

Key Points
Wdr26 is a critical regulator of nuclear condensation during red blood cell development in mammals and fish.
Wdr26 functions as a core subunit of an E3 ubiquitin ligase complex to promote the degradation of nuclear proteins in erythroblasts.

Abstract
Mammalian red blood cells lack nuclei. The molecular mechanisms underlying erythroblast nuclear condensation and enucleation, however, remain poorly understood. Here we show that Wdr26, a gene upregulated during terminal erythropoiesis, plays an essential role in regulating nuclear condensation in differentiating erythroblasts. Loss of Wdr26 induces anemia in zebrafish and enucleation defects in mouse erythroblasts because of impaired erythroblast nuclear condensation. As part of the glucose-induced degradation-deficient ubiquitin ligase complex, Wdr26 regulates the ubiquitination and degradation of nuclear proteins, including lamin B. Failure of lamin B degradation blocks nuclear opening formation leading to impaired clearance of nuclear proteins and delayed nuclear condensation. Collectively, our study reveals an unprecedented role of an E3 ubiquitin ligase in regulating nuclear condensation and enucleation during terminal erythropoiesis. Our results provide mechanistic insights into nuclear protein homeostasis and vertebrate red blood cell development.
Introduction
Maturation of mammalian red blood cells is a complex process involving production of hemoglobin, remodeling of cell membrane, and clearance of cellular organelles.1-3 A unique feature of mammalian erythrocytes is that they are devoid of nuclei, a phenomenon thought to accommodate the mature erythrocytes to the narrow lumen of capillaries.3 Erythroblast enucleation, or extrusion of the nucleus, requires a dramatic reduction in nuclear size, a process called nuclear condensation. The volume of mammalian erythroblast nuclei decreases about 8 times during terminal differentiation.3 Erythroblast nuclear condensation requires the clearance of a vast amount of nuclear proteins because the extruded nuclei are largely depleted of proteins.4 Hattangadi et al reported that Exportin 7 (Xpo7 or Ranbp16), a gene highly induced during terminal erythropoiesis, was required for the export of many nuclear proteins, including histones in late-stage erythroblasts.4 Besides being exported through the canonical nucleocytoplasmic RAN transport machinery, histones and other nuclear proteins may also exit nuclei through large openings transiently formed on the nuclear envelope of erythroblasts through the activation of caspase-3.5,6 The precise mechanisms underlying nuclear opening formation as well as nuclear protein degradation in differentiating erythroblasts, however, remain poorly understood.
The ubiquitin-proteasome system is a complex and highly regulated mechanism of intracellular protein degradation. Recently, the E2 ubiquitin-conjugating enzyme Ube2o was reported to promote elimination of ribosomes in terminally differentiating erythroblasts by targeting ribosomal proteins for degradation.7 In addition, a study in murine erythroblasts has implicated the E3 ubiquitin ligase Trim58 in the proteasome-dependent degradation of the microtubule motor dynein to facilitate enucleation.8 The erythroblast enucleation defect observed in this study, however, was later found to be caused by nonspecific effects of Trim58 short hairpin RNAs (shRNAs).9 Despite that the ubiquitin-proteasome system was first discovered in the circulating reticulocytes,10-12 the functional significance of this protein degradation system, especially the ubiquitin ligases, in differentiating erythroblasts remains unknown.
Through transcriptome profiling of maturing erythroblasts, we identified WD40 repeat protein 26 (Wdr26) as an erythroid-enriched gene. Wdr26 was shown to be part of the glucose-induced degradation-deficient (Gid) or Gid/C-terminal to LisH (CTLH) ubiquitin ligase complex.13,14 Despite its enriched expression in the erythroid tissue, the function of Wdr26 in hematopoiesis has not been reported. Here we analyzed the role of Wdr26 in terminal erythropoiesis by loss-of-function studies in mouse primary erythroblasts, mouse erythroleukemia (MEL) cells, and zebrafish. Our results demonstrate that Wdr26 functions as a core subunit of the Gid ubiquitin ligase complex to regulate the polyubiquitination and degradation of nuclear proteins, including lamin B. Wdr26-mediated lamin B degradation is essential for the formation of the transient nuclear opening required for rapid export and clearance of nuclear proteins. Our work reveals an unprecedented role of an E3 ubiquitin ligase in regulating nuclear condensation and enucleation during vertebrate terminal erythropoiesis.
Methods
Flow cytometry
In vitro-cultured erythroid cells were collected at 36 and 48 hours posterythropoietin-induced erythroid differentiation for analyses of nuclear size and enucleation, respectively. To analyze the enucleation, cells were stained with anti-mouse Ter-119 (BioLegend) and Hoechst 33342 (Cell Signaling Technology) followed by flow cytometry analysis using CytoFLEX cytometer (Beckman). To analyze the R1-R5 subpopulations, cells were stained with anti-mouse Ter-119 (BioLegend) and anti-mouse CD71 (BioLegend) followed by flow cytometry analysis.
Generation and phenotypic analysis of wdr26b-knockout zebrafish
Wdr26b was knocked out in zebrafish using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 technology.15 Peripheral blood cells from adult wdr26b−/− and wild-type fish were mounted onto slides by cytospin. Following a quick fixation in methanol, the cells were stained with Giemsa dye (Sigma).
To assess the tolerance to hypoxia, 2 wild-type and 2 wdr26b−/− fish were placed in a 25-cm2 flask filled with fish water. The flask was sealed with parafilm and the survival time for each fish was recorded. The dissolved oxygen levels were ∼7.5 and ∼1.7 mg/L at the beginning and end of the experiment, respectively. Three independent experiments were performed.
Generation of knockout MEL cell lines
The guide RNA (gRNA) oligos (supplemental Methods, available on the Blood Web site) were cloned into pX330 vector (Addgene).16 The gRNA plasmids were electroporated into MEL cells together with pEF1α plasmid that encodes a puromycin-resistance gene. The cells were selected with 5 μg/mL puromycin for 9 days and single clones were screened for deletion by polymerase chain reaction (PCR).
Heme staining and quantification
Heme was stained with o-dianisidine and quantified using fluorescence heme assay following the methods previously reported.17,18
RNA-seq analysis
RNA-sequencing (RNA-seq) libraries were constructed and subjected to Illumina sequencing. For RNA-seq in MEL cells before and after dimethyl sulfoxide (DMSO)-induced erythroid-like differentiation, the raw reads were mapped to mouse reference genome (GRCm38) by TopHat2.19 Genes upregulated after DMSO induction were identified by Cufflinks using fold change >1.5 and P < .05. For RNA-seq in wild-type and Wdr26-knockout MEL cells, the raw reads were aligned to mouse reference genome (mm10) by HISAT.20 Gene expression was analyzed by RSEM.21
Affinity purification and mass spectrometry analysis
The lysates of DMSO-induced MEL cells stably expressing Wdr26-FLAG or Gid8-FLAG were subjected to pull-down experiments using ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich) as previously described.22 For mass spectrometry analysis, the eluted proteins were digested with trypsin, followed by loading onto an analytical C18 column and analyzing with a TripleTOF 5600 System (AB SCIEX) fitted with a Nanospray III source (AB SCIEX). Peptides were identified by using MASCOT search engine (Matrix Science) against the UniProt mouse database.
In vitro ubiquitination assays
The ubiquitination reaction mix including 100 μM Ubiquitin (Boston Biochem), 100 nM human UBE1 (Boston Biochem), 1 μM human UBE2H (Boston Biochem), 5 μL purified Lamin B2, and 1 mM Mg-ATP was incubated with the protein pull-down fraction at 37°C for 1 hour. Following the reaction, samples were analyzed by immunoblotting with an anti-ubiquitin antibody (Abcam).
Results
Wdr26 is highly expressed in differentiating erythroblasts
To provide new insights into terminal erythropoiesis, we analyzed the transcriptomes of differentiating erythroid cells. An RNA-seq experiment was performed on MEL cells before and after erythroid-like differentiation induced by DMSO. A total of 348 protein-coding genes, including many known erythroid-enriched genes such as hemoglobin and heme synthesis genes, were significantly upregulated upon erythroid-like induction in MEL cells (supplemental Table 1). These genes were sorted based on their expression in differentiating mouse fetal erythroblasts and human primary CD34+ cells.23,24 We found that the expression of Wdr26, which encodes a WD40 repeat-containing protein, was upregulated in both human and mouse erythroblasts as well as in chemically induced erythroid-like cells (Figure 1A-B). These results were further validated by quantitative reverse-transcription PCR analyses in both MEL cells and mouse primary fetal erythroblasts (Figure 1C-D).

Wdr26 is upregulated during terminal erythropoiesis. (A) Heat maps showing the mRNA sequencing results of Wdr26 and control genes in MEL cells before and after DMSO-induced erythroid-like differentiation (left) and in R2-R5 subpopulations of primary mouse erythroblast23 (right, MPFE). MEL, mouse erythroleukemia cells; MPFE, mouse primary fetal liver erythroblasts. (B) Heat map for WDR26 and control genes in terminally differentiating human erythroblasts.24 Stages of erythroblasts shown include proerythroblast (ProE), early (EBaso) and late (LBaso) basophilic erythroblast, polychromatic erythroblast (Poly), and orthochromatic erythroblast (Ortho). (C-D) Quantitative reverse-transcription-PCR analyses of Wdr26 in differentiating MEL cells (C) and primary mouse erythroblasts isolated from E14.5 mouse fetal liver cells (D). Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (E) Western analysis of Wdr26 in multiple mouse tissues. (F) Wdr26 mRNA expression in different hematopoietic lineages isolated from adult mouse bone marrow. Error bars represent SEM from 3 replicates. **P < .01. (G-H) Western analysis of Wdr26 in (G) primary mouse erythroblasts and (H) differentiating MEL cells. SEM, standard error of the mean.
Wdr26 is upregulated during terminal erythropoiesis. (A) Heat maps showing the mRNA sequencing results of Wdr26 and control genes in MEL cells before and after DMSO-induced erythroid-like differentiation (left) and in R2-R5 subpopulations of primary mouse erythroblast23 (right, MPFE). MEL, mouse erythroleukemia cells; MPFE, mouse primary fetal liver erythroblasts. (B) Heat map for WDR26 and control genes in terminally differentiating human erythroblasts.24 Stages of erythroblasts shown include proerythroblast (ProE), early (EBaso) and late (LBaso) basophilic erythroblast, polychromatic erythroblast (Poly), and orthochromatic erythroblast (Ortho). (C-D) Quantitative reverse-transcription-PCR analyses of Wdr26 in differentiating MEL cells (C) and primary mouse erythroblasts isolated from E14.5 mouse fetal liver cells (D). Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (E) Western analysis of Wdr26 in multiple mouse tissues. (F) Wdr26 mRNA expression in different hematopoietic lineages isolated from adult mouse bone marrow. Error bars represent SEM from 3 replicates. **P < .01. (G-H) Western analysis of Wdr26 in (G) primary mouse erythroblasts and (H) differentiating MEL cells. SEM, standard error of the mean.
To reveal the endogenous expression pattern of Wdr26, we performed immunoblotting analysis on adult mouse tissues. Results showed that although the expression of Wdr26 was detected in multiple tissues including the spleen, brain, liver, and stomach, it was highly expressed in the hematopoietic tissue bone marrow (Figure 1E). Next, we surveyed the cell lineage-specific expression pattern of Wdr26 by fractionating the mouse bone marrow into 6 subpopulations using biotin-conjugated lineage-specific antibodies. Consistent with the transcriptomics results, Wdr26 was highly enriched in the Ter119+ erythroblasts (Figure 1F). To further determine the temporal expression of Wdr26 during terminal erythropoiesis, we derived different stages of erythroblasts by culturing mouse fetal erythroid progenitors with erythropoietin in vitro. Wdr26 was upregulated at 24 hours and remained at a high level until 48 hours after erythropoietin induction (Figure 1G). A similar stage-dependent pattern was observed in differentiating MEL cells (Figure 1H). Together, our results show that Wdr26 is highly expressed in terminally differentiating erythroblasts.
Mammalian hematopoietic gene expression is controlled by lineage-specific transcription factors such as the GATA-binding transcription factor Gata1 and the basic helix-loop-helix Tal1/Scl.25-27 Analysis of published chromatin immunoprecipitation-seq data using Validated Systematic Integration of hematopoietic epigenomes revealed binding sites for both Gata1 and Tal1 in the promoter region of Wdr26 (supplemental Figure 1),28,29 suggesting that it’s a target of these 2 hematopoietic transcription factors. Downstream from the Gata1/Tal1-regulatory site is a binding site for the CCCTC-binding factor CTCF (supplemental Figure 1), which is a known chromatin insulator with an essential role in regulating hematopoietic cell differentiation.30,31 Locations of these transcription regulatory sites correlate well with increased epigenetic chromatin signatures H3K4me3 and H3K27ac, both of which have been associated with enhancer function.23,32 These results suggest that Wdr26 is transcriptionally activated by Gata1, Tal1, and CTCF, providing potential mechanisms for the induction of Wdr26 during erythropoiesis.
Wdr26 is essential for terminal erythropoiesis
To elucidate the function of Wdr26 in erythropoiesis, we silenced it in mouse primary erythroblasts using shRNAs (Figure 2A; supplemental Figure 2A). Knockdown of Wdr26 resulted in severe defects in enucleation, as monitored by nuclear staining and flow cytometry (Figure 2B; supplemental Figure 2B). In addition, Drabkin’s assay revealed a significant reduction of hemoglobin production in Wdr26-silencing primary erythroblasts (Figure 2C; supplemental Figure 2C). Compared with control cells, Wdr26-deficient cells displayed reduced expression of erythroid markers including 5-aminolevulinate synthase 2 (Alas2), glycophorin A (Gypa), solute carrier family 4 member 1 (Slc4a1), ferrochelatase (Fech), and erythrocyte membrane protein band 4.1 (Epb4.1) (Figure 2D), and elevated expression of erythroid-repressed genes such as signal transducer and activator of transcription 5 (Stat5) and the proto-oncogenes cKit and Myb (supplemental Figure 2D). Further analysis revealed that the differentiation of Wdr26-silencing cells was mainly blocked at the basophilic erythroblast (R3) stage (Figure 2E; supplemental Figure 2E).

Loss of Wdr26 blocks terminal erythropoiesis. (A) Western analysis confirmed the silencing of Wdr26 by shRNAs in primary mouse erythroblasts. Knockdown of Wdr26 led to reduced (B) enucleation, (C) hemoglobin production, and (D) expression of erythroid-induced genes in mouse primary erythroid progenitors. Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (E) Silencing of Wdr26 led to increased R3 subpopulation and reduced R4 and R5 subpopulations in primary mouse erythroblasts. Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (F) Western analysis confirmed the knockout of Wdr26 in MEL cells. (G) Porphyrin fluorescence assay and (H) o-dianisidine staining showed reduced heme production in differentiating MEL cells. Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (I) Strategy to knock out wdr26b in zebrafish using 2 gRNA targeting sites (arrowheads) and verification by PCR using primers indicated as arrows. (J) Red blood cell number and (K) heme content in the peripheral blood of wdr26b−/− fish were reduced in comparison to the wild-type fish. Error bars represent SEM from 6 animals. **P < .01. (L) The wdr26b-knockout fish showed reduced heme content in the adult hematopoietic tissue kidney. Error bars represent SEM from 5 animals. **P < .01. (M) Survival curve of wild-type (n = 6) and wdr26b−/− (n = 6) fish under hypoxic condition.
Loss of Wdr26 blocks terminal erythropoiesis. (A) Western analysis confirmed the silencing of Wdr26 by shRNAs in primary mouse erythroblasts. Knockdown of Wdr26 led to reduced (B) enucleation, (C) hemoglobin production, and (D) expression of erythroid-induced genes in mouse primary erythroid progenitors. Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (E) Silencing of Wdr26 led to increased R3 subpopulation and reduced R4 and R5 subpopulations in primary mouse erythroblasts. Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (F) Western analysis confirmed the knockout of Wdr26 in MEL cells. (G) Porphyrin fluorescence assay and (H) o-dianisidine staining showed reduced heme production in differentiating MEL cells. Error bars represent SEM from 3 replicates. *P < .05, **P < .01. (I) Strategy to knock out wdr26b in zebrafish using 2 gRNA targeting sites (arrowheads) and verification by PCR using primers indicated as arrows. (J) Red blood cell number and (K) heme content in the peripheral blood of wdr26b−/− fish were reduced in comparison to the wild-type fish. Error bars represent SEM from 6 animals. **P < .01. (L) The wdr26b-knockout fish showed reduced heme content in the adult hematopoietic tissue kidney. Error bars represent SEM from 5 animals. **P < .01. (M) Survival curve of wild-type (n = 6) and wdr26b−/− (n = 6) fish under hypoxic condition.
To ascertain its role in mammalian erythropoiesis, we knocked out Wdr26 in mouse erythroleukemia cells by CRISPR/Cas9-based genome editing (Figure 2F; supplemental Figure 2F-G). Consistent with the phenotype in Wdr26-silencing mouse primary erythroblasts, o-dianisidine staining showed that loss of Wdr26 led to reduced hemoglobin production in MEL cells (Figure 2G-H).
The loss-of-function phenotype in Wdr26-deficient erythroblasts was further verified in zebrafish in vivo. The zebrafish genome has 2 Wdr26 paralogs, wdr26a and wdr26b, both of which show ∼80% identity to mouse Wdr26 at the protein level. We focused on analyzing the erythroid function of wdr26b because its expression in the adult hematopoietic tissue kidney was substantially higher than that of wdr26a (supplemental Figure 3A). Using the CRISPR/Cas9 system, we generated a wdr26b-knockout allele (wdr26bmut-164bp or wdr26b−/−) that contains a 164-bp deletion and a 9-bp random insertion in the first exon of wdr26b (Figure 2I; supplemental Figure 3B). The wdr26b−/− homozygous fish were able to grow into adulthood; however, they exhibited profound anemia, as demonstrated by significantly reduced red blood cell number and heme content in the peripheral blood (Figure 2J-K). The anemic phenotype was likely the result of defective erythropoiesis because the heme content in the hematopoietic tissue was also lower in wdr26b−/− fish (Figure 2L). To further assess the physiological consequence of anemia in wdr26b-deficient fish, we tested the tolerance of adult fish to hypoxia. Under hypoxic conditions, the wdr26b−/− fish were more susceptible and died more quickly than the wild-type fish (Figure 2M). This is likely a consequence of reduced erythrocyte production and thus compromised oxygen-carrying capacity. Together, the results from mouse erythroblasts, erythroleukemia cells, and zebrafish demonstrate that Wdr26 plays an essential role in erythropoiesis.
Wdr26 is required for nuclear condensation in differentiating erythroblasts
Although fish erythrocytes retain nuclei, their nuclear size is substantially smaller than that of erythroid progenitors.33 When analyzing fish peripheral blood, we found that deficiency of wdr26b caused overt changes in both size and shape of the erythrocyte nuclei (Figure 3A). Specifically, wdr26b−/− fish displayed swelling nuclei of near-round shape, which was in drastic contrast to the slim, rod-like shape in the wild-type fish (Figure 3A-B). These results imply that wdr26b promotes nuclear condensation during terminal erythropoiesis.
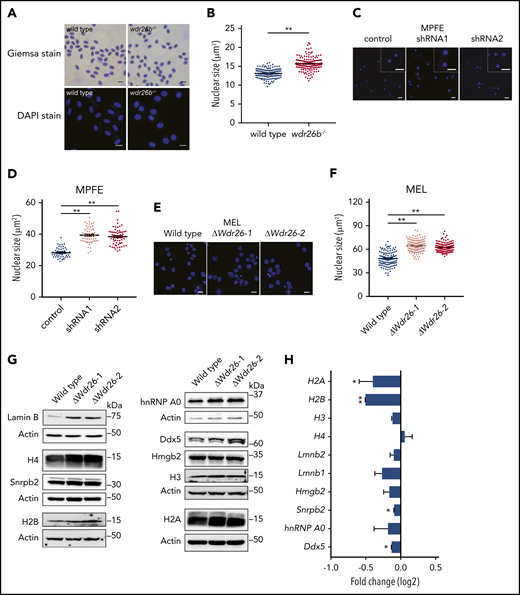
Deficiency of Wdr26 leads to enlarged nuclei with elevated protein abundance in differentiating erythroblasts. (A) Giemsa and DAPI staining of wild-type and wdr26b−/− zebrafish blood cells. Scale bars, 10 μm (Giemsa) and 5 μm (DAPI). (B) Quantification of nuclear size of zebrafish blood cells in panel A. Three pairs of wild-type and wdr26b−/− fish were analyzed. At least 100 cells were quantified for each genotype. **P < .01. (C) DAPI staining showed enlarged nuclei in Wdr26-silencing mouse primary erythroblasts. Scale bars, 20 μm. DAPI, 4′,6-diamidino-2-phenylindole; MPFE, mouse primary fetal liver erythroblast. (D) Quantitative analysis of the nuclear size of control and Wdr26-silencing mouse primary erythroblasts. More than 50 cells are shown for each shRNA. (E) DAPI staining showed enlarged nuclei in chemically induced Wdr26-knockout MEL cells. Scale bars, 10 μm. (F) Quantification of nuclear size in panel E. At least 100 cells were quantified for each clone. (G) Western analysis of nuclear proteins in DMSO-induced wild-type and Wdr26-knockout MEL cells. (H) mRNA expression of genes encoding the nuclear proteins shown in panel G. Error bars represent SEM from 2 replicates in an RNA-seq experiment. *P < .05, **P < .01.
Deficiency of Wdr26 leads to enlarged nuclei with elevated protein abundance in differentiating erythroblasts. (A) Giemsa and DAPI staining of wild-type and wdr26b−/− zebrafish blood cells. Scale bars, 10 μm (Giemsa) and 5 μm (DAPI). (B) Quantification of nuclear size of zebrafish blood cells in panel A. Three pairs of wild-type and wdr26b−/− fish were analyzed. At least 100 cells were quantified for each genotype. **P < .01. (C) DAPI staining showed enlarged nuclei in Wdr26-silencing mouse primary erythroblasts. Scale bars, 20 μm. DAPI, 4′,6-diamidino-2-phenylindole; MPFE, mouse primary fetal liver erythroblast. (D) Quantitative analysis of the nuclear size of control and Wdr26-silencing mouse primary erythroblasts. More than 50 cells are shown for each shRNA. (E) DAPI staining showed enlarged nuclei in chemically induced Wdr26-knockout MEL cells. Scale bars, 10 μm. (F) Quantification of nuclear size in panel E. At least 100 cells were quantified for each clone. (G) Western analysis of nuclear proteins in DMSO-induced wild-type and Wdr26-knockout MEL cells. (H) mRNA expression of genes encoding the nuclear proteins shown in panel G. Error bars represent SEM from 2 replicates in an RNA-seq experiment. *P < .05, **P < .01.
The nuclear condensation defects were also detected in Wdr26-deficient mouse erythroblasts. Silencing of Wdr26 in mouse primary erythroblasts led to increased nuclear size in comparison to control erythroblasts (Figure 3C-D). Consistently, the nuclei of Wdr26-knockout MEL cells were significantly larger than those in wild-type cells after DMSO-induced erythroid-like differentiation (Figure 3E-F). Because nuclear condensation is a prerequisite for enucleation during mammalian terminal erythropoiesis,3 these results suggest that the impaired enucleation observed in Wdr26-deficient erythroblasts are likely from defective nuclear condensation.
To determine whether the difference in the nuclear size was due to altered abundance of nuclear proteins, we performed immunoblotting analysis on major representative nuclear proteins including histones, nuclear membrane proteins, and heterogeneous nuclear ribonucleoproteins. Results showed that lamin B was severely elevated in Wdr26-knockout MEL cells in comparison to wild-type cells (Figure 3G). Additionally, most of the other nuclear proteins examined, including histones, heterogeneous nuclear ribonucleoprotein A0 (hnRNPA0), and DEAD box protein 5 (Ddx5), were more abundant in Wdr26-knockout cells (Figure 3G). Nuclear extraction and immunoblotting analysis further revealed that Wdr26 deficiency led to accumulation of major nuclear proteins within the nuclei (supplemental Figure 4). In contrast to the immunoblotting results, RNA-seq analysis did not detect increased expression of most of these genes in Wdr26-deficient cells (Figure 3H). Indeed, the messenger RNA (mRNA) levels of H2B, H3, Snrpb2, and Ddx5 were even reduced in Wdr26-knockout cells (Figure 3H). Together, these results suggest that Wdr26 deficiency impairs the export and degradation of nuclear proteins.
Wdr26 functions as a core subunit of the Gid ubiquitin ligase complex during terminal erythropoiesis
To investigate the molecular mechanism by which Wdr26 regulates erythropoiesis, we generated a MEL cell clone stably expressing FLAG-tagged Wdr26 and purified Wdr26 along with its interacting proteins using anti-FLAG M2 affinity agarose (Figure 4A). Analysis by mass spectrometry uncovered 6 subunits of the Gid ubiquitin ligase complex, including Ran binding protein 10 (Ranbp10), Ran binding protein 9 (Ranbp9), required for meiotic nuclear division 5 homolog A (Rmnd5a), Gid complex subunit 8 (Gid8), armadillo repeat containing 8 (Armc8), macrophage erythroblast attacher (Maea), and Gid complex subunit 4 (Gid4) (Figure 4B; supplemental Table 2). To corroborate this result, we used Gid8, a Wdr26-interacting Gid protein, as the bait to pull down proteins in differentiating MEL cells. Mass spectrometry showed that Gid8 interacts with Wdr26 and other members of the Gid complex in differentiating erythroblasts (Figure 4B; supplemental Table 3). Moreover, we found that similar to the erythroid-enriched pattern of Wdr26 expression, Ranbp10, Rmnd5a, and Gid8 were also highly expressed during terminal erythropoiesis (Figure 4C-D; supplemental Figure 5A).23,24 This suggests that these Gid genes are likely to function together with Wdr26 in erythroblasts. To further validate the interaction between Wdr26 and these Gid proteins, we performed coimmunoprecipitation analyses using Ranbp10-FLAG or Gid8-FLAG as the bait proteins. Both Ranbp10 and Gid8 were able to pull down the endogenous Wdr26 in chemically induced MEL cells (Figure 4E-F).
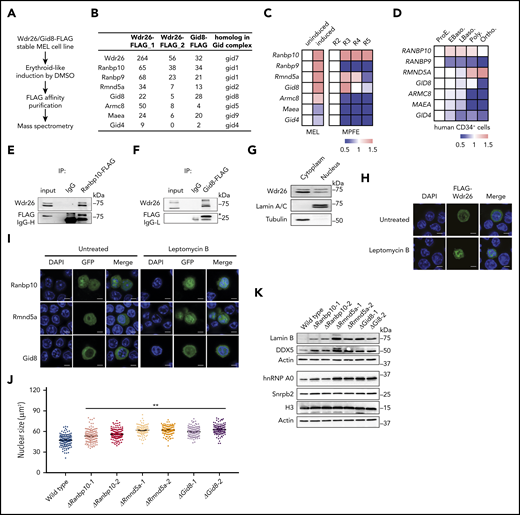
Wdr26 interacts with other Gid proteins to regulate nuclear condensation. (A) Schematic of protein pull-down assays and mass spectrometry pipeline. (B) The Gid proteins pulled down by Wdr26-FLAG or Gid8-FLAG in erythroid-like differentiating MEL cells. (C) mRNA expression of Gid genes in DMSO-induced MEL cells (left) and in R2-R5 subpopulations of primary mouse erythroblasts23 (right). (D) mRNA expression of GID genes in terminally differentiating human erythroblasts.24 Stages of erythroblasts shown are proerythroblast (ProE), early (EBaso) and late (LBaso) basophilic erythroblast, polychromatic erythroblast (Poly), and orthochromatic erythroblast (Ortho). (E-F) Validation of the interaction between Wdr26 and (E) Ranbp10 or (F) Gid8 in erythroid-induced MEL cells. (G) Western blot analysis revealed presence of Wdr26 in both the cytoplasm and nucleus of MEL cells. Lamin A/C and Tubulin were used as controls for subcellular fractionation. (H) Treatment of leptomycin B (60 nM) enhanced the localization of FLAG-Wdr26 in the nucleus of differentiating MEL cells. Scale bars, 5 µm. (I) Ranbp10, Rmnd5a, and Gid8 localized to the cytosol and nucleus; their nuclear localization was enhanced in the presence of 60 nM leptomycin B. (J) Knockout of Ranbp10, Gid8, or Rmnd5a in MEL cells resulted in increased nuclear size during erythroid-like differentiation. At least 100 cells were quantified for each clone. **P < .01. (K) Western analysis of nuclear proteins in DMSO-induced MEL cells that are deficient of Ranbp10, Gid8, or Rmnd5a.
Wdr26 interacts with other Gid proteins to regulate nuclear condensation. (A) Schematic of protein pull-down assays and mass spectrometry pipeline. (B) The Gid proteins pulled down by Wdr26-FLAG or Gid8-FLAG in erythroid-like differentiating MEL cells. (C) mRNA expression of Gid genes in DMSO-induced MEL cells (left) and in R2-R5 subpopulations of primary mouse erythroblasts23 (right). (D) mRNA expression of GID genes in terminally differentiating human erythroblasts.24 Stages of erythroblasts shown are proerythroblast (ProE), early (EBaso) and late (LBaso) basophilic erythroblast, polychromatic erythroblast (Poly), and orthochromatic erythroblast (Ortho). (E-F) Validation of the interaction between Wdr26 and (E) Ranbp10 or (F) Gid8 in erythroid-induced MEL cells. (G) Western blot analysis revealed presence of Wdr26 in both the cytoplasm and nucleus of MEL cells. Lamin A/C and Tubulin were used as controls for subcellular fractionation. (H) Treatment of leptomycin B (60 nM) enhanced the localization of FLAG-Wdr26 in the nucleus of differentiating MEL cells. Scale bars, 5 µm. (I) Ranbp10, Rmnd5a, and Gid8 localized to the cytosol and nucleus; their nuclear localization was enhanced in the presence of 60 nM leptomycin B. (J) Knockout of Ranbp10, Gid8, or Rmnd5a in MEL cells resulted in increased nuclear size during erythroid-like differentiation. At least 100 cells were quantified for each clone. **P < .01. (K) Western analysis of nuclear proteins in DMSO-induced MEL cells that are deficient of Ranbp10, Gid8, or Rmnd5a.
Next, we examined the subcellular distribution of Wdr26 along with Ranbp10, Rmnd5a, and Gid8 in MEL cells. Subcellular fractionation and immunoblotting analysis demonstrated that Wdr26 localized to both nuclear and nonnuclear fractions of MEL cells (Figure 4G), a pattern also confirmed by immunofluorescence analysis (Figure 4H). Furthermore, Wdr26 accumulated in the nucleus in the presence of the nuclear export inhibitor leptomycin B (Figure 4H), suggesting that Wdr26 shuttles between the cytoplasm and nucleus. Similarly, Ranbp10 and Rmnd5a also localized to both the cytoplasm and nucleus, whereas Gid8 mainly localized to the cytoplasm (Figure 4I). These 3 proteins also shuttle between the cytoplasm and nucleus because treatment with leptomycin B enhanced their nuclear localization (Figure 4I).
Given that the expression and localization of Ranbp10, Rmnd5a, and Gid8 closely resemble that of their partner Wdr26, these proteins may also involve in the regulation of nuclear condensation during terminal erythropoiesis. To test this hypothesis, we knocked out Ranbp10, Rmnd5a, and Gid8 in MEL cells (supplemental Figure 5B-C). After DMSO induction, the knockout clones displayed larger nuclei than wild-type cells (Figure 4J; supplemental Figure 5D). Consistent with the observation in Wdr26-knockout cells, deficiency of Ranbp10, Rmnd5a, or Gid8 also resulted in elevated abundance of nuclear proteins such as lamin B and Snrpb2 (Figure 4K; supplemental Figure 5E). Together, Wdr26 and its interacting proteins Ranbp10, Rmnd5a, and Gid8 may function together to regulate nuclear protein homeostasis and nuclear condensation during terminal erythropoiesis.
Wdr26 mediates the ubiquitination of lamin B during terminal erythropoiesis
The Gid complex was previously shown to be an E3 ubiquitin ligase that selectively degrades the gluconeogenic enzyme fructose-1, 6-bisphosphatase in yeast13,34 or the transcription factor Hbp1 in mammalian cells.14 Given that loss of Wdr26 caused increased abundance of nuclear proteins, we sought to assess whether Wdr26 and its partners mediate the ubiquitination of nuclear proteins in erythroblasts. Treatment with the proteasome inhibitor MG132 enhanced the ubiquitination levels of nuclear proteins (Figure 5A-B), implying that many nuclear proteins undergo proteolysis through the ubiquitin-proteasome pathway during erythroid differentiation. We found that the ubiquitination of nuclear proteins was decreased in Wdr26-knockout MEL cells (Figure 5A). Similarly, knockout of Rmnd5a, a component that bears ubiquitin ligase activity in the Gid complex,13,35 also led to reduced ubiquitination of nuclear proteins (Figure 5B). To examine whether the elevated abundance of nuclear proteins in Wdr26-knockout cells is attributed to their alleviated ubiquitination, we analyzed the ubiquitination status of these proteins. We found that ubiquitination of lamin B and H2A was decreased in MEL cells lacking Wdr26 (Figure 5C; supplemental Figure 6A). In contrast, most other proteins with elevated abundance in Wdr26-deficient cells, including H2B, hnRNP A0, and Ddx5, did not display notable difference in their ubiquitination levels between the wild-type and Wdr26-knockout cells (supplemental Figure 6B-E). By using the protein synthesis inhibitor cycloheximide (CHX), we confirmed that loss of Wdr26 stabilized lamin B protein (Figure 5D-E). These results were consistent with the finding that lamin B is degraded predominantly through the ubiquitin proteasome pathway during terminal erythropoiesis because MG132, but not the vacuolar H+-ATPase inhibitor bafilomycin A1, blocked its degradation (Figure 5F). Additionally, treatment with the nuclear export inhibitor leptomycin B did not alleviate the ubiquitination or degradation of lamin B (Figure 5G; supplemental Figure 6F), suggesting that clearance of lamin B is not dependent on the exportin-mediated pathway.

Wdr26 regulates lamin B ubiquitination in differentiating erythroblasts. (A-B) The ubiquitination level in the cytoplasm and nucleus of the chemically induced wild-type, (A) Wdr26-knockout, or (B) Rmnd5a-knockout MEL cells. The proteasome inhibitor MG132 (2.5 μM) was used to inhibit the degradation of ubiquitinated proteins. W, wild type; K, (A) Wdr26-knockout or (B) Rmnd5a-knockout. (C) Ubiquitination of Lamin B was alleviated in Wdr26-knockout MEL cells. *Lamin B. (D) Decreased rate of Lamin B degradation in Wdr26-knockout MEL cells. CHX (100 μg/mL) was used to inhibit protein synthesis. *Cleaved Lamin B. (E) Quantitative analysis of relative Lamin B protein level in panel D. Error bars represent SEM from 3 replicates. **P < .01. The Lamin B level in indicated time points was normalized to 0 hours. (F) Western analysis of Lamin B in DMSO-induced MEL cells treated with the protein synthesis inhibitor cycloheximide (CHX, 100 μg/mL) in combination with the proteasome inhibitor MG132 (2.5 μM) or the vacuolar H+ ATPase inhibitor bafilomycin A1 (Baf A1, 100 nM). (G) Treatment with leptomycin B (60 nM) did not alter the ubiquitination level of Lamin B in MEL cells. *Lamin B. (H) Ubiquitination of Lamin B by Wdr26-FLAG pull-down (PD) fraction in vitro. (I) Ubiquitination of Lamin B by Ranbp10-FLAG or Gid8-FLAG PD fractions derived from wild-type (with Wdr26) or Wdr26-konckout (without Wdr26) MEL cells. (J) Lysates from HEK293 cells transfected with FLAG-Wdr26 and HA-Lamin B were immunoprecipitated with anti-FLAG antibody or immunoglobulin G, and then immunoblotted with anti-HA or anti-FLAG antibodies (left). Ectopically expressed FLAG-Wdr26 was immunoprecipitated by endogenous Lamin B in HEK293 cells (right). *FLAG-Wdr26 (left) or Lamin B (right). (K) The N-terminal CTLH domain of Wdr26 interacts with Lamin B. HEK293 cells were transfected with FLAG-Wdr26-N (1-302 aa) or FLAG-Wdr26-C (303-641 aa). Immunoprecipitates by endogenous Lamin B were analyzed by immunoblotting with anti-FLAG antibodies. *Lamin B. (L) The C-terminal tail region of Lamin B interacts with Wdr26. The HA-tagged Lamin B-N (1-45 aa), Lamin B-CR (36-397 aa), or Lamin B-C (398-615 aa) constructs were transfected into HEK293 cells together with FLAG-Wdr26. The cell lysates were immunoprecipitated with anti-FLAG antibody and immunoblotted using anti-HA antibody. *FLAG-Wdr26. (M) Immunoblotting analysis of lamin B in the peripheral blood of wild-type and wdr26b−/− fish.
Wdr26 regulates lamin B ubiquitination in differentiating erythroblasts. (A-B) The ubiquitination level in the cytoplasm and nucleus of the chemically induced wild-type, (A) Wdr26-knockout, or (B) Rmnd5a-knockout MEL cells. The proteasome inhibitor MG132 (2.5 μM) was used to inhibit the degradation of ubiquitinated proteins. W, wild type; K, (A) Wdr26-knockout or (B) Rmnd5a-knockout. (C) Ubiquitination of Lamin B was alleviated in Wdr26-knockout MEL cells. *Lamin B. (D) Decreased rate of Lamin B degradation in Wdr26-knockout MEL cells. CHX (100 μg/mL) was used to inhibit protein synthesis. *Cleaved Lamin B. (E) Quantitative analysis of relative Lamin B protein level in panel D. Error bars represent SEM from 3 replicates. **P < .01. The Lamin B level in indicated time points was normalized to 0 hours. (F) Western analysis of Lamin B in DMSO-induced MEL cells treated with the protein synthesis inhibitor cycloheximide (CHX, 100 μg/mL) in combination with the proteasome inhibitor MG132 (2.5 μM) or the vacuolar H+ ATPase inhibitor bafilomycin A1 (Baf A1, 100 nM). (G) Treatment with leptomycin B (60 nM) did not alter the ubiquitination level of Lamin B in MEL cells. *Lamin B. (H) Ubiquitination of Lamin B by Wdr26-FLAG pull-down (PD) fraction in vitro. (I) Ubiquitination of Lamin B by Ranbp10-FLAG or Gid8-FLAG PD fractions derived from wild-type (with Wdr26) or Wdr26-konckout (without Wdr26) MEL cells. (J) Lysates from HEK293 cells transfected with FLAG-Wdr26 and HA-Lamin B were immunoprecipitated with anti-FLAG antibody or immunoglobulin G, and then immunoblotted with anti-HA or anti-FLAG antibodies (left). Ectopically expressed FLAG-Wdr26 was immunoprecipitated by endogenous Lamin B in HEK293 cells (right). *FLAG-Wdr26 (left) or Lamin B (right). (K) The N-terminal CTLH domain of Wdr26 interacts with Lamin B. HEK293 cells were transfected with FLAG-Wdr26-N (1-302 aa) or FLAG-Wdr26-C (303-641 aa). Immunoprecipitates by endogenous Lamin B were analyzed by immunoblotting with anti-FLAG antibodies. *Lamin B. (L) The C-terminal tail region of Lamin B interacts with Wdr26. The HA-tagged Lamin B-N (1-45 aa), Lamin B-CR (36-397 aa), or Lamin B-C (398-615 aa) constructs were transfected into HEK293 cells together with FLAG-Wdr26. The cell lysates were immunoprecipitated with anti-FLAG antibody and immunoblotted using anti-HA antibody. *FLAG-Wdr26. (M) Immunoblotting analysis of lamin B in the peripheral blood of wild-type and wdr26b−/− fish.
To directly demonstrate the E3 ubiquitin ligase activity of Wdr26-assocaited protein complex for lamin B, we performed in vitro protein ubiquitination assay in the presence of the E1 ubiquitin-activating enzyme UBE1 and the E2 ubiquitin conjugating enzyme UBE2H. Results showed that addition of the Wdr26 pull-down fraction substantially promoted the polyubiquitination of lamin B (Figure 5H). Similarly, both Ranbp10 and Gid8 pull-down fractions were able to enhance lamin B ubiquitination in vitro (Figure 5I). Absence of Wdr26 resulted in a dramatic decrease in lamin B ubiquitination by Ranbp10 or Gid8 pull-down proteins (Figure 5I), confirming the critical role of Wdr26 in ubiquitinating lamin B. Additionally, coimmunoprecipitation experiments showed that Wdr26 physically interacted with lamin B (Figure 5J). The interaction is likely to be mediated through the CTLH domain at the N-terminal region of Wdr26 and the C-terminal tail region of lamin B (Figure 5K-L).
We also substantiated the role of Wdr26 in regulating lamin B proteolysis in zebrafish in vivo. When wdr26b was knocked out, the fish showed elevated abundance of lamin B protein in the peripheral blood cells (Figure 5M; supplemental Figure 6G). Taken together, the biochemical and in vivo data unanimously support that Wdr26 regulates the ubiquitination and degradation of lamin B during vertebrate erythropoiesis.
Wdr26 facilitates nuclear opening formation and nuclear condensation in differentiating erythroblasts
Differentiating mammalian erythroblasts formed transient nuclear openings, a process involving degradation of lamin B, to accelerate the removal of nuclear proteins.5,6 Our results showed that many nuclear proteins accumulated in the nuclei of Wdr26-knockout cells (Figure 3G), but their ubiquitination level were not diminished by deficiency of Wdr26 (supplemental Figure 6B-E), implying that clearance of these proteins may be regulated indirectly by Wdr26, possibly through nuclear openings. Because Wdr26 regulates the degradation of lamin B, we investigated whether formation of the nuclear opening was affected by the loss of Wdr26. Immunofluorescence analysis showed that silencing of Wdr26 significantly reduced the nuclear opening events in mouse primary fetal erythroblasts (Figure 6A-C). To further validate that Wdr26 facilitates nuclear opening through promoting the lamin B degradation, we treated cells with the farnesyltransferase inhibitors tipifarnib and L744832, which inhibit farnesylation of lamins and thus their structural function.36 We found that farnesyltransferase inhibitors significantly rescued the nuclear condensation defect and enhanced the nuclear openings in Wdr26-knockdown erythroblasts (Figure 6D-G). Moreover, the enucleation defect was partially rescued by treatment with the farnesyltransferase inhibitors (Figure 6H). These data support a model that Wdr26 controls nuclear protein export through facilitating the formation of transient nuclear openings in differentiating erythroblasts.
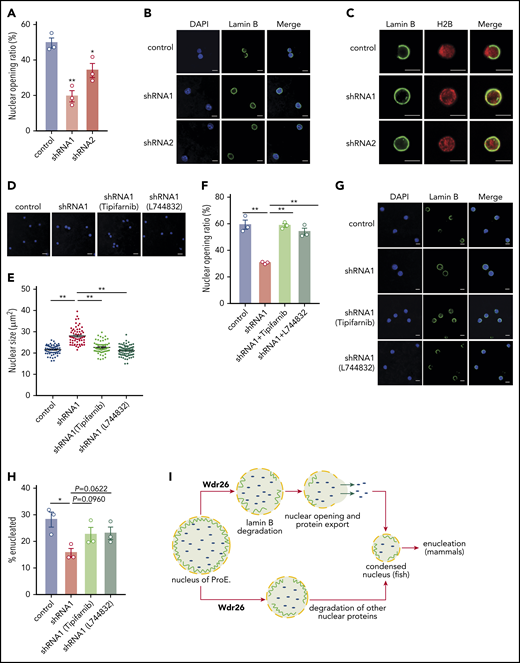
Silencing of Wdr26 results in reduced nuclear opening rate in differentiating erythroblasts. (A) Immunofluorescence assays with Lamin B antibody showed reduced nuclear opening ratio in Wdr26-knockdown primary mouse erythroblasts in comparison with the control shRNA cells at 48 hours after erythropoietin treatment. Error bars represent SEM from 3 replicates. At least 50 cells were quantified for each shRNA. *P < .05, **P < .01. (B) Representative images of panel A. Scale bars, 5 μm. (C) Immunofluorescence analyses of Lamin B and H2B in control and Wdr26-silencing primary mouse erythroblasts at 48 hours after erythropoietin treatment. Scale bars, 5 μm. (D-G) Treatment with farnesyltransferase inhibitors (1 μM Tipifarnib or L744832) rescued the defects of nuclear condensation and nuclear opening in Wdr26-silencing erythroblasts. Shown are representative images of (D) DAPI-stained nuclei and (G) Lamin B immunofluorescence, as well as the quantification of (E) nuclear size and (F) nuclear opening events. Error bars represent SEM from 3 replicates. At least 50 cells were quantified for each treatment condition. **P < .01. Scale bars, 5 μm. (H) Farnesyltransferase inhibitors partially rescued the enucleation defect in Wdr26-silencing erythroblasts. *P < .05. (I) The proposed role of Wdr26 in regulating nuclear protein degradation and nuclear condensation during vertebrate erythropoiesis. ProE, proerythroblast.
Silencing of Wdr26 results in reduced nuclear opening rate in differentiating erythroblasts. (A) Immunofluorescence assays with Lamin B antibody showed reduced nuclear opening ratio in Wdr26-knockdown primary mouse erythroblasts in comparison with the control shRNA cells at 48 hours after erythropoietin treatment. Error bars represent SEM from 3 replicates. At least 50 cells were quantified for each shRNA. *P < .05, **P < .01. (B) Representative images of panel A. Scale bars, 5 μm. (C) Immunofluorescence analyses of Lamin B and H2B in control and Wdr26-silencing primary mouse erythroblasts at 48 hours after erythropoietin treatment. Scale bars, 5 μm. (D-G) Treatment with farnesyltransferase inhibitors (1 μM Tipifarnib or L744832) rescued the defects of nuclear condensation and nuclear opening in Wdr26-silencing erythroblasts. Shown are representative images of (D) DAPI-stained nuclei and (G) Lamin B immunofluorescence, as well as the quantification of (E) nuclear size and (F) nuclear opening events. Error bars represent SEM from 3 replicates. At least 50 cells were quantified for each treatment condition. **P < .01. Scale bars, 5 μm. (H) Farnesyltransferase inhibitors partially rescued the enucleation defect in Wdr26-silencing erythroblasts. *P < .05. (I) The proposed role of Wdr26 in regulating nuclear protein degradation and nuclear condensation during vertebrate erythropoiesis. ProE, proerythroblast.
Discussion
Here we demonstrate that Wdr26, an erythroid-induced protein, plays a critical role in regulating nuclear condensation and enucleation during terminal erythropoiesis. Loss of Wdr26 leads to defects in erythroblast enucleation and differentiation in mammals and anemia in zebrafish.
As a prerequisite for enucleation in mammals, nuclear condensation involves export of massive amounts of proteins out of the nucleus for degradation.3,37 Two protein export pathways have been reported in differentiating erythroblasts, 1 is through the nucleocytoplasmic RAN transport machinery4 and the other is through the caspase-3-dependent nuclear opening.5 Consistently, we demonstrate that a subset of nuclear proteins such as lamin B and hnRNP A0 are eliminated through exportin-independent mechanism, whereas certain histones may be exported through the exportin-dependent pathway (supplemental Figure 6F). In this study, we show that Wdr26 promotes nuclear protein degradation and nuclear condensation during terminal erythropoiesis through 2 mechanisms (Figure 6I). First, Wdr26 regulates the polyubiquitination of a fraction of nuclear proteins, including lamin B and H2A, which are subsequently degraded by the ubiquitin-proteasome system. Second, Wdr26 controls the formation of the transient nuclear opening through lamin B proteolysis in differentiating erythroblasts. The nuclear opening mediates rapid release of major histones and other nuclear proteins for degradation in the cytosol.5,6 Because degradation of lamin B is required for the formation of the nuclear opening,5,6 accumulation of lamin B in Wdr26-deficient erythroblasts inevitably leads to impaired nuclear opening formation and nuclear condensation in differentiating erythroblasts.
Notably, Wdr26 deficiency did not impair the caspase-mediated cleavage of lamin B (Figure 5D; supplemental Figure 6H), indicating that this cleavage is independent of the Wdr26-mediated ubiquitination. On the other hand, the caspase-mediated cleavage is not required for lamin B ubiquitination as the Wdr26 protein complex was able to ubiquitinate the intact, full-length lamin B (Figure 5H). Therefore, it is likely that the caspase-mediated cleavage and Wdr26-mediated ubiquitination are 2 independent pathways responsible for degrading lamin B in developing erythroblasts.
Despite enucleation occurring almost exclusively in mammals, the nuclei of mature red blood cells are also highly condensed in other vertebrate species such as fishes and birds.33 We found that lack of wdr26 homolog in zebrafish induced profound anemia and impaired lamin B proteolysis as well as nuclear condensation, supporting a conserved role for Wdr26 in regulating vertebrate erythropoiesis.
Wdr26 is part of the Gid or GID/CTLH ubiquitin ligase complex.13,14 The Gid complex, first identified by a high-throughput interactome analysis in yeast,38 is an E3 ubiquitin ligase that mediates proteolytic degradation of gluconeogenic enzymes fructose-1, 6-bisphosphatase, and phospho-enol-pyruvate carboxykinase in yeast.13,34 Its human counterpart, the GID/CTLH complex, regulates cell proliferation by targeting the transcription factor Hbp1.14 Interestingly, 4 Gid members, Wdr26, Ranbp10, Rmnd5a, and Gid8, are highly expressed in mammalian hematopoietic tissues. Our results demonstrate that these Gid proteins regulate the polyubiquitination and degradation of nuclear proteins, including the histone protein H2A and the nuclear lamina component lamin B, in differentiating erythroblasts. The identification of Wdr26 and its partners as a critical E3 ubiquitin ligase in differentiating erythroblasts establishes a new paradigm of protein ubiquitination and degradation during terminal erythropoiesis.
Within the Gid complex, both Rmnd5a and Maea contain the RING finger domain, which may function to recruit E2 ubiquitin-conjugating enzyme and catalyze the transfer of ubiquitin to protein substrates.14,39 Previous studies suggested that Wdr26 may be a substrate adaptor or a scaffolding protein within E3 ubiquitin ligase complexes.40-42 Wdr26 comprises a CTLH domain and a WD40 domain, both of which coordinate protein–protein interactions.43,44 Our data support the model that Wdr26 is a substrate adaptor to coordinate the association between the nuclear substrates such as lamin B and the Gid ubiquitin ligase. Together, our study uncovers a critical role for Wdr26 in regulating lamin B ubiquitination, nuclear condensation, and enucleation, and sheds new insights into nuclear protein homeostasis and vertebrate hematopoiesis.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
RNA sequencing data have been deposited in the Gene Expression Omnibus database (accession numbers GSE133287 and GSE133343).
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Barry H. Paw for assistance and discussion and Harvey F. Lodish and Leonard I. Zon for advice and reading of the manuscript.
This work was supported by funding from the National Key Research and Development Program (2018YFA0507802 to C.C.), the National Natural Science Foundation of China (81770099 to J.S. and 31371435 to C.C.), the Zhejiang Natural Science Foundation (LR17C110001 to C.C.), the Hong Kong Health and Medical Research Fund (05160296), the Hong Kong Research Grants Council (21101218), the Shenzhen Science and Technology Innovation Fund (JCYJ20170413115637100 to J.S., JCYJ20170412152916724 to J.S., and JCYJ20170413141047772 to L.Z.), and the Sanming Project of Medicine in Shenzhen (SZSM201811092 to J.S.)
Authorship
Contribution: J.S. and C.C. conceived the project; R.Z., J.S., and C.C. wrote the manuscript; R.Z. performed most of the experiments and analyzed the data; C.M. participated in the primary erythroblast experiments; Z.Z., M.C., and H.F. contributed to the zebrafish experiments; X.Z. cloned deletion constructs for the immunoprecipitation analyses; L.Z. participated in the mass spectrometry experiments; and all authors discussed the results and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Caiyong Chen, College of Life Sciences, Zhejiang University, 866 Yuhangtang Rd, Hangzhou 310058, China; e-mail: chency@zju.edu.cn; and Jiahai Shi, Department of Biomedical Sciences, City University of Hong Kong, 83 Tat Chee Ave, Kowloon, Hong Kong, China; e-mail: jiahai.shi@cityu.edu.hk.


