Key Points
In normal Mks, calreticulin regulates the activation of SOCE by interacting with ERp57 and STIM1.
In CALR-mutated MPNs, defective interaction between mutant calreticulin and SOCE proteins promotes Mk proliferation.

Abstract
Approximately one-fourth of patients with essential thrombocythemia or primary myelofibrosis carry a somatic mutation of the calreticulin gene (CALR), the gene encoding for calreticulin. A 52-bp deletion (type I mutation) and a 5-bp insertion (type II mutation) are the most frequent genetic lesions. The mechanism(s) by which a CALR mutation leads to a myeloproliferative phenotype has been clarified only in part. We studied the interaction between calreticulin and store-operated calcium (Ca2+) entry (SOCE) machinery in megakaryocytes (Mks) from healthy individuals and from patients with CALR-mutated myeloproliferative neoplasms (MPNs). In Mks from healthy subjects, binding of recombinant human thrombopoietin to c-Mpl induced the activation of signal transducer and activator of transcription 5, AKT, and extracellular signal-regulated kinase 1/2, determining inositol triphosphate–dependent Ca2+ release from the endoplasmic reticulum (ER). This resulted in the dissociation of the ER protein 57 (ERp57)-mediated complex between calreticulin and stromal interaction molecule 1 (STIM1), a protein of the SOCE machinery that leads to Ca2+ mobilization. In Mks from patients with CALR-mutated MPNs, defective interactions between mutant calreticulin, ERp57, and STIM1 activated SOCE and generated spontaneous cytosolic Ca2+ flows. In turn, this resulted in abnormal Mk proliferation that was reverted using a specific SOCE inhibitor. In summary, the abnormal SOCE regulation of Ca2+ flows in Mks contributes to the pathophysiology of CALR-mutated MPNs. In perspective, SOCE may represent a new therapeutic target to counteract Mk proliferation and its clinical consequences in MPNs.
Introduction
Philadelphia-negative myeloproliferative neoplasms (MPNs) are a group of heterogeneous clonal disorders affecting the hematopoietic stem cell (HSC). MPNs are characterized by neoplastic proliferation of the myeloid lineage leading to an abnormally increased number of platelets in essential thrombocythemia, red blood cells in polycythemia vera, or megakaryocytes (Mks) with bone marrow fibrosis in primary myelofibrosis.1-3 To date, 3 major driver mutations have been identified, affecting respectively, the thrombopoietin (TPO) receptor gene (MPL), the intracellular Janus kinase 2 gene (JAK2), and the endoplasmic reticulum (ER) molecular chaperone calreticulin gene (CALR).1 CALR mutations affect the C terminus of the protein, causing the loss of the ER-retention motif KDEL and important changes in the charge of the C-terminal tail.4,5 More than 60 different mutations leading to frameshift (+1) in CALR exon 9 have been found, but a 52-bp deletion (type I mutation) and a 5-bp insertion (type II mutation) are the most common variants.5 CALR mutants form a molecular complex with c-Mpl determining a cytokine-independent JAK/signal transducer and activator of transcription (STAT) activation with increased Mk proliferation.6-9 Importantly, this mechanism seems to be shared by both CALR type I– and type II–mutated cells, and thus do not explain the phenotypic differences observed in patients. Indeed, a worse prognosis characterizes patients harboring CALR type I mutation in terms of thrombotic events, BM fibrosis, and leukemic transformation compared with CALR type II, which is preferentially associated with essential thrombocythemia and an indolent clinical course.5 Our group recently demonstrated that CALR type I Mks display significantly increased store-operated calcium (Ca2+) entry (SOCE), a key regulator of megakaryopoiesis,10 compared with CALR type II mutants.5 Interestingly, earlier studies reported that overexpression of CALR reduces SOCE activity in various cell types and that this effect is likely to be mediated by the negatively charged C-terminal domain, which is deleted in CALR type I mutants.11-13 ER protein 57 (ERp57) is a protein of the ER and a member of the protein disulfide isomerase family that is mobilized to the surface of activated platelets, regulating their function.14 Recent studies demonstrated that thiol isomerases, including ERp57, are synthesized by human and mouse Mks and then distributed to nascent platelets.15 Importantly, ERp57 has been reported to interact with stromal interaction molecule 1 (STIM1) and to modulate SOCE activity.16 Elf et al demonstrated that wild-type CALR presents strong and direct binding to ERp57, whereas mutant CALR does not.17 Accordingly, more recently, Pronier et al noticed that the CALR-mutant proteins show decreased binding affinities for ERp57 compared with wild-type CALR leading to consequent ERp57 mislocalization to the nucleus to enhance MPL transcription.18
Here, we investigated the mechanisms of interaction between CALR and SOCE in human Mks from healthy subjects and MPN patients harboring CALR mutations.
Methods
Mk differentiation from human cord and peripheral blood hematopoietic progenitor cells
Human cord blood (CB) was collected from the local CB bank following healthy pregnancies and deliveries with the informed consent of the parents. Human peripheral blood samples were obtained from healthy subjects and patients with MPNs after informed consent. Patient and healthy subjects characteristics are given in supplemental Table 1 (available on the Blood Web site). All samples were processed following the ethical committee of the Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Policlinico San Matteo Foundation and Gustave Roussy Hospital and the principles of the Helsinki Declaration. Hematopoietic progenitor cells were separated by immunomagnetic bead selection (Miltenyi Biotec, Bologna, Italy) and differentiated in Stem Span medium (STEMCELL Technologies Inc, Vancouver, BC, Canada), as previously described.19
[Ca2+]i measurements
Analysis of Ca2+ signaling was performed as previously described.5,20,21 Twelve-millimeter glass coverslips were coated with 100 µg/mL fibrinogen overnight at 4°C. At day 13 of culture, Mks were harvested and plated onto substrate-coated coverslips in 24-well plates (1 × 105 cells per well). After 60 minutes at 37°C and 5% CO2, Mks were loaded with 4 µM fura-2 AM in physiological salt solution (PSS) for a further 30 minutes. After washing in PSS, the coverslip was fixed to the bottom of a Petri dish, and the cells were observed using an upright epifluorescence Axiolab microscope (Carl Zeiss), equipped with a Zeiss ×63 Achroplan objective (water-immersion, working distance 2.0 mm, 0.9 numerical aperture). To analyze Ca2+ signaling in the presence of recombinant human TPO (rhTPO) (10-50-100 ng/mL), Mks were imaged in PSS or in Ca2+-free solution (Ø Ca2+) in the presence or absence of 10 µM cyclopiazonic acid (CPA), and pretreated (for 30 minutes) or not with 20 µM 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM), 20 µM Bis trifluoromethyl pyrazole (BTP-2), or 20 µM 2-aminoethoxy-diphenylborate (2-APB). None of these treatments affected Mk viability as routinely assessed by trypan blue exclusion assay (data not shown).
For the quantification of Ca2+ signaling, Mks were excited alternately at 340 and 380 nm, and the light emitted was detected at 510 nm. A first neutral density filter (1 or 0.3 optical density) reduced the overall intensity of the excitation light and a second neutral density filter (0.3 optical density) was coupled to the 380-nm filter to approximate the intensity of the 340-nm light; a round diaphragm was used to increase the contrast. The excitation filters were mounted on a filter wheel (Lambda 10; Sutter Instrument). Custom software, working in the LINUX environment, was used to drive the camera (Extended-ISIS camera; Photonic Science) and the filter wheel, and to measure and plot online the fluorescence of rectangular regions of interest, each 1 enclosing a single cell. The analyses were performed at room temperature. Intracellular calcium concentration ([Ca2+]i) was monitored by measuring, for each regions of interest, the ratio of the mean fluorescence emitted at 510 nm when exciting alternatively at 340 and 380 nm (termed ‘‘ratio’’); an increase in [Ca2+]i causes an increase in the ratio. For each cell, the signal amplitude was calculated as the difference between the basal 340-nm:380-nm ratio signal and the respective peak level of the agonist-induced transient. The plateau signal was calculated as the difference between the basal 340-nm:380-nm ratio signal and the average of ratios after treatment, recorded over the time of the analysis. Spontaneous Ca2+ spikes, recorded in complete absence of agonists, were classified as Ca2+ transients when values of the 340-nm:380-nm ratio over the basal were higher than 0.04.
Western blot analysis
Western blot experiments were performed as previously described.22,23 Mks were lysed with N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-glycerol lysis buffer (HEPES 50 mM, NaCl 150 mM, 10% glycerol, 1% Triton X-100, MgCl2 1.5 mM, EGTA 1 mM, NaF 10 mM, Na3VO4 1 mM, 1 µg/mL leupeptin, 1 µg/mL aprotinin). Lysis was performed on ice for 30 minutes and lysates clarified by centrifugation at 15 700g at 4°C for 15 minutes. Finally, protein concentration was measured by the bicinchoninic acid assay (Pierce, Milan, Italy). For immunoprecipitation experiments, cellular lysates were precleared by incubation with protein A-Sepharose and incubated overnight with 2 µg of the desired antibody at 4°C on a rotatory shaker. Lysates were then incubated with 100 µL of 50 mg/mL protein A-Sepharose on the rotatory shaker at 4°C. After 2 hours, beads were washed 3 times with lysis buffer, and samples were eluted with Laemmli buffer at 95°C for 5 minutes. Protein lysates were subjected to 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membrane (Bio-Rad, Milan, Italy).
Membranes were probed with affinity-purified primary antibodies, according to the manufacturer’s indications. Immunoreactive bands were detected by horseradish peroxidase–labeled secondary antibodies using enhanced chemiluminescence reagent (Merck Millipore, Milan, Italy). Prestained protein ladders were used to estimate the molecular weights (Bio-Rad). At least 3 different experiments were performed in the different tested conditions:
Study of the expression of store-operated Ca2+ entry effectors during Mk differentiation
To evaluate expression of SOCE effectors during megakaryopoiesis, HSCs (day 0) or Mks at days 7-10-13 of differentiation, all derived from the same CB, were lysed and subjected to western blot analysis as described in “Western blot analysis.” Samples were probed with antibodies against CALR, Orai1, STIM1, c-Mpl, and CD61, according to the manufacturer’s indications.
Study of store-operated Ca2+ entry effector interaction upon rhTPO stimulation
To evaluate SOCE activation in human Mks, at day 13 of culture, 1 × 106 cells per condition were cytokine starved for 24 hours and stimulated or not with 100 ng/mL rhTPO for 10 minutes. Mk lysates were then incubated with antibodies against calcium release-activated calcium modulator 1 (Orai1), STIM1, CALR, or ERp57 and immune-precipitated. Samples were probed with antibodies against Orai1, STIM1, transient receptor potential canonical 1 (TRPC1), CALR, and ERp57 according to the manufacturer’s indications. Cell lysates were probed with the same antibodies to ensure equal loading or β-ACTIN as input control.
Study of rhTPO-induced SOCE activation on signal transduction pathways in human Mks
At day 13 of culture, 1 × 106 Mks per condition were harvested and suspended in PSS or Ø Ca2+ solutions and pretreated (for 30 minutes) or not with BAPTA-AM (20 µM), BTP-2 (20 µM), or 2-APB (20 µM) for 30 minutes at 37°C and 5% CO2. Subsequently, Mks were stimulated or not with 100 ng/mL rhTPO or 100 ng/mL rhTPO supplemented with CPA (10 µM), at 37°C and 5% CO2. After an additional 10 minutes or at different time points (0-1-10-20-30-60 minutes), samples were lysed and subjected to western blot analysis. Samples were probed with antibodies against phospho-STAT5 (p-STAT5), total STAT5, phospho-Akt (p-Akt), total Akt, phospho–extracellular signal-regulated kinase 1/2 (ERK1/2; p-ERK1/2), and total ERK1/2 according to the manufacturer’s indications.
In situ proximity ligation assay
To examine protein-protein interaction, we used the Duolink in situ proximity ligation assay (PLA) system (Sigma-Aldrich, Milan, Italy), which uses a set of 2 secondary antibodies in which 1 is conjugated to a minus strand PLA probe, and the other is conjugated to a plus strand PLA probe. A total of 2 × 105 Mks, derived from human cord blood, at day 13 of culture, were harvested and suspended in Dulbecco's Modified Eagle Medium for 7 hours. Where indicated, samples were treated with 1 of the following compounds: 100 ng/mL rhTPO, 1 U/mL thrombin, 10 µM CPA, for 10 minutes at 37°C and 5% CO2. Subsequently, Mks were cytospun onto glass slides, fixed with 4% paraformaldehyde for 30 minutes, permeabilized with 0.5% Triton X-100 for 10 minutes, and then incubated with anti-STIM1 (1:100) and anti-CALR (1:100), anti-mutated CALR (1:20), or anti-ERp57 (1:100) for 1 hour at room temperature. The proximate binding of these antibodies was then detected using Duolink in situ PLA secondary antibodies following conditions recommended by the manufacturer. Fluorescent images were acquired by confocal microscopy (FV10i; Olympus). The number of fluorescent foci per single cell was quantified using NIH Image J software.
Statistical analysis
Values are expressed as mean plus or minus standard deviation or median and range. The Student t test or 1-way analysis of variance (ANOVA) followed by Bonferroni posttest were used to analyze experiments. P < .05 was considered statistically significant. All experiments were independently repeated at least 3 times.
Additional materials, solutions, description of patients, UT-7 culture, methods for immunofluorescence, and enzyme-linked immunosorbent assay are described in supplemental Materials and methods.
Results
TPO induces Ca2+ mobilization in human Mks
To study the effect of TPO on Ca2+ flows in physiologic megakaryopoiesis, CB-derived Mks were loaded with fura-2 AM and recorded during culture with 10 ng/mL or 50 ng/mL rhTPO. Mks displayed a dose-dependent increase of Ca2+ spike intensities when cultured with increasing concentrations of rhTPO (Figure 1A). It is known that the oscillatory discharges of Ca2+ produce a sufficiently ample drop in ER Ca2+ levels to activate SOCE, which provides the necessary Ca2+ influx capable of regulating megakaryopoiesis.10,20,21 Given this, to test the involvement of SOCE, Mks in culture were cytokine-starved for 7 hours and then stimulated with 100 ng/mL rhTPO and recorded for 10 minutes. This caused a significant increase in Ca2+ mobilization, which was significantly lowered in the absence of extracellular Ca2+, and entirely blocked by pretreating Mks with 20 µM BAPTA-AM, a potent intracellular Ca2+ chelator (Figure 1B). Analysis of the Ca2+ curves following 10 minutes of rhTPO stimulation demonstrated that the extent of both the initial Ca2+ peak and the plateau phase were significantly greater in the presence of extracellular Ca2+ (Figure 1C). Altogether, these data demonstrate the contribution of both intracellular and extracellular Ca2+ in determining the Ca2+ flows in Mks upon stimulation with rhTPO, thus suggesting SOCE activation.
![TPO evokes calcium flows in cultured Mks. (A) In physiological Ca2+ concentration (1.5 mM), chronic exposure to rhTPO (10 or 50 ng/mL) evokes Ca2+ spikes in mature Mks whereas starvation from rhTPO did not elicit any Ca2+ spike. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derives from 266 measurements from 4 independent experiments (P < .05). (B) In physiological Ca2+ concentration (1.5 mM), 10 minutes of stimulation with rhTPO (100 ng/mL) evokes an initial rise in [Ca2+] followed by a plateau that is lowered in the absence of extracellular Ca2+ (Ø Ca2+) and completely abolished by [Ca2+]i chelation by BAPTA-AM. (C) Statistical analysis of peak and plateau fluorescence intensities in Mks treated as in panel B. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 207 measurements from 3 independent experiments.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/2/10.1182_blood.2019001103/2/m_bloodbld2019001103f1.png?Expires=1769413114&Signature=YrKst-JiIQSbjAJ6g-5IHqd6xMwnJzTPhO6Zcf1-HHbXc5HxfeT8e2rNR1-r-ykRM8PRLw-J70o4gPFo21oT2WitT-fVkvoQaSArjNgFmRDaVG4xgmrXKbgTLgStVQV2rwlXNhL4gdg9ST~4gAw8ZVZxQqsHlhGQ~dNCP9YyJazAWjQLiTRmH8JCK8uQiXFbmF0ZHkzKR~D4oQjlpsKrj2xT6B4xJ72nWuNg~MxxvEMjlahB5nKsvm2aWZhjfP-sNCycgUrAjZ8drbs5cmOxFzThm~MIc84cQwKYlZl4GdIcUGvMFweM-Tk~J3XNYF9~H7bwAqycB0cqlQDFWMDKpw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TPO evokes calcium flows in cultured Mks. (A) In physiological Ca2+ concentration (1.5 mM), chronic exposure to rhTPO (10 or 50 ng/mL) evokes Ca2+ spikes in mature Mks whereas starvation from rhTPO did not elicit any Ca2+ spike. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derives from 266 measurements from 4 independent experiments (P < .05). (B) In physiological Ca2+ concentration (1.5 mM), 10 minutes of stimulation with rhTPO (100 ng/mL) evokes an initial rise in [Ca2+] followed by a plateau that is lowered in the absence of extracellular Ca2+ (Ø Ca2+) and completely abolished by [Ca2+]i chelation by BAPTA-AM. (C) Statistical analysis of peak and plateau fluorescence intensities in Mks treated as in panel B. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 207 measurements from 3 independent experiments.
TPO evokes calcium flows in cultured Mks. (A) In physiological Ca2+ concentration (1.5 mM), chronic exposure to rhTPO (10 or 50 ng/mL) evokes Ca2+ spikes in mature Mks whereas starvation from rhTPO did not elicit any Ca2+ spike. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derives from 266 measurements from 4 independent experiments (P < .05). (B) In physiological Ca2+ concentration (1.5 mM), 10 minutes of stimulation with rhTPO (100 ng/mL) evokes an initial rise in [Ca2+] followed by a plateau that is lowered in the absence of extracellular Ca2+ (Ø Ca2+) and completely abolished by [Ca2+]i chelation by BAPTA-AM. (C) Statistical analysis of peak and plateau fluorescence intensities in Mks treated as in panel B. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 207 measurements from 3 independent experiments.
c-Mpl downstream signaling activation is sustained by Ca2+ mobilization
The modulation of STAT5, AKT, and ERK1/2 signaling pathways is essential to achieve proper Mk differentiation and function.24,25 To study the impact of Ca2+ mobilization on downstream c-Mpl signaling pathways, CB-derived Mks were stimulated for 10 minutes with 100 ng/mL rhTPO in the absence of extracellular Ca2+ or in the presence of the intracellular Ca2+ chelator BAPTA-AM. In both cases, we observed a significant reduction in STAT5, AKT, and ERK1/2 activation compared with controls treated with only rhTPO (Figure 2A-B). We consistently observed a significant increase in c-Mpl downstream signaling pathways upon restoration of 1.5 mM extracellular Ca2+ in the medium of Mk initially stimulated for 10 minutes with 100 ng/mL rhTPO in the absence of extracellular Ca2+ (supplemental Figure 1). Finally, we used the CPA that explicitly blocks the sarco-ER Ca2+ ATPase activity preventing Ca2+ sequestration into the stores leading to their depletion.26,27 This maneuver is routinely used to lower ER Ca2+ levels and, consequently, to activate SOCE in nonexcitable cells. Mks treated with 10 µM CPA and 100 ng/mL rhTPO induced a significant increase in [Ca2+]i (Figure 2C) with a faster and sustained phosphorylation of STAT5, AKT, and ERK1/2 than rhTPO stimulation in the absence of CPA (Figure 2D). In all of these conditions, Mk viability and immunophenotype were not altered by the treatments (supplemental Figure 2). Therefore, these results strongly suggest that cytoplasmic Ca2+ amplifies c-Mpl signaling via SOCE activation.
![TPO-induced intracellular signaling depends on intracellular and extracellular calcium. (A) Western blot analysis of STAT5, AKT, and ERK1/2 phosphorylation (p-STAT5, p-AKT and p-ERK1/2 respectively) after 10 minutes stimulation with recombinant human thrombopoietin (rhTPO) (100 ng/mL) in the presence or not of Ethylene Glycol Tetraacetic Acid (EGTA). Total STAT5, AKT and ERK1/2 were stained to ensure equal loading. Band densitometry analysis derived from 3 independent experiments is shown (P < .05). (B) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 10 minutes of stimulation with rhTPO (100 ng/mL) in the presence or not of BAPTA-AM. Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading. Band densitometry analysis derived from 3 independent experiments is shown (P < .05). (C) In physiological calcium (Ca2+) concentration (1.5 mM), stimulation with rhTPO (100 ng/mL) evokes an initial rise in [Ca2+]i followed by a plateau, which is further increased by concomitant stimulation with CPA. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 82 measurements from 3 independent experiments (P < .05). (D) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 60, 600, 1200, 1800, or 3600 seconds of stimulation with rhTPO (100 ng/mL) in the presence or not of CPA. Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/2/10.1182_blood.2019001103/2/m_bloodbld2019001103f2.png?Expires=1769413114&Signature=3aD-E0~UjDNDG7fTd5iE79g3Y4iXZSTH4D4ebzeZ2ZOotGispXXzX-vtWtXcCuAVRL9n2xu7tKKWBe71v1SHVXQRWt1w9-E2mLaQOMOgkt1Bf~dLoAOsdPPYr~PVExgXvWXXWVwC4-LhmAYrPlWY5bu7Fh9CR70q8mr6LLQungUOpYSkEoSM3HTzSt3GNkAmOC93eRTMgcZf0iubBFj-uoh4R51GmCh4~i2i-aO2KoGyGynvq-jDvJ-YACE1XI50W5BdtKQhZWcd02QUuN9wmCPyhICb06kfG1ointV2dwtpA7-l6GFeiLRV4ko~btrUtrOtWr2cMloPy1vhpJxiLw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TPO-induced intracellular signaling depends on intracellular and extracellular calcium. (A) Western blot analysis of STAT5, AKT, and ERK1/2 phosphorylation (p-STAT5, p-AKT and p-ERK1/2 respectively) after 10 minutes stimulation with recombinant human thrombopoietin (rhTPO) (100 ng/mL) in the presence or not of Ethylene Glycol Tetraacetic Acid (EGTA). Total STAT5, AKT and ERK1/2 were stained to ensure equal loading. Band densitometry analysis derived from 3 independent experiments is shown (P < .05). (B) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 10 minutes of stimulation with rhTPO (100 ng/mL) in the presence or not of BAPTA-AM. Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading. Band densitometry analysis derived from 3 independent experiments is shown (P < .05). (C) In physiological calcium (Ca2+) concentration (1.5 mM), stimulation with rhTPO (100 ng/mL) evokes an initial rise in [Ca2+]i followed by a plateau, which is further increased by concomitant stimulation with CPA. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 82 measurements from 3 independent experiments (P < .05). (D) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 60, 600, 1200, 1800, or 3600 seconds of stimulation with rhTPO (100 ng/mL) in the presence or not of CPA. Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading.
TPO-induced intracellular signaling depends on intracellular and extracellular calcium. (A) Western blot analysis of STAT5, AKT, and ERK1/2 phosphorylation (p-STAT5, p-AKT and p-ERK1/2 respectively) after 10 minutes stimulation with recombinant human thrombopoietin (rhTPO) (100 ng/mL) in the presence or not of Ethylene Glycol Tetraacetic Acid (EGTA). Total STAT5, AKT and ERK1/2 were stained to ensure equal loading. Band densitometry analysis derived from 3 independent experiments is shown (P < .05). (B) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 10 minutes of stimulation with rhTPO (100 ng/mL) in the presence or not of BAPTA-AM. Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading. Band densitometry analysis derived from 3 independent experiments is shown (P < .05). (C) In physiological calcium (Ca2+) concentration (1.5 mM), stimulation with rhTPO (100 ng/mL) evokes an initial rise in [Ca2+]i followed by a plateau, which is further increased by concomitant stimulation with CPA. The statistical analysis of peak fluorescence intensities is shown by box plot depicting the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 82 measurements from 3 independent experiments (P < .05). (D) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 60, 600, 1200, 1800, or 3600 seconds of stimulation with rhTPO (100 ng/mL) in the presence or not of CPA. Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading.
TPO engages SOCE in Mks to strengthen c-Mpl downstream signaling
Stimulation of CB-derived Mks for 10 minutes with 100 ng/mL rhTPO induced the formation of the Orai1/TRPC1/STIM1 protein complex, indicative of the activated SOCE machinery (Figure 3A). Accordingly, in our experiments, stimulation of CB-derived Mks for 10 minutes with 100 ng/mL rhTPO determined a significant increase in inositol triphosphate (IP3) formation at a level comparable to 1 U/mL thrombin, used as positive control (Figure 3B). Notably, when stores were depleted by rhTPO stimulation in the absence of extracellular Ca2+, restoring the extracellular Ca2+ levels led to a marked Ca2+ inflow in Mk cytoplasm, suggesting the involvement of SOCE machinery. Moreover, IP3 receptor inhibition, by 20 µM 2-APB, prevented SOCE activation and significantly reduced c-Mpl downstream signaling in Mks (Figure 3C-D). Finally, in the presence of 20 µM BTP-2, a widely used and specific SOCE inhibitor, rhTPO (100 ng/mL)-stimulated Mk mobilization of [Ca2+]i was not followed by a sustained plateau phase, as recorded in the untreated control (Figure 3E). In these conditions, there was a slight increase in STAT5, AKT, and ERK1/2 phosphorylation compared with cytokine-starved Mks, but a significantly lower response compared with rhTPO stimulation in the absence of BTP-2 (Figure 3F). Neither 2-APB nor BTP-2 affected Mk viability and immunophenotype (supplemental Figure 2).
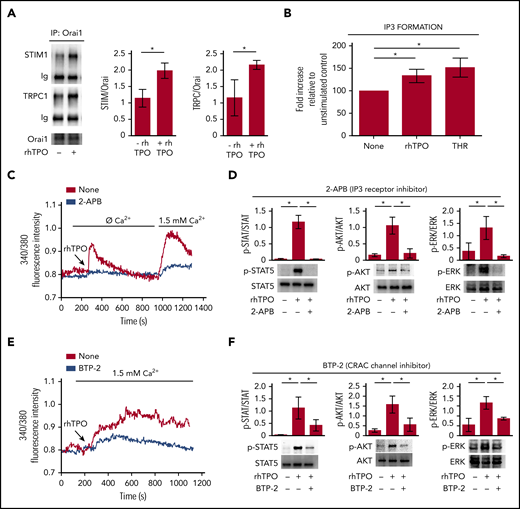
TPO-evoked calcium signals depend on SOCE mechanism. (A) Western blot analysis of STIM1 and TRPC1 coimmunoprecipitated with Orai1 after 10 minutes of stimulation with rhTPO (100 ng/mL). Band densitometry analysis of 4 independent experiments is shown (P < .05). (B) Enzyme-linked immunosorbent assay of IP3 formation in Mks after 10 minutes of stimulation with rhTPO (100 ng/mL). Thrombin stimulation (1 U/mL) was used as positive control (n = 7 independent experiments per condition; P < .05). (C) Representative analysis of Ca2+ flows in Mks after 10 minutes of stimulation with rhTPO (100 ng/mL), in the presence (blue line) or not (red line) of the IP3 receptor inhibitor 2-APB, in absence of extracellular Ca2+ (Ø Ca2+) and after addition of physiological extracellular Ca2+ concentration (1.5 mM). (D) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 10 minutes stimulation with rhTPO (100 ng/mL) in the presence or not of the IP3 receptor inhibitor 2-APB. Total STAT5, AKT, and ERK1/2 were stained to ensure the equal loading. The band densitometry analysis of 3 independent experiments is shown (P < .05). (E) Representative analysis of Ca2+ flows in Mks after 10 minutes of stimulation with rhTPO (100 ng/mL), in the presence (blue line) or not (red line) of the SOCE inhibitor BTP-2 (20 μM), in physiological extracellular Ca2+ concentration (1.5 mM). (F) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 10 minutes of stimulation with rhTPO (100 ng/mL) in the presence or not of BTP-2 (20 μM). Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading. The band densitometry analysis of 3 independent experiments is shown (P < .05).
TPO-evoked calcium signals depend on SOCE mechanism. (A) Western blot analysis of STIM1 and TRPC1 coimmunoprecipitated with Orai1 after 10 minutes of stimulation with rhTPO (100 ng/mL). Band densitometry analysis of 4 independent experiments is shown (P < .05). (B) Enzyme-linked immunosorbent assay of IP3 formation in Mks after 10 minutes of stimulation with rhTPO (100 ng/mL). Thrombin stimulation (1 U/mL) was used as positive control (n = 7 independent experiments per condition; P < .05). (C) Representative analysis of Ca2+ flows in Mks after 10 minutes of stimulation with rhTPO (100 ng/mL), in the presence (blue line) or not (red line) of the IP3 receptor inhibitor 2-APB, in absence of extracellular Ca2+ (Ø Ca2+) and after addition of physiological extracellular Ca2+ concentration (1.5 mM). (D) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 10 minutes stimulation with rhTPO (100 ng/mL) in the presence or not of the IP3 receptor inhibitor 2-APB. Total STAT5, AKT, and ERK1/2 were stained to ensure the equal loading. The band densitometry analysis of 3 independent experiments is shown (P < .05). (E) Representative analysis of Ca2+ flows in Mks after 10 minutes of stimulation with rhTPO (100 ng/mL), in the presence (blue line) or not (red line) of the SOCE inhibitor BTP-2 (20 μM), in physiological extracellular Ca2+ concentration (1.5 mM). (F) Western blot analysis of p-STAT5, p-AKT, and p-ERK1/2 after 10 minutes of stimulation with rhTPO (100 ng/mL) in the presence or not of BTP-2 (20 μM). Total STAT5, AKT, and ERK1/2 were stained to ensure equal loading. The band densitometry analysis of 3 independent experiments is shown (P < .05).
c-Mpl activation regulates CALR-STIM1 interaction in Mks
c-Mpl, STIM1, and Orai1 expression increased during Mk maturation from HSCs, whereas CALR was equally expressed at all of the observed time points (supplemental Figure 3). In cytokine-starved conditions, CALR formed a molecular complex with STIM1, as demonstrated by the presence of a positive signal revealed by in situ PLA. Following 10 minutes of stimulation with 100 ng/mL rhTPO, 10 μM CPA, or 1 U/mL thrombin that promotes SOCE activity, the proteins dissociated (Figure 4A). CALR-STIM1 dissociation upon 100 ng/mL rhTPO stimulation was further confirmed by immunoprecipitation assay in CB-derived Mks (Figure 4B). ERp57 is the CALR cochaperone known to bind STIM1 and thus to regulate SOCE.16 In cytokine-starved Mks, ERp57 immunoprecipitated with STIM1, whereas 10 minutes of stimulation with 100 ng/mL rhTPO led to ERp57 dissociation from STIM1 (Figure 4C).
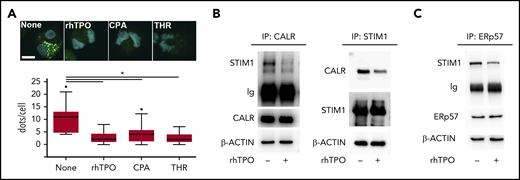
CALR-STIM1 binding is impaired in presence of CALR type I mutation. (A) Top, In situ PLA imaging of STIM1 and CALR interaction (green dots) in CB-derived mature Mks starved (None) or treated with 100 ng/mL rhTPO, 10 μM CPA, or 1 U/mL thrombin (THR). Nuclei are visualized with Hoechst 33258 (blue). Scale bar, 10 μm. Original magnification, ×60/NA1.2. Bottom, Quantification of dot number per single Mk (overall, data derive from 92 measurements; P < .05). (B) CB-derived Mks were treated (+) or not (−) for 10 minutes with rhTPO (100 ng/mL) and lysed. Lysates were immunoprecipitated (IP) with an anti-CALR or anti-STIM1 antibody and subjected to western blotting. Membranes were stained with antibodies against STIM1 and CALR, respectively, and reblotted with the same antibodies to ensure equal immunoprecipitation of the proteins of interest. (C) CB-derived Mks were treated (+) or not (−) for 10 minutes with rhTPO (100 ng/mL) and lysed. Lysates were immunoprecipitated with an anti-ERp57 antibody and subjected to western blotting. Membrane was stained with an antibody against STIM1 and reblotted with anti-ERp57 antibody to ensure equal immunoprecipitation of the protein. β-ACTIN was used as input control.
CALR-STIM1 binding is impaired in presence of CALR type I mutation. (A) Top, In situ PLA imaging of STIM1 and CALR interaction (green dots) in CB-derived mature Mks starved (None) or treated with 100 ng/mL rhTPO, 10 μM CPA, or 1 U/mL thrombin (THR). Nuclei are visualized with Hoechst 33258 (blue). Scale bar, 10 μm. Original magnification, ×60/NA1.2. Bottom, Quantification of dot number per single Mk (overall, data derive from 92 measurements; P < .05). (B) CB-derived Mks were treated (+) or not (−) for 10 minutes with rhTPO (100 ng/mL) and lysed. Lysates were immunoprecipitated (IP) with an anti-CALR or anti-STIM1 antibody and subjected to western blotting. Membranes were stained with antibodies against STIM1 and CALR, respectively, and reblotted with the same antibodies to ensure equal immunoprecipitation of the proteins of interest. (C) CB-derived Mks were treated (+) or not (−) for 10 minutes with rhTPO (100 ng/mL) and lysed. Lysates were immunoprecipitated with an anti-ERp57 antibody and subjected to western blotting. Membrane was stained with an antibody against STIM1 and reblotted with anti-ERp57 antibody to ensure equal immunoprecipitation of the protein. β-ACTIN was used as input control.
CALR mutants present altered CALR-STIM1 and ERp57-STIM1 interaction
In cytokine-starved conditions, we observed that samples from CALR type I and type II patients present a higher percentage of Mks with a Ca2+ periodic oscillatory activity compared with healthy controls and patients harboring a JAK2V617F mutation (Figure 5A-B). Confocal immunofluorescence analysis revealed the relocation of CALR mutants to the plasma membrane (Figure 5C). Concomitantly, we observed decreased interaction between CALR and STIM1 in MPN patients as compared with healthy subjects and JAK2V617F-mutated Mks (Figure 5D). Most importantly, the analysis of Mks, derived from the peripheral blood of an advanced case of myelofibrosis harboring CALR type I homozygous mutation, showed an almost complete dissociation of CALR type I from STIM1 (supplemental Figure 4).
![Altered CALR-ERp57-STIM1 binding in presence of CALR mutations. (A) Statistical analysis of the percentage of Mks presenting periodic Ca2+ spikes. Box plots depict the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 6 samples per subgroup (P < .05). (B) Representative analysis of spontaneous Ca2+ spikes in MPN Mk cultured in physiological Ca2+ concentration (1.5 mM) without addition of rhTPO. Samples were recorded for a minimum of 15 minutes. (C) Analysis of CALR localization in mature Mks from healthy controls (Ctrl) and MPN patients (CALR [green stain]; nuclei [blue stain]; scale bar, 15 μm; original magnification, ×60/NA1.2). (D) In situ PLA of STIM1 and CALR interaction. Quantification of dot number per single Mk (overall, data derive from 402 measurements; P < .05). (E) In situ PLA of STIM1 and ERp57 interaction. Quantification of dot number per single Mk (overall, data derive from 412 measurements; P < .05). Overall, data derive from 5 samples per subgroup.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/2/10.1182_blood.2019001103/2/m_bloodbld2019001103f5.png?Expires=1769413114&Signature=iL9P6EQUJ7pclxXlw7hQ0nsE78XyordPYX-ks9-mio3vHnEy5BoNn64e1JyFH9WfmLUH0D1jLupAjyddYTiq30tnPOEkYtXLOv1DseMNuRpFYYgV6YeOWW-rmDX29bnc474ImNteDEr3R~0Brj~LbQ2XCJtH7IEOwRyBopV6UKv933B-sk~Nr5Az7uSfZ6oVbhn6fGxo3x5GtELj83MyXGRFqneSC6ZSynQfTsCxbsQqqthns9XphMZ1h-QgzMoDkfgz1MazWU6AEEF6bwvv7fLSrSNc0wUQS9klIz4JEtdlNd~ztoJ9RnEVsGXLNYGIr51XpOKVNgKxunht0rQAFA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Altered CALR-ERp57-STIM1 binding in presence of CALR mutations. (A) Statistical analysis of the percentage of Mks presenting periodic Ca2+ spikes. Box plots depict the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 6 samples per subgroup (P < .05). (B) Representative analysis of spontaneous Ca2+ spikes in MPN Mk cultured in physiological Ca2+ concentration (1.5 mM) without addition of rhTPO. Samples were recorded for a minimum of 15 minutes. (C) Analysis of CALR localization in mature Mks from healthy controls (Ctrl) and MPN patients (CALR [green stain]; nuclei [blue stain]; scale bar, 15 μm; original magnification, ×60/NA1.2). (D) In situ PLA of STIM1 and CALR interaction. Quantification of dot number per single Mk (overall, data derive from 402 measurements; P < .05). (E) In situ PLA of STIM1 and ERp57 interaction. Quantification of dot number per single Mk (overall, data derive from 412 measurements; P < .05). Overall, data derive from 5 samples per subgroup.
Altered CALR-ERp57-STIM1 binding in presence of CALR mutations. (A) Statistical analysis of the percentage of Mks presenting periodic Ca2+ spikes. Box plots depict the upper and lower values (lowest and highest horizontal line, respectively), lower and upper quartile with median value (box), and outside values (dots). Overall, data derive from 6 samples per subgroup (P < .05). (B) Representative analysis of spontaneous Ca2+ spikes in MPN Mk cultured in physiological Ca2+ concentration (1.5 mM) without addition of rhTPO. Samples were recorded for a minimum of 15 minutes. (C) Analysis of CALR localization in mature Mks from healthy controls (Ctrl) and MPN patients (CALR [green stain]; nuclei [blue stain]; scale bar, 15 μm; original magnification, ×60/NA1.2). (D) In situ PLA of STIM1 and CALR interaction. Quantification of dot number per single Mk (overall, data derive from 402 measurements; P < .05). (E) In situ PLA of STIM1 and ERp57 interaction. Quantification of dot number per single Mk (overall, data derive from 412 measurements; P < .05). Overall, data derive from 5 samples per subgroup.
It was previously shown that CALR mutants lose the capacity to interact with ERp57, which relocates to the nucleus.17,18 In accordance, we showed that in CALR-mutated Mks ERp57-STIM1 interaction is significantly decreased, whereas it is normally retained in cytokine-starved Mks from healthy controls and JAK2V617F patients (Figure 5E).
Increased SOCE activity in CALR type I and type II Mks impacts cell proliferation
To analyze the impact of SOCE activation on Mk behavior, we treated CB-derived Mks with 100 ng/mL rhTPO in the presence or not of SOCE inhibition (20 µM BTP-2). Cell proliferation measured by both cell count and biochemical analysis of the proliferating cell nuclear antigen (PCNA), was significantly reduced by BTP-2 treatment, without affecting Mk viability and ploidy, suggesting that SOCE is an essential driver of Mk proliferation (supplemental Figures 5 and 6). Consistently, Mks from CALR type I and type II patients demonstrated a higher proliferation rate than healthy controls, as demonstrated by the significantly increased number of Mks counted at the end of differentiation compared with the starting number of HSCs and PCNA expression (Figure 6A-B). Inhibition of SOCE with 20 µM BTP-2 was able to significantly restrict the proliferation of CALR-mutated Mks cultured for the last 3 days of differentiation in cytokine-starved conditions, providing evidence that targeting intracellular Ca2+ signaling may counteract CALR type I– and type II–induced constitutive cell proliferation (Figure 6C-F). Finally, in CALR type I mutant UT-7 cells, where endogenous levels of CALR wild type were downmodulated by short hairpins RNAs, CALR type I conferred a constitutively increased proliferation capacity, in cytokine-starved conditions, which was dampened by blocking SOCE with 20 µM BTP-2 (supplemental Figure 7).

Proliferation of CALR type I Mks is counteracted by SOCE inhibition. (A) Cell proliferation assay of Mk culture from healthy controls (Ctrl) and CALR type I and type II patients (n = 8 per subgroup; P < .05). (B) Representative western blot analysis of PCNA expression in mature Mks from healthy control (ctrl) and CALR type I and type II patients. β-ACTIN was used to ensure equal loading. The band densitometry analysis of 3 independent experiments is shown (P < .05). (C) Cell proliferation assay of cytokine-starved CALR type I Mks in presence or not of the SOCE inhibitor BTP-2 (20 μM) (n = 3; P < .05). (D) Western blot analysis of PCNA expression in cytokine-starved CALR type I Mks cultured in absence (−) or presence (+) of 20 µM BTP-2. β-ACTIN serves as loading control. The band densitometry analysis of 3 independent experiments is shown (P < .05). (E) Cell proliferation assay of cytokine-starved CALR type II Mks in presence or not of BTP-2 (20 μM) (n = 3; P < .05). (F) Western blot analysis of PCNA expression in cytokine-starved CALR type II Mks cultured in absence (−) or presence (+) of 20 µM BTP-2. β-ACTIN serves as loading control. The band densitometry analysis of 3 independent experiments is shown (P < .05).
Proliferation of CALR type I Mks is counteracted by SOCE inhibition. (A) Cell proliferation assay of Mk culture from healthy controls (Ctrl) and CALR type I and type II patients (n = 8 per subgroup; P < .05). (B) Representative western blot analysis of PCNA expression in mature Mks from healthy control (ctrl) and CALR type I and type II patients. β-ACTIN was used to ensure equal loading. The band densitometry analysis of 3 independent experiments is shown (P < .05). (C) Cell proliferation assay of cytokine-starved CALR type I Mks in presence or not of the SOCE inhibitor BTP-2 (20 μM) (n = 3; P < .05). (D) Western blot analysis of PCNA expression in cytokine-starved CALR type I Mks cultured in absence (−) or presence (+) of 20 µM BTP-2. β-ACTIN serves as loading control. The band densitometry analysis of 3 independent experiments is shown (P < .05). (E) Cell proliferation assay of cytokine-starved CALR type II Mks in presence or not of BTP-2 (20 μM) (n = 3; P < .05). (F) Western blot analysis of PCNA expression in cytokine-starved CALR type II Mks cultured in absence (−) or presence (+) of 20 µM BTP-2. β-ACTIN serves as loading control. The band densitometry analysis of 3 independent experiments is shown (P < .05).
Discussion
c-Mpl signaling is essential for Mk differentiation. TPO binding to c-Mpl leads to the activation of multiple downstream signaling pathways, including the phosphoinositide-3-kinase/AKT, MAPK, and ERK1/2 pathways.28 AKT and ERK1/2 kinases are known to be crucial for the regulation of Mk maturation and platelet production.21,25,29 In different human Mk cell lines, rhTPO treatment induces a rapid and sustained activation of ERK1/2.30-32 In human CB-derived CD34+ progenitor cultures, the MAPK inhibitor PD98059 induces cell proliferation and delayed Mk differentiation in the presence of TPO.33 Cell proliferation is related to Ca2+ signaling inside of the cells, due to Ca2+ fluctuation from and to the ER.34 SOCE is a pivotal regulator of Ca2+ flux inside of the cell and is activated by the interplay between STIM1, an ER Ca2+ sensor, and Orai1, a cell membrane Ca2+ channel.10
In this article, we thought to investigate more precisely the physiological regulation of Ca2+ mobilization by TPO and the effect of its alteration in CALR type I and II MPNs. We showed that rhTPO elicits dose-dependent cytosolic Ca2+ flows in cultured Mks from human CB. Importantly, exploiting a human TPO-dependent leukemia cell line, UT-7/TPO,35 it was demonstrated that rhTPO induces a significant accumulation of IP3 with consequent Ca2+ mobilization from the intracellular stores.36 Similarly, we demonstrate that the treatment of human CB-derived Mks with rhTPO stimulates IP3 formation and induces ER Ca2+ depletion via activation of the IP3 receptor and subsequent Ca2+ flows from SOCE activation. These data confirmed our previous work showing that emptying the IP3-sensitive Ca2+ reservoir led to the formation of a complex between STIM1, Orai1, and TRPC1 and to the resulting activation of SOCE in human Mks.20 This global increase of intracellular Ca2+ strengthened c-Mpl downstream signaling. Thus, stimulation of Mks with rhTPO, in the absence of extracellular Ca2+ or the presence of the intracellular Ca2+ chelator BAPTA-AM, led to a significant decrease in STAT5, AKT, and ERK1/2 phosphorylation compared with untreated controls. The role of Ca2+ in sustaining chemokine and cytokine-dependent activation of intracellular responses has been described in different cell types.37-40 Furthermore, Ca2+ is emerging as an essential regulator of Mk function.10 Some of us demonstrated that SOCE activators and Ca2+ mobilization activate signaling cascades that trigger proplatelet formation.20,21,41 Additionally, Mks express glutamate-gated Ca2+-permeable N-Methyl-d-aspartic acid receptors that support the proliferation of leukemic megakaryoblastic cell lines by promoting Ca2+ entry from the extracellular environment.42 Furthermore, inhibition of the TRPC6, determining a decrease in Ca2+ influx, resulted in a reduction in cell proliferation of CD34+-derived Mks.43
Here, we show that cytoplasmic Ca2+ spikes are regulated by CALR-STIM1 interaction. Our data demonstrate that the complex between CALR and STIM1 in cytokine-starved Mks, dissociates after rhTPO stimulation. Additionally, the interaction between CALR and STIM1 seemed to be mediated by the CALR cochaperone ERp57 that dissociated from STIM1 after rhTPO stimulation. It has been demonstrated that overexpression of CALR affects both store depletion and store-operated Ca2+ influx in different cell types, thus supporting the notion that changing the levels of CALR can alter ER Ca2+ homeostasis within ranges that are relevant to control SOCE.11-13 It is well known that ER luminal oxidoreductase ERp57 is an associated cochaperone of CALR, which docks onto the extended proline-rich arm of the protein.44,45 Importantly, ERp57 binds to the ER luminal domain of STIM1 serving as a physiological brake for the initiation of SOCE, as demonstrated by the presence of an accelerated SOCE in ERp57−/− cells.16
In Mks derived from CALR type I and II MPN patients, we observed a decreased association between CALR and STIM1. Importantly, we also observed that in Mks, from a CALR type I homozygous primary myelofibrosis patient, CALR relocated to the plasma membrane, while completely dissociating from STIM1. This process seemed to be mediated by the CALR cochaperone ERp57 because, in CALR type I and type II mutant Mks, STIM1-ERp57 interaction was constitutively decreased compared with healthy controls. Importantly, Elf et al showed that CALR mutants fail to interact with ERp57,17 whereas Pronier et al demonstrated that the CALR-mutant proteins show decreased binding affinities for ERp57 compared with wild-type CALR, leading to consequent-enriched recruitment of ERp57 to the c-Mpl promoter into the nucleus.18
We previously showed an increased mobilization from ER of Ca2+ in type I–mutated Mks compared with type II–mutated Mks followed with an increased uptake of extracellular Ca2+ after CPA induction.5 In this work, we observed an increase of spontaneous Ca2+ periodic oscillatory activity in CALR-mutated Mks compared with controls and JAK2V617F Mks. This alteration of Ca2+ flows was the consequence of the decreased interaction between CALR mutants and STIM1 in Mks.
In healthy Mks, inhibition of SOCE significantly restricted Mk proliferation induced by increasing doses of rhTPO. Most importantly, CALR type I– and II–mutated Mks displayed increased proliferation capacity compared with healthy controls that was corrected by SOCE inhibition. Overall, our data suggest an additional mechanism to the constitutive activation of c-Mpl by CALR type I and II mutants.6,7,46,47 We propose that the relocation of CALR mutants to the TPO receptor determines a decreased formation of the CALR-ERp57-STIM1 complex which, in turn, leads to altered regulation of Ca2+ flux; increased activation of STAT5, AKT, and ERK1/2; and Mk proliferation. It is proven that Ca2+ oscillations play a fundamental role in regulating many cell processes, including cell proliferation and metabolism.48 As a consequence, changes in the levels or activity of Ca2+ regulatory proteins have a profound impact on cancer development and progression.49 Thus, modulating Ca2+ regulatory pathways is a promising therapeutic approach.49-51
In conclusion, our data demonstrate that the loss of function of the CALR mutant in binding ERp57 and STIM1 determines constitutive activation of SOCE. This altered regulation of SOCE further sustains the MPL activation and subsequent phosphorylation of STAT5, AKT, and ERK1/2, inducing Mk proliferation. Further investigation is needed to understand whether this altered cell function is determined by the decrease in CALR levels in the ER or by the presence of CALR mutants. Nevertheless, the abnormal regulation of Ca2+ flows may represent a new therapeutic target to counteract myeloproliferation in CALR-mutated MPNs (Figure 7).
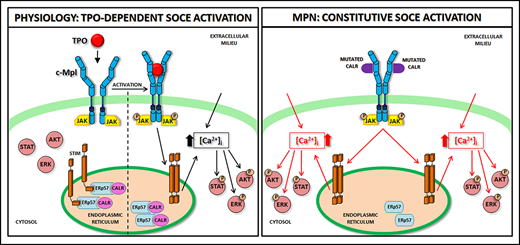
![Schematic representation of SOCE activation in physiologic and pathologic megakaryopoiesis. In physiologic conditions (A), upon binding with TPO, c-Mpl promotes phosphorylation of downstream signaling molecules (STAT, AKT, ERK) together with IP3 formation and consequent mobilization of the IP3-sensitive Ca2+ pool. In Mks with repleted ER, STIM is localized in an inactive configuration in the ER membrane, and forms a complex with CALR and ERp57. Depletion of Ca2+ stores triggers Ca2+ release from the ER through IP3 receptors (IP3Rs) and consequent Ca2+ dissociation from STIM-CALR-ERp57 complex. Consequently, STIMs oligomerize and translocate next to the plasma membrane. Then, STIM binding to Orai and TRPC results in the opening of these channels and extracellular Ca2+ entry. The increased cytosolic Ca2+ concentration ([Ca2+]i) in turn sustains the long-lasting phosphorylation of c-Mpl downstream pathways. In pathologic conditions (B), the TPO-independent activation of c-Mpl by mutant CALR, which leaves the ER and consequently loses its binding to STIM and ERp57, leads to a constitutive activation of SOCE, which results in increased Mk proliferation.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/2/10.1182_blood.2019001103/2/m_bloodbld2019001103f7.png?Expires=1769413114&Signature=NmbFYObi3C2ElKh0HxIZbP-QohGM~Dfk6~4v5O~mXdvmiSWZKFzIs61UhnHzL-pv7Ctsp20TSAQPT1QBOWhUCMyMSViaOEkNRfOhxTxyVuxym~NlhhrdhSslvnjPYmIPXONXN6jN82f1X07sPO4skelgX8OHPDoVaHb2~RvyFgQATBh0aJnH6PkSJ1R~EO8d~pw2E1fy-oi7cDkCW02c2Oz9vtpkEyQFz6SlQ0a8gtMTbLMIl4jaC2X8bdfpA2th~EMRAc9kV2-2NmI2-ALrNqjjigImvGGULCv9y4yqNyEc1lRn01Tfq1VAuRZw-AUn8diGb4LMM-gstzJqx~dDFA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematic representation of SOCE activation in physiologic and pathologic megakaryopoiesis. In physiologic conditions (A), upon binding with TPO, c-Mpl promotes phosphorylation of downstream signaling molecules (STAT, AKT, ERK) together with IP3 formation and consequent mobilization of the IP3-sensitive Ca2+ pool. In Mks with repleted ER, STIM is localized in an inactive configuration in the ER membrane, and forms a complex with CALR and ERp57. Depletion of Ca2+ stores triggers Ca2+ release from the ER through IP3 receptors (IP3Rs) and consequent Ca2+ dissociation from STIM-CALR-ERp57 complex. Consequently, STIMs oligomerize and translocate next to the plasma membrane. Then, STIM binding to Orai and TRPC results in the opening of these channels and extracellular Ca2+ entry. The increased cytosolic Ca2+ concentration ([Ca2+]i) in turn sustains the long-lasting phosphorylation of c-Mpl downstream pathways. In pathologic conditions (B), the TPO-independent activation of c-Mpl by mutant CALR, which leaves the ER and consequently loses its binding to STIM and ERp57, leads to a constitutive activation of SOCE, which results in increased Mk proliferation.
Schematic representation of SOCE activation in physiologic and pathologic megakaryopoiesis. In physiologic conditions (A), upon binding with TPO, c-Mpl promotes phosphorylation of downstream signaling molecules (STAT, AKT, ERK) together with IP3 formation and consequent mobilization of the IP3-sensitive Ca2+ pool. In Mks with repleted ER, STIM is localized in an inactive configuration in the ER membrane, and forms a complex with CALR and ERp57. Depletion of Ca2+ stores triggers Ca2+ release from the ER through IP3 receptors (IP3Rs) and consequent Ca2+ dissociation from STIM-CALR-ERp57 complex. Consequently, STIMs oligomerize and translocate next to the plasma membrane. Then, STIM binding to Orai and TRPC results in the opening of these channels and extracellular Ca2+ entry. The increased cytosolic Ca2+ concentration ([Ca2+]i) in turn sustains the long-lasting phosphorylation of c-Mpl downstream pathways. In pathologic conditions (B), the TPO-independent activation of c-Mpl by mutant CALR, which leaves the ER and consequently loses its binding to STIM and ERp57, leads to a constitutive activation of SOCE, which results in increased Mk proliferation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Cesare Perotti for supplying human CB.
This work was supported by the Cariplo Foundation (2013-0717 and 2017-0920) (A.B. and C.A.D.B.); US National Institutes of Health, National Institute of Biomedical Imaging and Bioengineering grant R01 EB016041-02 and National Heart, Lung, and Blood Institute grant R01 HL134829 (A.B.); Associazione Italiana per la Ricerca sul Cancro (AIRC) IG 2016 18700 (A.B.); “Special Program Molecular Clinical Oncology 5x1000” AIRC–Gruppo Italiano Malattie Mieloproliferative (AGIMM; detailed description of the AGIMM project is available at www.progettoagimm.it) (G.B. and M.C.); AIRC 5x1000 call “Metastatic disease: the key unmet need in oncology” to the MYeloid NEoplasms Research Venture Airc (MYNERVA) project, #21267 (M.C.); Fondazione Regionale Ricerca Biomedica (FRRB; project no. 2015-0042) (M.C.); Italian Ministry of Health (Ricerca Finalizzata Giovani Ricercatori GR-2016-02363136) (V.A.); INCAPLbio 2015 (I.P. and C.M.); and la Ligue Nationale Contre le Cancer (I.P. and C.M.).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authorship
Contribution: C.A.D.B. and V.A. designed research studies, conducted experiments, acquired and analyzed data, and wrote the manuscript; C.M., D.P., P.M.S., and D.L. conducted experiments, acquired and analyzed data, and edited the manuscript; F.M. provided technical support for calcium analysis, analyzed data, and edited the manuscript; E.R., D.C., U.G., A.I., G.B., V.R., I.P., and M.C. provided reagents and patient samples, analyzed data, and edited the manuscript; and A.B. supervised the project, designed research studies, acquired and analyzed data, provided reagents, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alessandra Balduini, Department of Molecular Medicine, University of Pavia, via Forlanini 6, 27100, Pavia, Italy; e-mail: alessandra.balduini@unipv.it.