Key Points
NGS provides a more accurate picture of BCR-ABL1 mutation status in CML patients with failure or warning responses to TKI therapy.
TKI-resistant low-level mutations undergo selective expansion if the TKI is not changed or an inappropriate TKI or TKI dose is chosen.

Abstract
In chronic myeloid leukemia (CML) patients, tyrosine kinase inhibitors (TKIs) may select for drug-resistant BCR-ABL1 kinase domain (KD) mutants. Although Sanger sequencing (SS) is considered the gold standard for BCR-ABL1 KD mutation screening, next-generation sequencing (NGS) has recently been assessed in retrospective studies. We conducted a prospective, multicenter study (NEXT-in-CML) to assess the frequency and clinical relevance of low-level mutations and the feasibility, cost, and turnaround times of NGS-based BCR-ABL1 mutation screening in a routine setting. A series of 236 consecutive CML patients with failure (n = 124) or warning (n = 112) response to TKI therapy were analyzed in parallel by SS and NGS in 1 of 4 reference laboratories. Fifty-one patients (22 failure, 29 warning) who were negative for mutations by SS had low-level mutations detectable by NGS. Moreover, 29 (27 failure, 2 warning) of 60 patients who were positive for mutations by SS showed additional low-level mutations. Thus, mutations undetectable by SS were identified in 80 out of 236 patients (34%), of whom 42 (18% of the total) had low-level mutations somehow relevant for clinical decision making. Prospective monitoring of mutation kinetics demonstrated that TKI-resistant low-level mutations are invariably selected if the patients are not switched to another TKI or if they are switched to a inappropriate TKI or TKI dose. The NEXT-in-CML study provides for the first time robust demonstration of the clinical relevance of low-level mutations, supporting the incorporation of NGS-based BCR-ABL1 KD mutation screening results in the clinical decision algorithms.
Introduction
Despite the striking efficacy of tyrosine kinase inhibitors (TKIs) for the treatment of chronic myeloid leukemia (CML),1 a proportion of patients do not achieve an optimal response and require treatment optimization.2 To this purpose, three generations of molecules are nowadays available.3 A variety of mechanisms may underlie lack or loss of response to TKIs, but acquisition of point mutations in the BCR-ABL1 kinase domain (KD) is, at present, the only actionable one.4 Imatinib and second-generation TKIs (2GTKIs) are known to have a well-defined spectrum of sensitive and resistant mutants.5 Failure to turn off BCR-ABL1 activity and achieve a rapid and deep clearance of mutant cells would not only undermine clinical response but also fuel the acquisition of additional mutations.6-8 This, in some patients, might result in a clonal complexity (the so-called compound mutants) that has been shown to be much more difficult to address therapeutically.9 Screening for mutations is thus recommended both by the European LeukemiaNet (ELN)10 and the National Comprehensive Cancer Network11 in case of failure and warning; that is, whenever a change of therapy is necessary or to be considered.
Sanger sequencing (SS) is the current gold standard for diagnostic BCR-ABL1 KD mutation screening. In recent years, however, next-generation sequencing (NGS) has entered routine diagnostic workflows in hematology and oncology, as it has proven to be a powerful and robust technology.12 A series of studies investigating the use of NGS for BCR-ABL1 KD mutation screening have shown that, in a proportion of patients, mutations detectable by SS may just be the tip of the iceberg.13-19 The greater sensitivity of NGS and the clonal nature of sequencing may provide a more accurate picture of mutation status and, in case of multiple mutations, may often disentangle the complexity of clonal configurations.20 In longitudinal analyses, NGS was capable to pick emerging mutations up to several months earlier16,17 and, in patients who relapsed on second-line dasatinib or nilotinib therapy, trace TKI-resistant mutations back to the switchover sample.13 In all the studies, low-level mutations (down to 1%) were found to be invariably selected whenever the patients received the TKI that these mutations were known to be resistant to, suggesting that mutations detectable by NGS may have the same clinical relevance as those routinely identified by less sensitive, SS-based approaches.
However, almost all the published data suffer from the limitation of deriving from retrospective analyses of relatively small series of patients, who were selected for having acquired mutations on TKI therapy. Hence, the frequency and clinical relevance of low-level mutations detectable by NGS still need to be assessed prospectively and in larger series of patients. Additionally, the implementation of routine, NGS-based BCR-ABL1 KD mutation screening in a molecular diagnostic laboratory network has never been attempted. We thus undertook a prospective study (NEXT-in-CML) aimed to (1) assess the feasibility, cost, and turnaround times of routine, NGS-based BCR-ABL1 KD mutation screening; (2) investigate the frequency of low-level mutations detectable by NGS in a consecutive series of CML patients with failure or warning response to TKI therapy; and (3) follow the kinetics of low-level mutations over time and in relation to the selective pressure exerted by treatment.
Materials and methods
Patients and study design
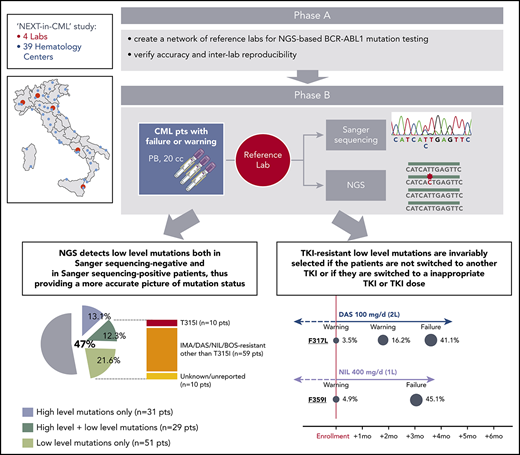
The NEXT-in-CML study consisted of 2 consecutive phases. In the first phase (phase A), 4 expert laboratories (Bologna, Orbassano, Naples, and Catania) that were already engaged in SS-based BCR-ABL1 KD mutation screening within the national LabNet network shared a common protocol for NGS-based BCR-ABL1 KD mutation screening and established an optimized pipeline of data analysis. Accuracy and interlaboratory reproducibility of the NGS assay were subsequently assessed (see supplemental Methods, available on the Blood Web site, for details). In the second phase (phase B), 236 consecutive CML patients were enrolled by 39 hematology centers all over Italy. Inclusion criteria were (1) documented failure or warning response to any TKI, any line, according to the 2013 ELN recommendations; and (2) positivity for either b2a2 or b3a2 BCR-ABL1 fusion transcripts. Patient disposition is detailed in Table 1. A 20-mL peripheral blood sample was shipped to 1 of the 4 reference laboratories, and BCR-ABL1 KD mutation screening was performed in parallel by SS and by NGS. For each patient, 2 separate reports were generated. Clinicians were free to choose whether to include NGS results in their decision algorithms or whether to adopt a “wait-and-watch” strategy, with the option to perform NGS-based prospective monitoring of low-level mutation kinetics. Clinical data were collected for each patient at baseline and during follow-up, up to 12 months. This study (ref. 113/2014/U/Tess) was approved by the ethical committee of the Sant’Orsola Malpighi Hospital, Bologna (which was the promoter and sponsor institution) and the ethical committees of all the other participating centers. Research was conducted in accordance with the Declaration of Helsinki.
SS-based screening of the BCR-ABL1 KD
SS-based screening of the BCR-ABL1 KD was performed on an ABI PRISM 3730 (Applied Biosystems, Foster City, CA) as previously described.21 Briefly, after RNA extraction from white blood cells and reverse transcription (RT) to complementary DNA, three overlapping amplicons were generated by nested polymerase chain reaction (PCR) that encompassed a region of the KD corresponding to residues 201 through 524. High-fidelity enzymes were used both for RT and for PCR.
NGS-based screening of the BCR-ABL1 KD
NGS-based screening of the BCR-ABL1 KD was performed on a Roche GS Junior instrument (Roche Applied Science) using RNA as input material as well, according to an amplicon-based approach setup and optimized first in-house (Bologna laboratory; reported in Soverini et al14 ) and subsequently within the framework of the IRON II (Interlaboratory Robustness of NGS) international study network.22 The protocol is detailed in supplemental Methods. Read alignment to the ABL1 reference sequence (GenBank accession number NM_005157.5), variant calling at nucleotide positions corresponding to amino acids 235 through 498 (KD), annotation, and filtration were performed from .fastq files with the Amplicon Suite software (SmartSeq s.r.l, Novara), which was implemented to maximize the reliability of variant calls based on an algorithm integrating specific acceptability criteria and estimation of error rates at each nucleotide position calculated using a retrospective data set of patients and donors provided by the Bologna laboratory.
Detailed standard operating protocols for wet laboratory and bioinformatics analyses were prepared and circulated among all laboratories to ensure uniformity of procedures. Acceptability criteria are detailed in supplemental Methods. For each sequence variant not immediately recognized as being resistant to one or more TKIs, the Catalogue of Somatic Mutations in Cancer (COSMIC) database v87 (https://cancer.sanger.ac.uk/cosmic) and an in-house database of >1,500 TKI-resistant patients with Philadelphia chromosome–positive leukemia analyzed between 2003 and 2015 were interrogated to obtain information about whether the mutation had been previously found or reported. As a periodical quality control, randomly selected positive and negative samples were reprocessed starting from the RT step.
Definitions
Failure and warning to first and second-line TKI therapy were defined according to the 2013 ELN recommendations. In the absence of specific definitions, failure and warning to subsequent lines were defined using the same criteria as for second line. High-level mutations are herein defined as mutations identified in ≥20% of BCR-ABL1 transcripts (hence detectable both by NGS and by SS). Low-level mutations are herein defined as mutations identified in 3% to 20% of BCR-ABL1 transcripts by NGS and undetectable by SS.
Results
Accuracy and interlaboratory reproducibility of NGS-based BCR-ABL1 KD mutation screening
In the first phase of the study (phase A), identical batches of 32 blinded complementary DNA samples were analyzed in parallel by each laboratory in 4 sequencing runs of 8 samples each. Results are detailed in supplemental Methods. One hundred and twenty-six out of 128 (98.4%) total samples could successfully be sequenced. A median of 3085 (range, 2121-4486) high-quality reads were generated for each amplicon of each sample (supplemental Figure 1). Comparison of observed vs expected mutations is shown in supplemental Table 1. Out of 292 expected mutations, 270 (92%) were collectively called across the 4 laboratories. The 22 mutations that ≥1 laboratories failed to detect were between 1% and 3%. A total of 11 false-positive calls likely to result from PCR or sequencing errors were observed; all were <3%. A very good accuracy in mutation quantitation was achieved for all but 1 mutation (supplemental Figure 2). Taken together, results of the first phase of the study confirmed the accuracy and interlaboratory reproducibility of the results of the NGS-based BCR-ABL1 KD mutation screening protocol, laying the ground for the second phase. Three percent was taken as lower detection limit, and in all subsequent sequencing runs, all variant calls <3% were filtered out.
Feasibility, turnaround times, and cost of NGS-based BCR-ABL1 KD mutation screening
The average turnaround time from sample to results for SS-based analysis was 6 working days (range, 4-14). The average turnaround time for NGS-based analysis was 11 working days (range, 7-24). Twelve samples (5%) were not evaluable by either method because of insufficient RNA quality and/or quantity. Eight percent of samples analyzed by SS had to be repeated because of unsuccessful amplification or poor-quality sequencing traces. Eleven percent of samples analyzed by NGS had to be repeated because they failed to meet the acceptability criteria at one of the critical control points during library preparation or because of insufficient depth of coverage for any of the 6 amplicons. The costs for reagents and disposable plasticware necessary to carry out all protocol steps from RNA extraction to sequencing (in case of an 8-sample run) were estimated to be ∼180 Eur per sample on a GS Junior (Roche), 90 Eur per sample on a MiSeq (Illumina), and 100 Eur per sample on an Ion Personal Genome Machine (Thermo Fisher). For comparison, the costs for reagents and disposable plasticware necessary for SS-based analysis were estimated to be ∼200 Eur per sample. Technician hands-on time for a batch of 8 samples (in case no automated solution is used in any of the steps of the workflow) was estimated to be 13.5 hours for the NGS workflow and 8 hours for the SS workflow. Analysis of sequencing results was estimated to require 2.5 hours for NGS and 6 hours for SS; in neither case are bioinformatics skills required, since a variety of open-source and licensed softwares are available to assist in NGS read alignment to the reference sequence and variant calling.
Frequency of low-level mutations and clinical correlates
In the second phase of the study (phase B), 236 consecutive CML patients with a nonoptimal response to TKI therapy (ie, candidates for BCR-ABL1 KD mutation screening according to ELN recommendations) were enrolled and analyzed in parallel by SS and NGS. One hundred and twenty-four patients had a response defined as failure, and 112 patients had a response defined as warning (Table 1). The mutation status of each of the 236 patients enrolled is detailed in supplemental Table 2. Sixty out of 236 patients (25%) were positive for any mutation by SS, and 111 out of 236 patients (47%) were positive for any mutation by NGS. Low-level (median 7.3%; range, 3.1% to 17.7%) mutations were detected in 51 patients negative for mutations by SS, but 29 patients positive for high-level mutations by SS were found to carry additional low-level mutations by NGS (Figure 1A). Eighty out of 236 (34%) patients were thus positive for ≥1 (up to 4) low-level mutations, for a total of 128 low-level mutations detected by NGS. A low-level T315I was detected in 10 patients (7 failures, 3 warnings); 59 additional patients had ≥1 low-level mutation known to be associated with resistance to imatinib or 2GTKIs other than T315I (Figure 1A). The remaining 10 patients had only low-level mutations with an unknown resistance profile (ie, mutations neither included in our internal mutation database nor reported in the COSMIC database in association with any TKI resistance in Philadelphia chromosome–positive leukemias), although some of them were in the COSMIC database because they had been found in sequencing studies of other neoplastic conditions. To exclude the possibility that these mutations could be technical artifacts, sample analysis was repeated twice in 2 different laboratories. Besides the 10 patients harboring a low-level T315I, 38 harbored low-level mutations known to confer resistance to ≥1 2GTKI.
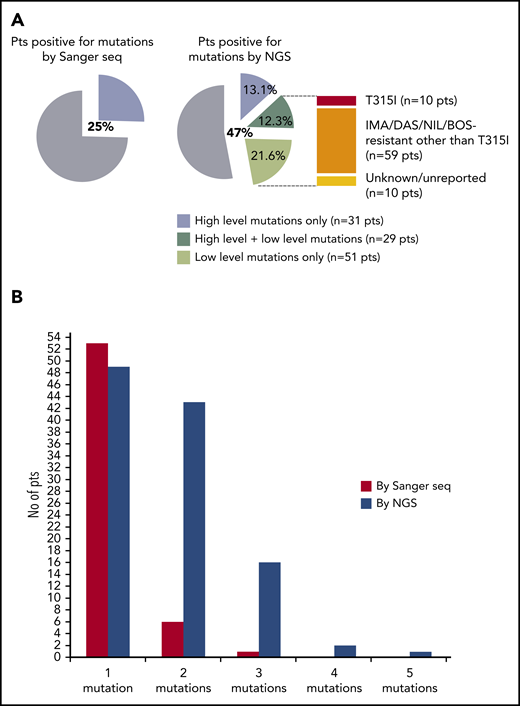
Comparison between SS and NGS results. (A) Percentage of patients positive for mutations by SS and by NGS. Among patients positive for mutations by NGS, 31 (13.1%) had high-level mutations only (≥20%; detectable by SS too); 29 (12.3%) had both ≥1 high-level mutations and ≥1 low-level mutations (≤20%; detectable by NGS only); 51 (21.6%) had only low-level mutations. A low-level T315I was detected in 10 patients; 59 additional patients had ≥1 low-level mutations known to be associated with resistance to imatinib or 2GTKIs other than the T315I (ie, Y253H; E255K/V; V299L; F317L/V/I/C; F359V/I/C). The remaining 10 patients had only low-level mutations with an unknown resistance profile and/or not listed in the COSMIC database. (B) Patients positive for 1 or multiple mutations as assessed by SS vs NGS. BOS, bosutinib; DAS, dasatinib; IMA, imatinib; NIL, nilotinib; pts, patients; seq, sequencing.
Comparison between SS and NGS results. (A) Percentage of patients positive for mutations by SS and by NGS. Among patients positive for mutations by NGS, 31 (13.1%) had high-level mutations only (≥20%; detectable by SS too); 29 (12.3%) had both ≥1 high-level mutations and ≥1 low-level mutations (≤20%; detectable by NGS only); 51 (21.6%) had only low-level mutations. A low-level T315I was detected in 10 patients; 59 additional patients had ≥1 low-level mutations known to be associated with resistance to imatinib or 2GTKIs other than the T315I (ie, Y253H; E255K/V; V299L; F317L/V/I/C; F359V/I/C). The remaining 10 patients had only low-level mutations with an unknown resistance profile and/or not listed in the COSMIC database. (B) Patients positive for 1 or multiple mutations as assessed by SS vs NGS. BOS, bosutinib; DAS, dasatinib; IMA, imatinib; NIL, nilotinib; pts, patients; seq, sequencing.
Breakdown of mutation frequency as assessed by SS vs NGS by level of nonresponse (failure or warning) and by line of therapy is shown in Table 2.
Patients positive for mutations by SS who turned out to carry additional low-level mutations by NGS were almost exclusively failures (27/29 [93%]), had more frequently acquired (20/29 [69%]) vs primary resistance, were more frequently on second- or subsequent-line therapy (20/29 [69%]), and more frequently had intermediate or high Sokal risk (21/27 [78%] cases with available risk information). Patients who had low-level mutations only were more frequently warnings (29/51 [57%]) and more frequently had intermediate or high Sokal risk (35 of 48 [73%] cases with available risk information), and many of them (35/51 [69%]) were receiving a reduced TKI dose or were known to have experienced problems of nonadherence to therapy.
A total of 62 patients had ≥2 mutations as assessed by NGS (Figure 1B). Nevertheless, only 6 compound mutations (E255K+T315I; E255K+F317L; T315I+L387M; T315I+M244V; T315I+M244V+V289A; T315I+E255V) were found in 5 patients (3 of whom had progressed to AP or blastic phase). Patients with ≥2 mutations more frequently exhibited a failure response than patients with 1 mutation (47 vs 21; Fisher’s P = .008). Moreover, of the 14 patients in AP or blastic phase, 10 were in the group of patients with ≥2 mutations, whereas only 1 was in the group with 1 mutation. Of the 13 patients with additional cytogenetic abnormalities associated with poor prognosis (+8, +Philadelphia, +19, +21, +17/inv17, 11q23, −7, 7q−, and abnormalities of chromosome 3), 8 were in the group with ≥2 mutations, whereas only 2 were in the group with 1 mutation. Additionally, follow-up data revealed that patients with ≥2 mutations more frequently failed subsequent-line treatment than patients with 1 mutation (33 vs 10; Fisher’s P = .0004).
Dynamics of low-level mutations
Five patients (2.2%) were lost to follow-up, leaving 231 evaluable cases. In 85 patients (failures, n = 65; warnings, n = 20) positive for mutations by SS and/or NGS, the therapy was changed to another TKI; 7 patients subsequently underwent allogeneic stem cell transplant. In the remaining patients (warnings, n = 21) positive for mutations by NGS, the therapy was not immediately changed, but more frequent patient monitoring was planned. In order to monitor the kinetics of low-level mutations over time and in relation to therapeutic choices, follow-up samples were collected and analyzed for 10 patients in the first group and for all the patients in the second group. Low-level mutations that were known to be resistant to the TKI the patient was receiving at the time of enrolment remained consistently detectable and invariably increased in burden whenever the treatment was not changed or was changed to a TKI (or to a TKI dose; see UPN 141) not active against those mutations (Figure 2). In particular, all the 21 warning patients who remained on the same TKI despite having low-level mutations known to be resistant to that TKI vs 19 out of 69 warning patients without evidence of mutations experienced subsequent treatment failure. On the other hand, low-level mutations not expected to confer resistance to the TKI the patient was receiving did not necessarily require treatment change to become undetectable at subsequent follow-up evaluations. To exclude the possibility of technical artifacts, the original RNA samples harboring such mutations were resequenced (starting from the RT step) in a different laboratory.
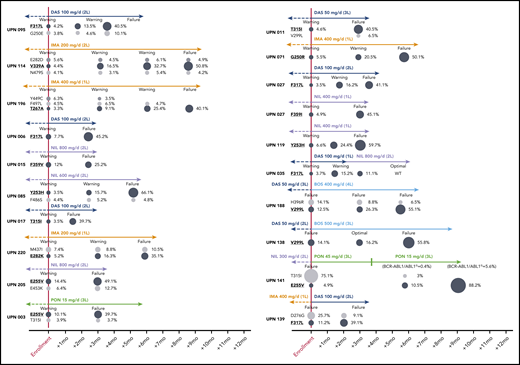
Prospective NGS-based monitoring of the kinetics of TKI-resistant low-level mutations (highlighted in bold and underlined) in 16 representative patients with warning and 4 representative patients with failure at the time of enrollment. Patient details can be found in supplemental Table 2. To define “optimal response,” failure and warning in patients receiving third-line TKI therapy and beyond, the same ELN 2013 criteria as for second-line had to be used. UPN138: after 3 months of bosutinib (to which the patient was switched before NGS results were made available), BCR-ABL1/ABL1IS levels had increased (from 1.3% to 5.7%), yet according to ELN 2013, the response was formally definable as optimal. UPN141: after the switch to ponatinib 45 mg/d, the patient regained complete hematological response and showed a rapid decline of BCR-ABL1/ABL1IS levels, so the dose was reduced to 15 mg/d for safety concerns. However, the 50% inhibitory concentration of the E255V mutant is higher than the average plasma concentration achievable with 15 mg/d, which explains the outgrowth of the E255V-positive clone. Similar E255V expansion under ponatinib 15 mg/d was observed in UPN003. L, line; PON, ponatinib.
Prospective NGS-based monitoring of the kinetics of TKI-resistant low-level mutations (highlighted in bold and underlined) in 16 representative patients with warning and 4 representative patients with failure at the time of enrollment. Patient details can be found in supplemental Table 2. To define “optimal response,” failure and warning in patients receiving third-line TKI therapy and beyond, the same ELN 2013 criteria as for second-line had to be used. UPN138: after 3 months of bosutinib (to which the patient was switched before NGS results were made available), BCR-ABL1/ABL1IS levels had increased (from 1.3% to 5.7%), yet according to ELN 2013, the response was formally definable as optimal. UPN141: after the switch to ponatinib 45 mg/d, the patient regained complete hematological response and showed a rapid decline of BCR-ABL1/ABL1IS levels, so the dose was reduced to 15 mg/d for safety concerns. However, the 50% inhibitory concentration of the E255V mutant is higher than the average plasma concentration achievable with 15 mg/d, which explains the outgrowth of the E255V-positive clone. Similar E255V expansion under ponatinib 15 mg/d was observed in UPN003. L, line; PON, ponatinib.
Discussion
We here report the results of the first multicenter prospective study of routine NGS application to BCR-ABL1 KD mutation screening.
Our study first of all demonstrates that BCR-ABL1 KD mutation screening by NGS is feasible, robust, and reproducible and can be successfully implemented in national networks of reference laboratories. Several NGS-based screening protocols, including ours, had been reported to achieve a lower detection limit of 1% in single-center experiences. However, after the interlaboratory control round, it was evidenced that some false-positive and false-negative results may occur (between 1% and 3%). Thus, a 3% threshold for variant frequency was adopted in the NEXT-in-CML study. This is in line with the threshold identified by Kizilors et al,19 who have developed and validated a similar assay on the same platform (Illumina MiSeq), obtaining the ISO15189 accreditation. The lower limit of SS, in contrast, is reported to vary between 15% and 25% depending on the level of background noise signal (which varies from sample to sample) and the experience of the operator, since the detection of small mutant peaks essentially relies on visual inspection of the sequencing traces. In this study, we thus investigated the prevalence and the kinetics of “low-level” mutations, specifically defined as mutations detectable in a proportion of BCR-ABL1 transcripts ranging from 3% to 20%.
What are the clinical advantages of NGS over SS? By prospectively comparing NGS and SS results in a relatively large, consecutive cohort of CML patients with nonoptimal response to TKI therapy, the NEXT-in-CML study first of all showed that approximately half of the patients positive for mutations by SS had additional low-level mutations detectable by NGS, and this was almost exclusively a feature of patients with a failure response (27/29 patients). We acknowledge that the line of therapy and the remaining available and/or suitable TKI options may influence the extent to which mutation status may practically guide treatment selection. In one third of the cases, however, the low level T315I, Y253H, E255K, F359V/I/C, F317L or V299L mutations might actually have influenced the selection of the subsequent TKI. Moreover, it is important to bear in mind that positivity for mutations as well as mutation complexity are per se two useful pieces of information in that they identify a subset of patients at higher risk to relapse and develop additional mutations.6-8,23
Second, our study showed that in patients with Failure of TKI therapy who have no evidence of mutations by SS, variants may be identified that in more than half of the cases have frequencies just slightly below the level of SS detection. Out of 78 cases of Failure scored negative by SS analysis, 22 (28%) were found to have one or multiple low level mutations detectable by NGS. Seventeen of 43 such mutations had a frequency ranging between 10% and 17%, and 10 additional mutations had a frequency ranging between 7% and 10%. In the great majority of the cases, low-level mutations could be recognized as poorly sensitive to the TKI the patient was receiving at the time of analysis. Five patients only had mutations for which no association with TKI resistance could be found either in our in-house mutation database or in COSMIC (although 4 mutations had entries for other tumor types in COSMIC). Interestingly, all 5 patients had ≥2 mutations. It may be hypothesized that, as a consequence of clonal cooperation, a mutant that per se would not be so markedly insensitive to treatment may somehow, at least temporarily, survive and expand to a certain extent, although it will never be able to achieve dominance. Another explanation takes into account the possibility that these mutations are simply passenger events.
Third, our study showed that 26% and 35% of the patients who have a warning response level to first- or second-line TKI therapy, respectively, and are negative for mutations by SS analysis do harbor low-level mutations detectable by NGS. Overall, 24 out of 98 (21%) warning patients negative for mutations by SS had low-level mutations known to be resistant to the TKI they were receiving. Three patients had a low-level T315I. Patients with a response level defined as warning are considered to be in a sort of “gray zone.” For such patients, the 2013 ELN recommendations advise only to perform more frequent monitoring, essentially because of the lack of any criterion (except for the presence of a mutation) to predict who will ultimately turn into a failure and who will rather turn into an optimal responder. Considering that all warning patients who remained on the same TKI despite having low-level mutations known to be resistant to that TKI subsequently experienced treatment failure, we can conclude that detection of emerging mutations by NGS in warning patients should support proactive TKI switch.
Lastly and most importantly, our study provides robust evidence supporting the clinical relevance of low-level mutations detectable by NGS by showing that those known to be resistant to the TKI the patient is receiving invariably expand if the selective pressure is not released. For ethical reasons, NGS results were communicated to physicians, who were left the choice whether to use SS or NGS results for clinical decision making. As a consequence, 21 warning patients were not immediately switched to another TKI, and longitudinal prospective follow-up samples were collected to monitor mutation kinetics. Clonal selection could be observed in all 16 cases that had a low-level mutation known to be resistant to the ongoing TKI and ultimately led to TKI failure after 3 to 12 months. Clonal selection was also observed in 4 patients who were switched to a TKI not active against (one of) the low-level mutations detected by NGS (in all cases, the TKI was changed before NGS results were made available). A few cases had low-level mutations not expected to confer resistance to the TKI the patient was receiving, and these mutations did not necessarily require treatment change to become undetectable at subsequent follow-up evaluations. Since the recently developed error-corrected sequencing strategies suggest that low-level mutations that disappear may be technical artifacts,24 the original RNA samples were reprocessed in a different laboratory to ensure they indeed harbored true mutations.
In conclusion, NGS-based BCR-ABL1 KD mutation screening in a relatively large, prospective series of unselected CML cases with failure or warning response to TKI therapy showed evidence of mutations undetectable by SS in 80 out of 236 patients (34%), of whom 42 (18% of the total patient population) had low-level mutations somehow relevant for clinical decision making (eg, mutations that, based on in vitro 50% inhibitory concentration data and consolidated clinical evidence, were known to confer resistance to ≥1 of the 2GTKIs that could have been selected for subsequent-line therapy). We also showed that low-level mutations tend to expand when the TKI is not changed or when an inappropriate TKI, or TKI dose, is chosen. Our study was not designed to assess whether an NGS-driven therapeutic change would result in significantly superior event-free or overall survival. However, based on the above evidences, it can reasonably be hypothesized that avoiding a (most probably) ineffective TKI and preventing the outgrowth of (further) mutant populations would increase the probability to improve response while avoiding patient exposure to unnecessary toxicities (treatment with some 2GTKIs has been associated with serious adverse events) and optimizing the use of patient’s and/or Health System’s financial resources (2GTKIs and ponatinib are very expensive). All of this can be achieved using a technology that will become more and more widely available as a “natural” evolution of SS and that is not associated with significantly higher costs (provided that the samples are centralized in reference laboratories). In the pre-NGS era, the added value of greater sensitivity in mutation screening had already found support in a series of studies by Parker et al, who had designed multiplexed primer extension assays coupled with mass spectrometry-based identification of the extended nucleotides (Sequenom MassArray) allowing to scan for 31 different mutations.23,25-27 The MassArray, however, is an expensive high-throughput platform definitely more suited to large-scale research studies than to routine diagnostic testing. Digital PCR represents an attractive alternative to search for definite mutations. Digital PCR has the potential to become a cheaper and faster approach and ensure even greater sensitivity than NGS. At present, validated commercial assays have been developed for a very limited set of mutations (T315I, Y253H, and E255K), but more individual assays, as well as multiplex solutions, might become available in the future. If so, further studies will be warranted to investigate whether digital PCR may complement or even substitute for NGS.
At present, however, the NEXT-in-CML study results and the feasibility of robust and reproducible NGS testing in an expert laboratory network setting support the adoption of routine NGS-based BCR-ABL1 KD mutation screening wherever technical resources, standardized protocols, and dedicated, well-trained personnel are available.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The following additional investigators participated in the study: Attilio Olivieri, Anna Rita Scortechini (Clinica di Ematologia, Ospedale Regionale di Torrette, Ancona), Robin Foà, Massimo Breccia (Cattedra di Ematologia, Università “La Sapienza," Roma), Alessandro Rambaldi, Tamara Intermesoli (Divisione di Ematologia, Ospedali Riuniti, Bergamo), Emanuele Angelucci, Emilio Usala (Divisione di Ematologia, Ospedale Oncologico “A. Businco," Cagliari), Michele Rizzo (Unità Operativa di Ematologia, Presidio Ospedaliero S.Elia, Caltanissetta), Stefano Molica, Luciano Levato (Divisione di Ematologia, Ospedale Regionale Pugliese, Catanzaro), Antonio Cuneo, Francesco Cavazzini (Unità Operativa di Ematologia, Azienda Ospedaliero-Universitaria Arcispedale Sant'Anna, Ferrara), Alberto Bosi, Antonella Gozzini (Divisione di Ematologia, Policlinico Careggi, Firenze), Silvana Franca Capalbo (Unità Operativa Complessa di Ematologia, Ospedali Riuniti Foggia), Marco Gobbi, Maurizio Miglino (Dipartimento di Medicina Interna, Cattedra di Ematologia, IRCCS San Martino, Genova), Patrizia Tosi, Anna Lia Molinari (Unità Operativa di Oncologia ed Oncoematologia, Ospedale Infermi, Rimini), Monica Crugnola (Ematologia e Centro Trapianti midollo osseo, Azienda Ospedaliero-Universitaria Parma), Atelda Romano, Andrea Mengarelli (IFO IRCCS Istituto Nazionale Tumori Regina Elena), Giuseppe Pietrantuono (Dipartimento di Onco-Ematologia, IRCCS Centro di riferimento Oncologico della Basilicata, Rionero in Vulture), Carmine Selleri (Unità Operativa di Ematologia e Centro Trapianti, Azienda Ospedaliero-Universitaria “Ruggi D’Aragona," Salerno), Claudio Fozza (Clinica Ematologica, Università di Sassari), Renato Fanin, Mario Tiribelli (Clinica Ematologica ed Unità di Terapie Cellulari Carlo Melzi, Azienda Ospedaliero Universitaria Udine), Diamante Turri (Unità Operativa Complessa di Ematologia, Ospedali Riuniti Villa Sofia-Cervello, Palermo), Clementina Caracciolo (Divisione di Ematologia con Trapianto, Policlinico P. Giaccone, Palermo), and Elisabetta Novella (Divisione di Ematologia, Ospedale San Bortolo, Vicenza).
Authorship
Contribution: S. Soverini., L.B., M.M., C.D.B., G.M., G.S., F.P., P.V., A.S., S. Stella, S.E., M. Baccarani, and G.R. designed and performed the study; the remaining authors enrolled 3 or more patients and provided clinical information; and all authors critically reviewed the manuscript and gave final approval for submission.
Conflict-of-interest disclosure: S. Soverini received honoraria from Incyte Biosciences, Novartis, and Bristol-Myers Squibb; M. Baccarani received honoraria from Novartis, Incyte Biosciences, and Pfizer; S.G. received speaker fees from Pfizer, Novartis, and Incyte Biosciences; A.I. received honoraria from Novartis, Pfizer, and Incyte Biosciences; G.R., F. Castagnetti, G.G., F.S., and E.A. received honoraria from Novartis, Bristol-Myers Squibb, Pfizer, and Incyte Biosciences; P.V. received honoraria from Astra Zeneca, Celgene, Incyte Biosciences, Italfarmaco, Novartis, Pfizer, Tesaro, and Teva and research funding from Novartis and Pfizer. The remaining authors declare no competing financial interests.
The current affiliation for G.M. is Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, IRCCS, Meldola, Italy.
Correspondence: Simona Soverini, Institute of Hematology “L. e A. Seràgnoli,” Via Massarenti 9, 40138 Bologna, Italy; e-mail: simona.soverini@unibo.it.
For the full study protocol, please contact simona.soverini@unibo.it. Individual participant data will not be shared.