and aging led us to identify TNF-α as the key proinflammatory cytokine promoting platelet hyperreactivity in aged mice. In addition to this, we present evidence that the previously reported high levels of TNF-α in old mice, older frail humans, and patients with MPNs contribute to the development of platelet hyperreactivity and that this phenotype is associated with platelet mitochondrial dysfunction. Finally, TNF-α blockade reversed aging-associated platelet hyperreactivity, thrombosis, and mitochondrial changes. Taken together, our findings provide new evidence to suggest that elevated systemic levels of TNF-α in aging promote megakaryocyte reprogramming, platelet mitochondrial dysfunction, and thrombosis.
Materials and methods
Reagents
Additional information about reagents and methods are provided in supplemental Table 1 (available on the Blood Web site).
Platelet preparation
Mouse platelets were obtained as previously described.19 This preparation yielded a platelet purity of >98% as assessed by flow cytometry. In some experiments, platelets were purified using anti-mouse direct lineage depletion beads (Miltenyi Biotec; supplemental Figure 1C). Washed human platelets were prepared as previously described.20
Flow cytometry of murine and human platelets
Flow cytometry of platelets was determined as previously published with modifications using the reagents described in supplemental Table 1.21 Whole blood from humans was stimulated with adenosine diphosphate (ADP; 1 μM) for 20 minutes at 37°C followed by incubation with PAC-1 and CD41 for 20 minutes at room temperature. Samples were then immediately fixed and run using a FACScan (BD Biosciences) or a Gallios (Beckman Coulter). Data were analyzed using FlowJo v9 or with Kaluza (Beckman Coulter) software.
Platelet bioenergetics
Immunoblots
Washed platelets were lysed using radioimmunoprecipitation assay buffer. Protein concentrations were determined using a bicinchoninic acid assay.
Microfluidics assays
Microfluidics experiments were performed on a collagen surface at 650 s−1 as previously described.24
Transmission electron microscopy
Details are described in supplemental Methods.
Megakaryocyte ploidy analysis
Murine bone marrow megakaryocytes were processed and analyzed as previously described.25
scRNA-seq of bone marrow megakaryocytes
Bone marrow was obtained from murine femurs, tibiae, and hips. Samples were enriched for megakaryocytes using a bovine serum albumin (BSA) gradient as previously described.26 This step was followed by positive selection by anti-mouse CD61 beads (Miltenyi Biotec). Cells were captured using the 10X Genomics system. Cells were processed and sequenced using the 10X Chromium single-cell system and Illumina HiSeq 4000 platform at the University of Colorado Genomics and Sequencing Core Facility for a read depth of 50 000 reads per cell. From this, pathway analysis was performed using Ingenuity Pathway Analysis (IPA; Qiagen Bioinformatics) on the differential transcriptional profiles seen in the cell clusters.
Sample preparation for metabolomics
Sample processing and analyses were performed as previously published.27 Details are provided in supplemental Methods.
Statistics
Statistical analyses were performed using the unpaired 2-tailed Student t test or analysis of variance (ANOVA), as appropriate (GraphPad Software v7.0). For multiple group comparisons, ANOVA, the Mann-Whitney, or the Sidak post hoc test was performed. Data were expressed as mean plus or minus standard error of the mean (SEM). Statistical significance was set at a 2-tailed P < .05.
Results
Old mice have elevated levels of plasma TNF-α and exhibit thrombocytosis and anemia
Plasma cytokines from C57BL/6J young (2-3 months) and old (>18 months) were quantified by multiplex analysis. Old mice had significantly elevated plasma levels of TNF-α (Figure 1A), thrombocytosis (Figure 1B), and anemia (Figure 1C) whereas total leukocyte numbers did not differ between groups (Figure 1D). The levels of IL-1β and thrombin-antithrombin complexes were similar between young and old mice, suggesting the lack of active thrombin-dependent coagulation in this model of aging (supplemental Figure 1A-B).
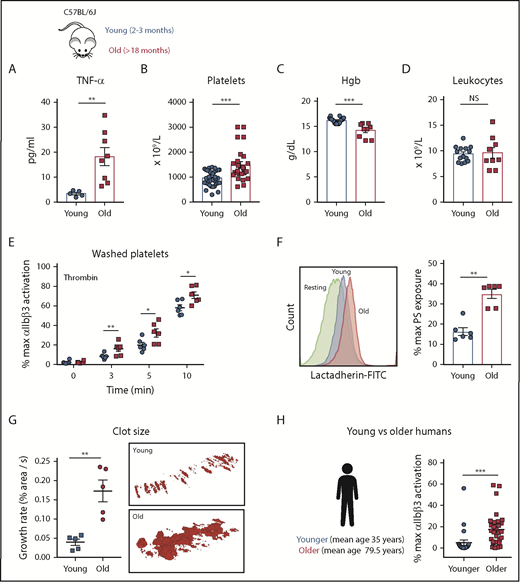
Aging associates with elevated TNF-⍺ plasma levels and platelet hyperreactivity in mice and humans. (A) TNF-⍺ plasma levels from heathy young (8-12 weeks) and old (>72 weeks) C57BL/6J mice (n = 6-8; mean plus or minus SEM). Hematological profile of (B) platelets, (C) hemoglobin (Hgb), and (D) leukocytes from healthy young and old C57BL/6J mice (n = 8-20 mice per group; mean plus or minus SEM). (E) Platelet activation analysis by flow cytometry of washed platelets from young (8-12 weeks) and old (>72 weeks) mice stimulated with thrombin (0.1 IU/mL) as determined by activation of the ⍺IIbβ3 integrin (n > 3 per experiment with 3-5 biological replicates). Mann-Whitney test. (F) Platelet phosphatidylserine (PS) exposure 5 minutes after activation with thrombin (0.1 IU/mL) of washed platelets from young (8-12 weeks) and old (>72 weeks) C57BL/6J mice (n = 6; mean plus or minus SEM) using bovine fluorescein isothiocyanate (FITC)-lactadherin. Student t test. (G) Assessment of platelet adhesion and clot formation over collagen-coated slides under physiological flow conditions (650 s−1) of reconstituted whole blood. Normal pooled murine plasma and erythrocytes were mixed with washed platelets from either young (8-12 weeks) and old (>72 weeks) mice at the same concentration. Representative TEM of clots formed over collagen-coated slides by TEM (n = 5-6; mean plus or minus SEM). Mann-Whitney test. (H) Activation profile of the ⍺IIbβ3 integrin in human washed platelets from younger (mean age, 35.0 ± 10.1 years; n = 25) and older (mean, 79.5 ± 8.8 years; n = 25) healthy volunteers. Mann-Whitney test. *P < .05; **P < .01; and ***P < .001. max, maximum; NS, not significant.
Aging associates with elevated TNF-⍺ plasma levels and platelet hyperreactivity in mice and humans. (A) TNF-⍺ plasma levels from heathy young (8-12 weeks) and old (>72 weeks) C57BL/6J mice (n = 6-8; mean plus or minus SEM). Hematological profile of (B) platelets, (C) hemoglobin (Hgb), and (D) leukocytes from healthy young and old C57BL/6J mice (n = 8-20 mice per group; mean plus or minus SEM). (E) Platelet activation analysis by flow cytometry of washed platelets from young (8-12 weeks) and old (>72 weeks) mice stimulated with thrombin (0.1 IU/mL) as determined by activation of the ⍺IIbβ3 integrin (n > 3 per experiment with 3-5 biological replicates). Mann-Whitney test. (F) Platelet phosphatidylserine (PS) exposure 5 minutes after activation with thrombin (0.1 IU/mL) of washed platelets from young (8-12 weeks) and old (>72 weeks) C57BL/6J mice (n = 6; mean plus or minus SEM) using bovine fluorescein isothiocyanate (FITC)-lactadherin. Student t test. (G) Assessment of platelet adhesion and clot formation over collagen-coated slides under physiological flow conditions (650 s−1) of reconstituted whole blood. Normal pooled murine plasma and erythrocytes were mixed with washed platelets from either young (8-12 weeks) and old (>72 weeks) mice at the same concentration. Representative TEM of clots formed over collagen-coated slides by TEM (n = 5-6; mean plus or minus SEM). Mann-Whitney test. (H) Activation profile of the ⍺IIbβ3 integrin in human washed platelets from younger (mean age, 35.0 ± 10.1 years; n = 25) and older (mean, 79.5 ± 8.8 years; n = 25) healthy volunteers. Mann-Whitney test. *P < .05; **P < .01; and ***P < .001. max, maximum; NS, not significant.
Platelets from old mice and older humans are hyperreactive
Platelets from old mice exhibited increased activation of platelet integrin αIIbβ3 (by JON/A binding) with thrombin (Figure 1E) and exposed higher amounts of phosphatidylserine (PS) (Figure 1F). Similar hyperreactivity was observed for convulxin and ADP by fluorescence-activated cell sorter (FACS) and platelet aggregometry (supplemental Figure 2A-B). To determine the functional relevance of these differences, we proceeded to assess the ability of platelets ex vivo to adhere to collagen and form aggregates under physiologic flow conditions using a microfluidics system.28,29 In these experiments, washed pooled erythrocytes from young C57B/6J mice were mixed with pooled plasma from young and old C57B/6J mice, followed by the addition of a constant number of platelets from either young or old mice to control for the platelet count difference between young and old mice. Platelets from old mice adhered faster to collagen and formed larger thrombi (Figure 1G) than platelets from young mice, indicating that the phenotype of hyperreactive platelets in aging mice functionally results in enhanced thrombus formation under physiologic flow conditions (supplemental Video 1). Finally, platelets from older adults (n = 21-25; mean age, 79.5 ± 8.8 years) exhibited significantly higher levels of αIIbβ3 integrin activation (PAC-1 binding) by ADP (Figure 1H) as well as formation of platelet-leukocyte aggregates (supplemental Figure 4D) in comparison with younger healthy adults (n = 20-25; mean age, 35 ± 5 years) despite the fact that 60% of patients in the older group were on aspirin (Table 1).
Expanded megakaryocyte lineage hematopoiesis in aging mice
To gain a better understanding of whether the differences in platelet numbers and activation profiles between young and old mice were driven by changes arising from hematopoietic progenitor cells, we performed extensive immunophenotyping of the bone marrow compartment as previously described.30 Consistent with prior studies, we observed significantly elevated numbers of long- and short-term hematopoietic stem cells30,31 and (Figure 2A) megakaryocyte progenitors (MkPs; Figure 2B), a subset of the multipotent progenitor 2 (MPP2)32 population (overall not significantly different) in the bone marrow of old mice, when compared with young mice. A decreased MPP431,33 population is likely the reflection of decreased lymphoid output observed in old mice. Moreover, ploidy was significantly higher in megakaryocytes from old mice (Figure 2C), consistent with our observation of the presence of thrombocytosis in old mice (Figure 1B). Interestingly, plasma thrombopoietin levels were significantly lower in old mice and likely represent the elevated platelet mass observed in old mice as their platelet counts are significantly higher (supplemental Figure 3A). Analysis of in vivo clearance of biotinylated platelets and platelet recovery did not show significant differences between the life span of platelets in young and old mice (supplemental Figure 3B-C). Young and old mice were injected with recombinant thrombopoietin and the response was similar as both groups exhibited significant elevation of the platelet count (supplemental Figure 3D). These results indicate that the thrombocytosis associated with aging in mice is likely due to age or inflammation-related changes in megakaryopoiesis and thrombopoiesis.
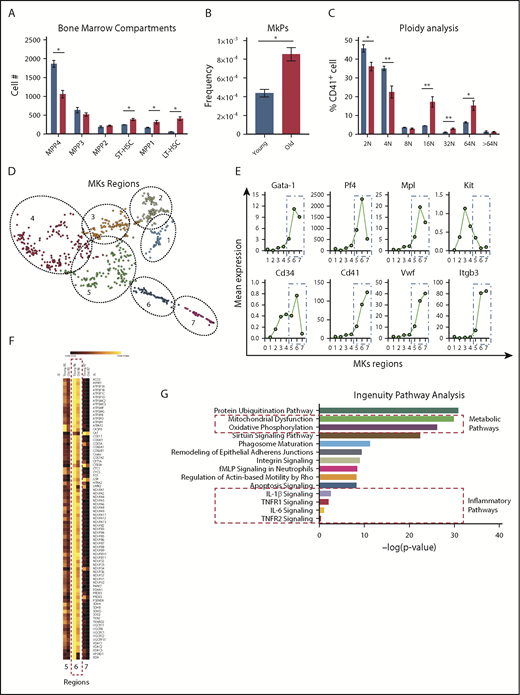
Analysis of the bone marrow compartments between young and old mice show significant cellular and transcriptomic differences. (A) Age-associated differences of the hematopoietic bone marrow compartments from young and old mice (n = 5 mice per group; mean plus or minus SEM). (B) Old mice exhibit higher numbers of megakaryocyte (MK)-committed progenitors (MkPs) (n = 5 mice per group; mean plus or minus SEM). (C) Megakaryocyte ploidy analysis of bone marrow megakaryocytes from young and old mice (n = 5 mice per group; mean plus or minus SEM). (D) scRNA-seq of native bone marrow megakaryocytes from young and old mice (n = 4-5 mice per group) identifies 7 clusters of cells at different maturation stages. (E) Identification of megakaryocyte maturation regions (MK regions 1-7) based on megakaryocyte-specific transcripts. (F) Representative heat map of the mitochondrial dysfunction IPA gene pathway across the 7 megakaryocyte regions. (G) IPA identifies significant differentially regulated mitochondrial, metabolic, and inflammatory gene pathways in the transcriptome of young and old bone marrow megakaryocytes. *P < .05; **P < .01; and ***P < .001. Two-tailed t test and Mann-Whitney test. LT-HSC, long-term hematopoietic stem cell; fMLP, N-Formylmethionyl-leucyl-phenylalanine; ST-HSC, short-term hematopoietic stem cell.
Analysis of the bone marrow compartments between young and old mice show significant cellular and transcriptomic differences. (A) Age-associated differences of the hematopoietic bone marrow compartments from young and old mice (n = 5 mice per group; mean plus or minus SEM). (B) Old mice exhibit higher numbers of megakaryocyte (MK)-committed progenitors (MkPs) (n = 5 mice per group; mean plus or minus SEM). (C) Megakaryocyte ploidy analysis of bone marrow megakaryocytes from young and old mice (n = 5 mice per group; mean plus or minus SEM). (D) scRNA-seq of native bone marrow megakaryocytes from young and old mice (n = 4-5 mice per group) identifies 7 clusters of cells at different maturation stages. (E) Identification of megakaryocyte maturation regions (MK regions 1-7) based on megakaryocyte-specific transcripts. (F) Representative heat map of the mitochondrial dysfunction IPA gene pathway across the 7 megakaryocyte regions. (G) IPA identifies significant differentially regulated mitochondrial, metabolic, and inflammatory gene pathways in the transcriptome of young and old bone marrow megakaryocytes. *P < .05; **P < .01; and ***P < .001. Two-tailed t test and Mann-Whitney test. LT-HSC, long-term hematopoietic stem cell; fMLP, N-Formylmethionyl-leucyl-phenylalanine; ST-HSC, short-term hematopoietic stem cell.
scRNA-seq of primary bone megakaryocytes reveals that metabolic, mitochondrial, and inflammatory signaling pathways are altered in aging
To test the hypothesis that an aged proinflammatory bone marrow environment in old mice alters the megakaryocyte transcriptome, we defined the transcriptional changes in megakaryocytes from old mice at the single-cell level. Transcripts used to identify the megakaryocyte populations were: Gata-1, Pf4, Mpl, KiT, Cd34, Cd41, Vwf, and Itgb3. We identified 7 discrete groups of cells within the megakaryocyte population that may represent megakaryocytes at different maturation stages based on the transcript expression of these 8 genes (Figure 2D). Canonical megakaryocyte-specific transcripts such as Cd41, Vwf, and Itgb3 increased significantly from groups 5 to 7, suggesting that these megakaryocytes represent highly transcriptionally active cells undergoing thrombopoiesis (Figure 2E). Further analysis of these pathways revealed that most of the transcriptional differences are more evident within the megakaryocyte region 6 as depicted in the heat map of the mitochondrial dysfunction pathway (Figure 2F). Two of the top pathways identified by IPA analyses when comparing old with young mice included mitochondrial dysfunction and oxidative phosphorylation. In addition, TNF-signaling pathways (TNF-R1 and TNF-R2 pathways) were overrepresented in megakaryocytes from old mice, demonstrating a significant TNF effect on the megakaryocyte transcriptome (Figure 2G). The highest upregulated transcript in old megakaryocytes, class 1A aldehyde dehydrogenase (Aldh1a; which is highly abundant in hematopoietic precursors), was also significantly expressed in platelets from old mice (supplemental Figure 3D-E).
Platelets from old mice show increased mitochondrial mass
Based on the results from the transcriptome and IPA analyses, we next examined the mitochondria content and function in platelets from young and old mice. Transmission electron microscopy (TEM) showed significantly higher number of mitochondria in platelets from old mice (Figure 3A-B). These results were confirmed by quantifying platelet mitochondrial mass by flow cytometry using Mitotracker Green FM (Figure 3C) and by immunoblotting of the mitochondrial-specific protein, Tom20 (Figure 3D). This difference in mitochondrial mass was independent of platelet size (supplemental Figure 4A-B).
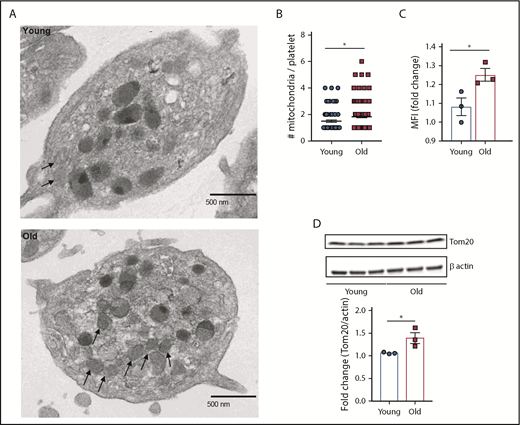
Platelets from old mice exhibit significantly higher mitochondrial mass. (A-B) Quantification of platelet mitochondrial mass by TEM, flow cytometry (C), and by (D) immunoblotting of the mitochondrial protein Tom20 (n = 3-6 mice per group; mean plus or minus SEM). MFI, mean fluorescence intensity.
Platelets from old mice exhibit significantly higher mitochondrial mass. (A-B) Quantification of platelet mitochondrial mass by TEM, flow cytometry (C), and by (D) immunoblotting of the mitochondrial protein Tom20 (n = 3-6 mice per group; mean plus or minus SEM). MFI, mean fluorescence intensity.
Platelets from aged mice show increased OCR upon activation and altered metabolic pathways and energy dynamics
To determine functional implications of age-dependent increases in mitochondrial mass, we analyzed platelet mitochondrial bioenergetics by Seahorse analysis. Platelets from old mice had an almost doubled maximum oxygen consumption rate (OCR) upon activation with 0.1 U/mL thrombin, compared with platelets from young mice (Figure 4A). These differences in OCR upon platelet activation indicate that mitochondrial respiration of platelets from old mice is different and potentially the result of increased mass. In light of the Seahorse data, we used liquid chromatography–mass spectrometry-based metabolomics to better resolve the metabolic profiles in platelets isolated from young and old mice, either at baseline or after ex vivo stimulation with thrombin (Figure 4B). Partial least squares discriminant analysis revealed that activation contributes to 35.6% of the variation between groups along principal component 1, whereas 15.4% of the variation is explained by age-associated differences alone along principal component 2 (Figure 4C). Based on increased mitochondrial content and OCR, we first looked at tricarboxylic acid (TCA) cycle intermediates. Interestingly, the levels of these metabolites did not differ significantly as a factor of age or activation status (supplemental Figure 5). However, metabolic rewiring in glycolysis and the pentose phosphate pathway was apparent (Figure 4D), where late-stage glycolytic metabolites pyruvate and lactate were basally lower in old platelets in conjunction with elevated levels of 6-phosphogluconolactone, 6-phosphogluconate, and erythrose-4-phosphate (Figure 4D-E). Despite unchanged TCA cycle and depressed state glycolysis in older platelets, adenylates adenosine 5′-monophosphate (AMP), ADP, and adenosine triphosphate (ATP) were all higher at baseline levels in old platelets (Figure 4F). These results concur with the Seahorse data (Figure 4A), which demonstrated that old platelets have a higher ATP-linked respiration capacity upon activation and suggested higher basal ATP pools. Taken together, these data suggest altered energetics in old platelets as reflected by higher levels of baseline ADP and ATP that could also be key players of the platelet hyperreactivity of old mice.
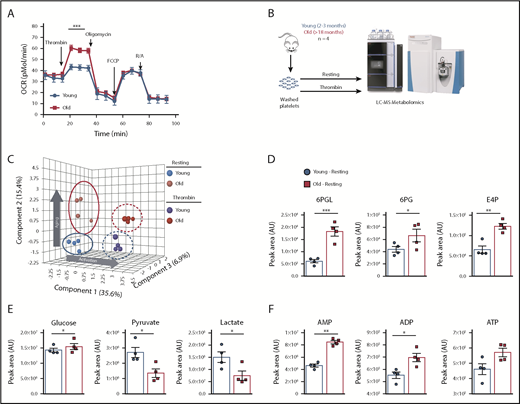
Platelets from young and old mice have a distinctive mitochondrial respiration and metabolome. (A) Bioenergetics Seahorse analysis of platelets from young and old mice shows that platelets from old mice have significantly higher respiration upon activation with 0.1 U/mL thrombin (n = 4-6 mice per group; mean plus or minus SEM). (B) Platelet metabolomics experimental approach. (C) Partial least squares discriminant analysis (PLS-DA) of platelets from young and old mice at resting and activated state shows distinctive metabolomic profiles of platelets due to age and activation (n = 4 mice per group; plus or minus SEM). (D) Pentose phosphate pathway metabolites of platelets from old mice are elevated at baseline and show preferential glycolytic metabolism at baseline. (E) TCA cycle upstream metabolites glucose, pyruvate, and lactate between platelets of young and old mice. (F) Differences in the adenylates AMP, ADP, and ATP between platelets from young and old mice. *P < .05; **P < .01; and ***P < .001. Student t test and Mann-Whitney test. AU, arbitrary unit; FCCP, carbonylcyanide p-trifluoromethoxyphenylhydrazone; LC-MS, liquid chromatography–mass spectrometry; R/A, rotenone/antimycin A.
Platelets from young and old mice have a distinctive mitochondrial respiration and metabolome. (A) Bioenergetics Seahorse analysis of platelets from young and old mice shows that platelets from old mice have significantly higher respiration upon activation with 0.1 U/mL thrombin (n = 4-6 mice per group; mean plus or minus SEM). (B) Platelet metabolomics experimental approach. (C) Partial least squares discriminant analysis (PLS-DA) of platelets from young and old mice at resting and activated state shows distinctive metabolomic profiles of platelets due to age and activation (n = 4 mice per group; plus or minus SEM). (D) Pentose phosphate pathway metabolites of platelets from old mice are elevated at baseline and show preferential glycolytic metabolism at baseline. (E) TCA cycle upstream metabolites glucose, pyruvate, and lactate between platelets of young and old mice. (F) Differences in the adenylates AMP, ADP, and ATP between platelets from young and old mice. *P < .05; **P < .01; and ***P < .001. Student t test and Mann-Whitney test. AU, arbitrary unit; FCCP, carbonylcyanide p-trifluoromethoxyphenylhydrazone; LC-MS, liquid chromatography–mass spectrometry; R/A, rotenone/antimycin A.
Systemic and chronic exposure to TNF-α promotes platelet hyperreactivity and increased mitochondrial mass
To determine the contribution of systemic inflammation by TNF-α on platelet hyperreactivity, independently of other aging-associated changes, we used a model of aseptic systemic inflammation by injecting young mice (8-12 weeks of age) daily with recombinant murine TNF-α for a total of 20 days (or vehicle as control; Figure 5A). As expected, plasma levels of TNF-α in mice injected with TNF-α were significantly higher than control mice (supplemental Figure 6A). These systemic levels of TNF-α approached the levels observed in old mice, where TNF-α levels are endogenously elevated (Figure 1A). TNF-α treatment increased activation of integrin αIIbβ3 in response to either convulxin or thrombin (Figure 5B-C). In a model of platelet-dependent clot formation under ex vivo physiologic flow conditions, whole blood from mice injected with TNF-α adhered faster and formed larger thrombi compared with vehicle control conditions (Figure 5D). Additionally, we used a model of chronically elevated levels of TNF-α, the TNFΔARE mice that carry a deletion in the TNF adenylate-uridylate–rich elements (AREs), which results in elevated systemic levels of TNF-α since embryogenesis and development of arthritis.34-37 Similar to TNF-treated young mice (and old mice), platelets from TNFΔARE mice exhibit increased activation of the integrin αIIbβ3 (supplemental Figure 6B), increased platelet-leukocyte aggregates (supplemental Figure 4C), and significantly higher mitochondrial mass by FACS and immunoblotting (supplemental Figure 6C-D). Moreover, due to the previously established redundancy in signaling between TNF-α and IL-1β, the previously reported elevations of IL-1β in very old mice (>22 months) and older humans,4,38-42 and the differentially regulated IL-1β gene pathways observed in megakaryocytes from old mice, we tested whether exogenous administration of IL-1β (also for 20 days) promoted platelet hyperreactivity. In contrast to TNF-α, IL-1β did not increase platelet hyperreactivity as determined by the microfluidics assay or by flow cytometry (Figure 5F-G). Moreover, mice lacking functional TNF receptors (p55/p75 KO mice)43 do not exhibit platelet hyperreactivity after injections with daily recombinant TNF-α at 3 minutes after activation with low-dose thrombin (0.1 U/mL) and convulxin (50 ng/mL) by FACS and by microfluidics (Figure 5H-I). Together, these results indicate that whether acute or chronic, TNF-α exposure selectively increases platelet reactivity.
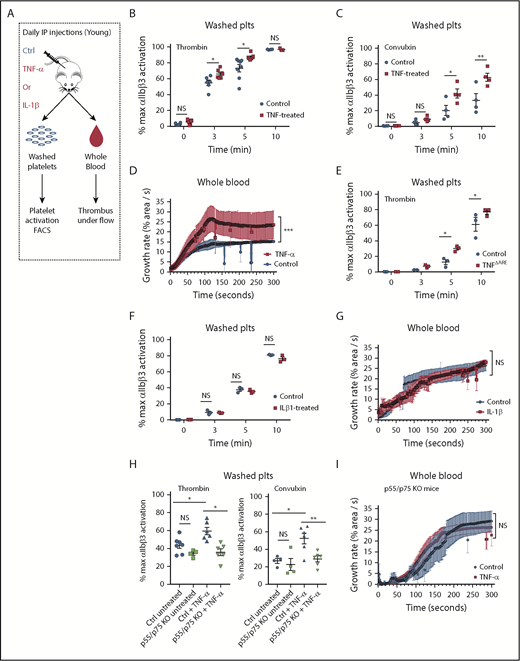
Platelets from young mice exposed to TNF-α in vivo for 20 consecutive days become hyperreactive. (A) Experimental approach. Representative flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets (plts) from young mice treated with TNF-⍺ or control (ctrl) for 20 days after activation with (B) thrombin (0.1 U/mL) or (C) convulxin (50 ng/mL), (n = 4-6; mean plus or minus SEM). (D) Whole blood microfluidics assay. Platelet adhesion to collagen-coated slides under flow of young mice treated with TNF-⍺ or control for 20 days (n = 4; mean plus or minus SEM). (E) Representative flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets from TNFΔARE or littermate controls (n = 3; mean plus or minus SEM). (F) Representative flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets from young mice treated with IL-1β or control for 20 days (n = 3; mean plus or minus SEM). (G) Whole blood microfluidics assay. Platelet adhesion to collagen-coated slides under flow of young mice treated with IL-1β or control for 20 days (n = 4; mean plus or minus SEM). (H) Flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets from young C57BL/6J and p55/p75 KO mice treated with TNF-⍺ or control for 20 days after activation with thrombin (0.1 U/mL) or convulxin (50 ng/mL; n = 4-6; mean plus or minus SEM). (I) Microfluidics analysis of whole blood flowed over collagen-coated slides of young C57BL/6J and p55/p75 KO mice treated with TNF-⍺ or control for 20 days (n = 4; mean plus or minus SEM). *P < .05; **P < .01; and ***P < .001. Student t test and Mann-Whitney test. IP, intraperitoneal.
Platelets from young mice exposed to TNF-α in vivo for 20 consecutive days become hyperreactive. (A) Experimental approach. Representative flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets (plts) from young mice treated with TNF-⍺ or control (ctrl) for 20 days after activation with (B) thrombin (0.1 U/mL) or (C) convulxin (50 ng/mL), (n = 4-6; mean plus or minus SEM). (D) Whole blood microfluidics assay. Platelet adhesion to collagen-coated slides under flow of young mice treated with TNF-⍺ or control for 20 days (n = 4; mean plus or minus SEM). (E) Representative flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets from TNFΔARE or littermate controls (n = 3; mean plus or minus SEM). (F) Representative flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets from young mice treated with IL-1β or control for 20 days (n = 3; mean plus or minus SEM). (G) Whole blood microfluidics assay. Platelet adhesion to collagen-coated slides under flow of young mice treated with IL-1β or control for 20 days (n = 4; mean plus or minus SEM). (H) Flow cytometry assessment of the activation of the ⍺IIbβ3 integrin of washed platelets from young C57BL/6J and p55/p75 KO mice treated with TNF-⍺ or control for 20 days after activation with thrombin (0.1 U/mL) or convulxin (50 ng/mL; n = 4-6; mean plus or minus SEM). (I) Microfluidics analysis of whole blood flowed over collagen-coated slides of young C57BL/6J and p55/p75 KO mice treated with TNF-⍺ or control for 20 days (n = 4; mean plus or minus SEM). *P < .05; **P < .01; and ***P < .001. Student t test and Mann-Whitney test. IP, intraperitoneal.
The megakaryocyte transcriptome of old mice is similar to the transcriptome of megakaryocytes chronically exposed to TNF-α
To test the hypothesis that a proinflammatory TNF-α–driven bone marrow environment alters the megakaryocyte transcriptome similarly to that of old age mice, we performed bulk RNA-seq of native bone marrow megakaryocytes from young and old mice, bone marrow megakaryocytes from young mice exposed to exogenous TNF-α (20-day injections), and bone marrow megakaryocytes from TNFΔARE mice. Megakaryocytes were isolated and sorted by FACS as previously described.25 Principal component analysis identified that the transcriptome of megakaryocytes from old mice (where TNF-α levels are increased) was similar to the transcriptome of megakaryocytes from young mice treated with TNF-α as well as to the transcriptome of megakaryocytes from TNFΔARE mice (supplemental Figure 6E). This demonstrates that experimentally raising systemic TNF-α levels in young mice, either by exogenous injections or by constitutive overexpression by a transgene (TNFΔARE), results in megakaryocyte transcriptional profiles that are similar to those observed in old mice. In addition, genes that encode for proteins involved in platelet activation show significantly differential expression in the scRNA-seq analysis of megakaryocytes from young and old mice (supplemental Figure 6F).
Patients with MPNs exhibit significantly high levels of systemic TNF-α and elevated mitochondrial mass
MPN is an aging-associated clonal hematological disease characterized by marked elevations in TNF-α levels, thrombocytosis, and an increased incidence of thromboembolic events. Therefore, we examined platelet mitochondrial mass in patients with MPNs and compared them to controls. As previously reported, MPN (Jak2 V617F+) patients had significantly high levels of TNF-α (Figure 6A). In addition, platelet mitochondrial mass was significantly higher in platelets from patients with Jak2 V617F+ MPN when compared with healthy age-matched controls (Figure 6B-C; supplemental Figure 7B). These differences were also significant when comparing a larger group of younger healthy controls with all MPN patients (supplemental Figure 7A).
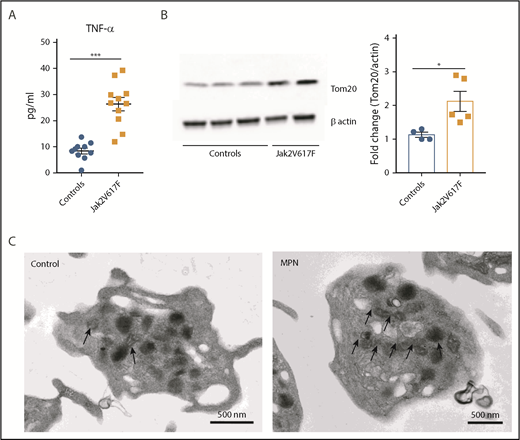
Jak2 V617F+ MPN is associated with increased circulating levels of TNF-α and higher platelet mitochondrial mass. (A) Plasma TNF-⍺ levels of healthy controls and Jak2 V617F+ MPN patients. (B) Representative immunoblot (left) and biochemical quantification (right) of the mitochondrial protein Tom20 between healthy controls and Jak2 V617F+ MPN patients. (C) Representative TEM images of platelets from healthy controls and Jak2 V617F+ MPN patients with black arrows depicting the significantly higher mitochondrial content in platelets from Jak2 V617F+ MPN patients (n = 4-12; mean plus or minus SEM). *P < .05; **P < .01; and ***P < .001. Student t test and Mann-Whitney test.
Jak2 V617F+ MPN is associated with increased circulating levels of TNF-α and higher platelet mitochondrial mass. (A) Plasma TNF-⍺ levels of healthy controls and Jak2 V617F+ MPN patients. (B) Representative immunoblot (left) and biochemical quantification (right) of the mitochondrial protein Tom20 between healthy controls and Jak2 V617F+ MPN patients. (C) Representative TEM images of platelets from healthy controls and Jak2 V617F+ MPN patients with black arrows depicting the significantly higher mitochondrial content in platelets from Jak2 V617F+ MPN patients (n = 4-12; mean plus or minus SEM). *P < .05; **P < .01; and ***P < .001. Student t test and Mann-Whitney test.
In vivo neutralization of TNF-α decreases platelet hyperreactivity and restores mitochondrial mass in aged mice
Finally, to determine whether specifically blocking systemic TNF-α levels reverses platelet hyperreactivity and the observed mitochondrial phenotype, we performed in vivo neutralization studies using a monoclonal anti–TNF-α antibody. Young and old mice were injected for 10 days, which is equivalent to 2.5 platelet half-lives (Figure 7A). Plasma TNF-α levels were significantly decreased in old mice treated with the neutralizing antibody when compared with the old mice treated with vehicle control (Figure 7B), indicating that our pharmacological blockade had a significant effect. In vivo neutralization of TNF-α in old mice resulted in a significant decrease in activation of integrin αIIbβ3 (Figure 7D) and normalized platelet adhesion and clot formation under flow, back to levels similar to those observed in young mice (Figure 7E). Moreover, TNF-α blockade in old mice caused decreased mitochondrial mass to levels compared with the ones of young mice (Figure 7F). All of these effects were independent of platelet numbers as the platelet counts were not significantly altered with neutralization of TNF-α (Figure 7C). Platelet adhesion and clot formation over collagen remained unchanged when p55/p75 KO43 (deficient for both TNF-α receptors) mice were injected with TNF-α for 20 days (Figure 5I). In addition, platelet mitochondrial mass was similar between WT and p55/p75 KO mice, demonstrating that functional TNF-α receptors are required for the development of platelet hyperreactivity and the mitochondrial phenotype due to elevated TNF-α levels (supplemental Figure 8A-B).
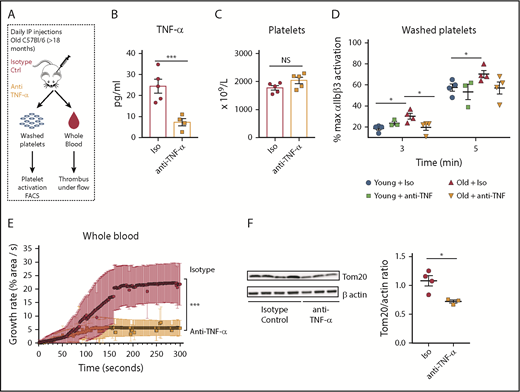
In vivo neutralization of TNF-α decreases platelet hyperreactivity and restores mitochondrial mass in aged mice. (A) Experimental approach. (B) Decreased plasma levels of TNF-⍺ in old mice treated with anti–TNF-⍺ antibody (n = 4-5 mice per group; mean plus or minus SEM). (C) Platelet counts did not significantly change between old mice treated with anti–TNF-⍺–neutralizing antibodies or isotype control (n = 5 mice per group; mean plus or minus SEM). (D) Inhibition of the ⍺IIbβ3 integrin by anti–TNF-⍺–neutralizing antibodies as measured by FACS (n = 4 mice per group; mean plus or minus SEM). (E) Platelets from old mice treated with by anti–TNF-⍺–neutralizing antibodies show significantly decreased platelet adhesion over collagen-coated slides under flow at 650 s−1 (n = 4-5 mice per group; mean plus or minus SEM). (F) Mitochondrial mass assessment after anti–TNF-⍺–neutralizing antibodies (n = 3-4 mice per group; mean plus or minus SEM). *P < .05; **P < .01; and ***P < .001. Student t test and ANOVA.
In vivo neutralization of TNF-α decreases platelet hyperreactivity and restores mitochondrial mass in aged mice. (A) Experimental approach. (B) Decreased plasma levels of TNF-⍺ in old mice treated with anti–TNF-⍺ antibody (n = 4-5 mice per group; mean plus or minus SEM). (C) Platelet counts did not significantly change between old mice treated with anti–TNF-⍺–neutralizing antibodies or isotype control (n = 5 mice per group; mean plus or minus SEM). (D) Inhibition of the ⍺IIbβ3 integrin by anti–TNF-⍺–neutralizing antibodies as measured by FACS (n = 4 mice per group; mean plus or minus SEM). (E) Platelets from old mice treated with by anti–TNF-⍺–neutralizing antibodies show significantly decreased platelet adhesion over collagen-coated slides under flow at 650 s−1 (n = 4-5 mice per group; mean plus or minus SEM). (F) Mitochondrial mass assessment after anti–TNF-⍺–neutralizing antibodies (n = 3-4 mice per group; mean plus or minus SEM). *P < .05; **P < .01; and ***P < .001. Student t test and ANOVA.
Discussion
Although the increased thrombotic risk of aging is likely multifactorial, accumulating evidence indicates that systemic inflammation plays a critical role. Yet, the mechanisms by how inflammation increases thrombotic risk are poorly understood. To address the effect of aging and inflammation on platelet reactivity, we used mouse models and relevant human correlates to elucidate the contribution of aging-associated inflammation to platelet hyperreactivity and thrombosis. Our findings show, using several models, that elevated TNF-α levels associated with aging is sufficient to induce platelet hyperreactivity, increased platelet procoagulant potential, and thrombus formation. Our data also demonstrate that TNF-α induces molecular and metabolic reprogramming associated with increased platelet mitochondrial mass and platelet hyperactivity. Moreover, we demonstrated that in vivo treatment of old mice with a monoclonal anti–TNF-α antibody significantly decreases platelet reactivity.
We propose that TNF-α contributes to the risk of cardiovascular disease by promoting platelet hyperreactivity due to reprogramming of inflammatory, mitochondrial, and metabolic pathways in megakaryocytes. As Henry et al previously reported, old mice have high levels of TNF-α in both plasma and bone marrow. TNF-α levels were markedly higher (approximately fivefold) in the bone marrow compartment,4 an observation that suggests that increased TNF-α levels in the bone marrow of old mice may exert a higher effect on megakaryopoiesis and thrombopoiesis by inducing changes in resident megakaryocytes that might generate platelets with a hyperreactive profile during aging. Consistent with this, bulk RNA-seq analysis showed clear clustering of megakaryocytes from old, TNFΔARE–, and TNF-α–treated mice in a principal component analysis, suggesting that megakaryocytes from old mice are more transcriptionally homogenous and associate to TNF-α exposure.
Likewise, our novel scRNA-seq analysis of freshly isolated bone marrow megakaryocytes shows significantly differentially regulated inflammatory (TNF and IL1B gene pathways) as well as mitochondrial and metabolic pathways. In addition, and as an important validation step of our novel scRNA-seq analysis, the top differentially upregulated transcript in megakaryocytes from old mice, ALDH1A, is significantly highly expressed in platelets from old mice. Furthermore, ALDH1A is a highly expressed protein in hematopoietic precursors and plays an important role in cellular detoxification, suggesting that its elevation might represent a compensatory mechanism aimed at protecting aged megakaryocytes and platelets from oxidative stress.
In addition, we observed that platelets from old mice exhibit higher mitochondrial mass and respiration upon activation with thrombin. Furthermore, increased mitochondrial mass was also observed in TNFΔARE and TNF-α–treated mice. Interestingly, neutralization of TNF in vivo in aged mice resulted in decreased platelet hyperreactivity and mitochondrial mass, suggesting a link between TNF-α exposure and modulation of mitochondrial mass in platelets.
Metabolomics analysis revealed that platelets from old mice have a different metabolome when compared with young mice. These metabolic differences are represented by significantly higher amounts of the adenylates AMP, ADP, and ATP at baseline in old platelets, suggesting that these platelets have a higher potential to provide ATP upon activation and to enhance this activation in an ADP-dependent manner. Interestingly, decreased lactate and pyruvate with elevated pentose phosphate pathway intermediaries 6PGL, 6PG, and E4P in platelets from old mice suggest the presence of differential metabolic reprogramming either due to age-associated modifications, TNF-α–driven inflammation, or both. Moreover, this metabolic reconfiguration of the pentose phosphate pathway has been observed as a response to oxidative stress or accelerated cellular proliferation and opens new avenues for future research in megakaryocytes and platelets.44,45
Although the metabolic and mitochondrial differences observed in old mice may represent compensatory or adaptive responses to chronic inflammation, these changes also appear to confer platelets with a dysregulated activation profile that would significantly increase the thrombotic risk in aging and other inflammatory conditions. During activation, platelets are capable of releasing mitochondria, therefore, hyperreactive platelets with larger mitochondrial mass have the potential of exacerbating inflammatory responses by releasing higher amounts of mitochondrial DNA and other inflammatory mitochondrial components.46-50
Taken together, our findings identify previously unrecognized aspects of the functional and metabolic changes, along with related prothrombotic responses, induced by TNF-α in megakaryocytes and platelets during aging. Our results build upon and extend prior work studying oxidative and metabolic mechanistic factors that participate in the development of platelet hyperreactivity. Dayal et al reported that platelets from old mice can generate high levels of hydrogen peroxide (H2O2) that contribute to the aging-associated platelet hyperreactivity in mice.51 Similarly, Yang et al showed that activation of mammalian target of rapamycin complex 1 in platelets from old mice is associated with platelet hyperreactivity. Moreover, they showed that H2O2 can effectively activate mammalian target of rapamycin complex 1 in platelets from younger mice.52
We recognize that differences in mitochondrial mass between platelets from young and old mice could be the result of abnormal clearance of damaged mitochondria through mitophagy, dysregulation of the mitochondrial fusion and fission pathways, or the result of increased mitochondrial biogenesis as compensatory mechanism to overcome the distinctive metabolic and oxidative changes of megakaryocytes and platelets from old mice. It has been proposed that the increase in mitochondria numbers observed during aging in other cells might be the result of decreased autophagy,53 observations that warrant further investigation and represent undergoing efforts in our laboratory.
Based on these data, in parallel studies, we also examined platelets from patients with Jak2 V617F MPNs, a hematological disease characterized by high systemic TNF-α levels, platelet hyperreactivity, clonal hematopoiesis, and an increased incidence of thrombosis, and showed that the number of mitochondria per platelet is also increased. Interestingly, these findings confirm observations published over 50 years ago by researchers studying morphological differences in platelets from patients with MPNs54-56 and may help drive further research aimed at elucidating the interplay between TNF-α–associated inflammation, mitochondrial dynamics, and the pathophysiology of thrombotic events in MPNs.
In addition, work from other groups has linked TNF-associated pathologies such as HIV and ovarian cancer with increased atherothrombotic risk and changes in mitochondrial mass in CD8+ T cells and platelets from patients with HIV and ovarian cancer, respectively.57-59
The cellular source of elevated TNF-α in aging and MPN appears to be hematopoietic, and specifically from monocytes.60 Recent reports demonstrated that, in murine models of aging, monocytes are the main source of bone marrow and systemic elevations of TNF-α61 ; Hearps et al showed increased baseline intracellular TNF-α in monocytes of older humans.62 Finally, it has been recently shown that monocytes are a significant source of TNF-α in patients with MPNs upon activation of Toll-like receptors.14
In summary, our study demonstrates the effect of aging-associated inflammation by TNF-α on platelet hyperreactivity. We have also shown that megakaryocytes from aged or inflamed mice are transcriptionally reprogrammed and generate platelets that exhibit increased mitochondrial mass, mitochondrial dysfunction, and a distinctive metabolic profile. These discoveries have the potential to lead to the development of new therapeutic interventions aimed at modulating platelet hyperreactivity by targeting TNF-α (or its downstream pathways) and, therefore, decreasing the morbidity and mortality associated with inflammatory conditions such as RA, CVD, and aging.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by: the Pediatric Scientist Development Program of the National Institutes of Health Eunice Kennedy Shriver National Institute of Child Health and Human Development, grant K12HD00050 (P.D.-C.); the 2018 American Society of Hematology Research Training Award for Fellows (P.D.-C.); National Heart, Lung, and Blood Institute grants R01HL120728 (J.D.P.), HL142804 (M.T.R.), HL112311, and HL126547; grant H30MC24049 of the Mountain States Hemophilia Network (J.D.P.); the Postle Chair of Pediatric Cancer and Blood Disorders (J.D.P.); National Cancer Institute grants R01CA180175 (J.D.) and F30CA210383-01 (K.H.); National Institute on Aging grant AG04802 (M.T.R.); National Heart, Lung, and Blood Institute grants 1R35HL139726-01 (E.N.-G.) and T32 HL007171 (A.A.); and American Heart Association grant 19POST34380396 (A.A.). Additional support was received by the National Center for Research Resources of the National Institutes of Health under award number 1S10RR026802-01, the Flow Cytometry Core at the University of Utah, and the Cancer Center Shared Resources at the University of Colorado (National Cancer Institute grant 2-P30-CA46934).
Authorship
Contribution: P.D.-C., J.D., M.T.R., and J.D.P. designed research studies; B.M., S.A., D.B., K.A., R.A.C., E.M., E.M.P., A.A., E.N.-G., T.N., L.S., N.A.S., K.H., A.C., Z.P.-R., G.H., and E.W. conducted experiments and acquired data; A.D., N.C., G.D.T., and K.J. analyzed data; and P.D.-C., J.D., M.T.R., and J.D.P. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jorge Di Paola, School of Medicine, University of Colorado, 12800 E 19th St, Aurora, CO 80015; e-mail: jorge.dipaola@ucdenver.edu; or Matthew T. Rondina, University of Utah, 50 N Medical Dr, Salt Lake City, UT 84132; e-mail: matthew.rondina@hsc.utah.edu.