

Key Points
Inhibition of the menin-MLL interaction targets FLT3 mutations transcriptionally via MEIS1 in NPM1-mutant and MLL-rearranged leukemias.
Combined menin-MLL and FLT3 inhibition is a synergistic therapeutic opportunity in these leukemia subtypes with concurrent FLT3 mutation.

Abstract
The interaction of menin (MEN1) and MLL (MLL1, KMT2A) is a dependency and provides a potential opportunity for treatment of NPM1-mutant (NPM1mut) and MLL-rearranged (MLL-r) leukemias. Concomitant activating driver mutations in the gene encoding the tyrosine kinase FLT3 occur in both leukemias and are particularly common in the NPM1mut subtype. In this study, transcriptional profiling after pharmacological inhibition of the menin-MLL complex revealed specific changes in gene expression, with downregulation of the MEIS1 transcription factor and its transcriptional target gene FLT3 being the most pronounced. Combining menin-MLL inhibition with specific small-molecule kinase inhibitors of FLT3 phosphorylation resulted in a significantly superior reduction of phosphorylated FLT3 and transcriptional suppression of genes downstream of FLT3 signaling. The drug combination induced synergistic inhibition of proliferation, as well as enhanced apoptosis, compared with single-drug treatment in models of human and murine NPM1mut and MLL-r leukemias harboring an FLT3 mutation. Primary acute myeloid leukemia (AML) cells harvested from patients with NPM1mutFLT3mut AML showed significantly better responses to combined menin and FLT3 inhibition than to single-drug or vehicle control treatment, whereas AML cells with wild-type NPM1, MLL, and FLT3 were not affected by either of the 2 drugs. In vivo treatment of leukemic animals with MLL-r FLT3mut leukemia reduced leukemia burden significantly and prolonged survival compared with results in the single-drug and vehicle control groups. Our data suggest that combined menin-MLL and FLT3 inhibition represents a novel and promising therapeutic strategy for patients with NPM1mut or MLL-r leukemia and concurrent FLT3 mutation.
Introduction
Acute myeloid leukemia (AML) is a neoplastic disease of hematopoietic progenitor cells with acquired genetic abnormalities characterized by impaired differentiation and clonal expansion.1,2 Comprehensive sequencing studies of AML samples have provided a broad list of recurrent cytogenetic and mutational abnormalities that are considered potential driver events of AML pathogenesis. These studies confirmed nucleophosmin 1 (NPM1) and the fms-related tyrosine kinase 3 (FLT3) as the most commonly mutated genes in AML and discovered frequent genetic abnormalities in genes encoding epigenetic regulators of transcription, with about two-thirds of AML cases being affected.3-5 Genetic abnormalities represent the basis for the current World Health Organization classification and for prediction of the outcome of AML.1,6
Despite our growing understanding of its pathogenesis, AML remains a therapeutic challenge. Curative treatment efforts still rely on intensive chemotherapy as a backbone.6,7 Currently, only 35% to 40% of the younger and 5% to 15% of the elderly patients (>60 years) can be cured, and the outcome of patients who are not eligible for intensive treatment is even more dismal.2 Hope arises from the introduction of mechanism-based agents that target AML-specific genetic abnormalities or epigenetic vulnerabilities.7,8 Since 2017, eight novel drugs have been approved by the U.S. Food and Drug Administration for the treatment of AML, and numerous novel agents are currently under clinical investigation, many of them targeting epigenetic mechanisms.9,10
One promising therapeutic opportunity for tackling an epigenetic vulnerability in AML is to inhibit the interaction between the histone methyltransferase MLL and the chromatin-associated oncogenic cofactor menin.11 This approach was initially proposed for the treatment of MLL-rearranged (MLL-r) leukemias, in which a chromosomal translocation leads to the formation of an oncogenic MLL-fusion protein.12,13 These fusion proteins require the interaction with menin for chromatin binding to activate leukemogenic gene expression that includes the MEIS1, PBX3, and MEF2C transcription factors and to drive the leukemic transformation of hematopoietic stem or progenitor cells.14-19 Both can be reversed by pharmacological inhibition of the menin-MLL interaction,11,20-25 and 2 small-molecule inhibitors are currently entering clinical trials (NCT04067336 and NCT04065399).
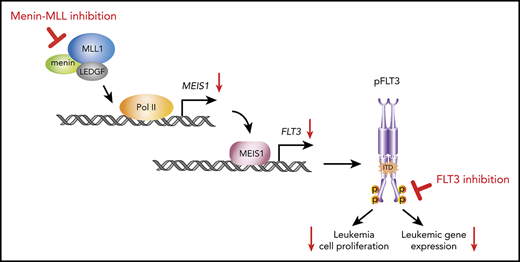
We have recently demonstrated that the menin-MLL interaction is also a dependency in the NPM1-mutant (NPM1mut) leukemias, which represent the most common AML subtype in adults. These leukemias lack an MLL-fusion protein but express a leukemogenic gene program, including MEIS1, HOX, PBX3, and FLT3 that is very similar to the program expressed in MLL-r leukemias.26 As in MLL-r leukemias, small-molecule inhibition of this interaction reversed expression of these genes and showed dramatic antileukemic activity against human and murine models of NPM1mut leukemias in vitro and in vivo.26 These findings were recently confirmed by other groups that used medicinal chemistry to develop menin-MLL inhibitors with better drug-like properties.22,27,28
Of note, we and others found FLT3 a putative transcriptional MEIS1 target to be among the genes most dramatically downregulated by menin-MLL inhibition in NPM1mut AML.26 This finding attracted our interest, because activated mutations within FLT3 are important leukemic drivers. FLT3 mutations occur across all AML subgroups, including MLL-r leukemias (10%), and are particularly common in NPM1mut AML (60%).29-32 An internal tandem duplication (ITD), the most common type of FLT3 mutation, is associated with adverse treatment outcome and, if present at a high allelic ratio, also converts the relatively favorable NPM1mut AML subtype into an intermediate prognostic category.6,33-36 Also, FLT3 mutations represent an important therapeutic target and midostaurin, a first-generation multitargeted inhibitor of FLT3 phosphorylation has been shown to improve survival rates of patients with AML who have de novo FLT3 mutations, when given in combination with intensive chemotherapy.37 Next-generation FLT3 inhibitors with higher potency and selectivity, such as quizartinib, gilteritinib, and crenolanib, induce high remission rates in relapsed/refractory patients with FLT3-ITD+ AML as single agents.38-40 Gilteritinib was recently approved for this indication in the United States and Europe, but none of the currently used FLT3 inhibitors has induced long-term remissions as single agents.38,39 The development of a curative treatment is therefore currently focused on the exploration of these compounds in combination treatment regimens.41
FLT3-transcription is consistently downregulated upon menin-MLL inhibition in NPM1mut AML, and we therefore hypothesized that combining these agents with FLT3 inhibitors would enhance their therapeutic efficacy. In this study, we demonstrated that combined menin-MLL and FLT3 inhibition has enhanced on-target activity against activating FLT3 mutations in MLL-r or NPM1mut AML cells and shows remarkably superior activity compared with single-drug treatment in vivo.
Materials and methods
Cell culture
All human AML cell lines used in this study were authenticated by Multiplex Cell Authentication by Multiplexion (Heidelberg, Germany), as described previously,42 and maintained in standard conditions, as described.26,43 The murine Npm1CA/+Flt3ITD/+ leukemia model, as well as murine retrovirally transformed Mll-Af9-cells and their culture, have been described.26,44,45
Primary AML blast cells and coculture assay
Primary human NPM1mutFLT3ITD AML samples were obtained from patients treated at the University Medical Center, Mainz, under Institutional Review Board–approved protocols and in accordance with the Declaration of Helsinki. The coculture treatment assays were performed as reported previously26,43 and are also described in the supplemental Methods, available on the Blood Web site.
In vitro studies
In vitro drug treatment, cell viability assays, annexin-V staining, RNA isolation, cDNA synthesis, quantitative real-time-PCR (qRT-PCR), chromatin-immunoprecipitation (ChIP), western blot analysis, flow cytometry, viral transduction, and colony-forming unit assays were performed according to standard procedures.26,43 Drug synergism was calculated using the Chou-Talalay Method.46 A detailed description is provided in the supplemental Methods.
RNA sequencing and analysis
For gene expression analysis, OCI-AML3, MOLM13, and MV411 cells were treated in 3 independent experiments. Normalization on synthetic RNA spike-in controls, as proposed by Lovén and colleagues,47 was performed as described.26,47 See the supplemental Methods for RNA preparation, normalization details, and sequencing analysis.
AML xenograft model
Six- to 10-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/ SzJ (NSG) mice were purchased from the Translational Animal Research Center at the University Medical Center, Mainz. For in vivo experiments, they were injected via the tail vein with 5 × 106 MV411 cells. Animals were randomized into the following treatment groups: vehicle (25% DMSO, 25% PEG400, and 50% phosphate buffered saline), MI-503 (50 mg/kg; twice daily intraperitoneally), quizartinib (10 mg/kg; PO, once daily), or a combination of both drugs. For assessment of leukemia burden, the mice were euthanized after drug treatment. Harvested bone marrow cells were analyzed for human CD45 expression by flow cytometry. For survival analysis, treatment was initiated on day 12 after transplantation and continued until day 45, with a 2-day treatment break to allow partial recovery from local irritation at the injection sites. Moribund animals were euthanized when they displayed signs of terminal leukemic disease. All mouse experiments were approved by the National Investigation Office Rheinland-Pfalz.
Results
MEIS1 and FLT3 transcription is uniformly suppressed by menin-MLL inhibition in MLL-rearranged and NPM1mut leukemia
Because we found MEIS1 and its putative transcriptional target FLT3 to be dramatically downregulated upon menin-MLL inhibition in our previous study, we wanted to explore whether MEIS1 and FLT3 are uniformly suppressed after pharmacological disruption of the menin-MLL interaction in MLL-r and NPM1mut leukemias.
We first confirmed the selective inhibitory effects of menin-MLL inhibition on cell proliferation in a selection of MLL-r and NPM1mut human and murine leukemia cells, with and without activating FLT3 mutations, using the small-molecule inhibitor MI-503 (MLL-r: MOLM13 [FLT3-ITD], MV411 [FLT3-ITD], OCI-AML2 [wild-type (wt) FLT3] and murine Mll-Af9 [wtFlt3]; and NPM1mut: OCI-AML3 [wtFLT3], murine Npm1CA/+Flt3ITD/+). In accordance with previous reports20,26 the inhibitory effects of MI-503 on proliferation were assessed after 7 and 11 days, because this compound affects cell growth with a latency of several days, most likely as a consequence of differentiation induction (supplemental Figure 1). As expected, we observed a profound dose-dependent reduction in proliferation of all MLL-r and NPM1mut cells, whereas leukemia cells lacking MLL-r or NPM1mut showed only minor responses to very high concentrations of the drug (human APL cell lines: NB4; HL-60; murine Hoxa9-Meis1-transformed cells), with 50% inhibitory concentrations (IC50) of >1500 nM (Figure 1A-B; supplemental Figure 1A-C).
![Gene and protein expression changes upon menin-MLL inhibition in NPM1mut and MLL-r AML. (A) Human (left) and murine (right) AML cells were treated for 11 days with MI-503. Viable (4′,6-diamidino-2-phenylindole [DAPI]–negative) cells were assessed by flow cytometry, and IC50 values were calculated with GraphPad Prism software. (B) Summary of IC50 values (MI-503), MLL-rearrangement, and NPM1 and FLT3 mutation status in the AML cells assessed. (C) Venn diagram showing downregulated genes identified by RNA-seq (more than twofold decrease; adjusted P < .05), in NPM1mut OCI-AML3, MLL-r MOLM13, and MV411 cells after MI-503 treatment (2.5 µM) compared with the DMSO control. (D) Volcano plots of RNA-seq data obtained from OCI-AML3, MOLM13, and MV411 cells treated with MI-503 (2.5 µM). FLT3 and selected MLL-fusion targets are labeled. (E) FLT3 and MEIS1 mRNA expression in human and murine leukemia cells after 4 days of MI-503 treatment (2.5 µM), as assessed by qRT-PCR. ChIP was performed with antibodies against menin (F) or MLL1 (G) and IgG as the negative control, followed by qPCR to detect a sequence within the MEIS1 gene body or SOX2 as negative control. Cells were treated with MI-503 (2.5 µM) or vehicle control for 4 days. (H) FLT3 protein (cell surface) expression assessed by flow cytometry in human and murine NPM1mut and MLL-rearranged AML cells after MI-503 treatment (2.5 µM for 4 or 7 days, as indicated). Representative histograms of 3 independent experiments are shown. Bar graphs in panels A and E-G represent the mean of 3 independent experiments, each performed in technical triplicate. Bar graph in panel H showing Npm1CA/+Flt3ITD/+ cells represents 2 independent experiments performed in technical triplicate. Error bars represent standard deviation.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f1.png?Expires=1771105444&Signature=oneVG5FXhm9cJ~ZZzHbylGS4Sq0uMTXQSkRkmfb51eE6h97B6FI90Igd8MX9NdbOZh~LNq5Mp2gz9yf9W45W2l5y2YFcW1U2GhLg8~R0VBK3ldHS2pbg9PynfJNgeWKkauL9a2rvkqjRuNZqDtMDssvPHsdJRknNZSSQJpSfx7v7hC7fdmT~G3UZMjNxDRiNc6dW9YY3vWbZiSuusz3z9nf3TlEUhNxwINQZJRY1pX0iicnjKWsG0UsIxe0LT3mnovcKEncj-lRWhTe6S78RZ6slVvQlucZvAlZ7Dwj8B3sSYE7th8A7x7bXd0BcyFCYu7lD-n5mK7xH1TAFUCKb8Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Gene and protein expression changes upon menin-MLL inhibition in NPM1mut and MLL-r AML. (A) Human (left) and murine (right) AML cells were treated for 11 days with MI-503. Viable (4′,6-diamidino-2-phenylindole [DAPI]–negative) cells were assessed by flow cytometry, and IC50 values were calculated with GraphPad Prism software. (B) Summary of IC50 values (MI-503), MLL-rearrangement, and NPM1 and FLT3 mutation status in the AML cells assessed. (C) Venn diagram showing downregulated genes identified by RNA-seq (more than twofold decrease; adjusted P < .05), in NPM1mut OCI-AML3, MLL-r MOLM13, and MV411 cells after MI-503 treatment (2.5 µM) compared with the DMSO control. (D) Volcano plots of RNA-seq data obtained from OCI-AML3, MOLM13, and MV411 cells treated with MI-503 (2.5 µM). FLT3 and selected MLL-fusion targets are labeled. (E) FLT3 and MEIS1 mRNA expression in human and murine leukemia cells after 4 days of MI-503 treatment (2.5 µM), as assessed by qRT-PCR. ChIP was performed with antibodies against menin (F) or MLL1 (G) and IgG as the negative control, followed by qPCR to detect a sequence within the MEIS1 gene body or SOX2 as negative control. Cells were treated with MI-503 (2.5 µM) or vehicle control for 4 days. (H) FLT3 protein (cell surface) expression assessed by flow cytometry in human and murine NPM1mut and MLL-rearranged AML cells after MI-503 treatment (2.5 µM for 4 or 7 days, as indicated). Representative histograms of 3 independent experiments are shown. Bar graphs in panels A and E-G represent the mean of 3 independent experiments, each performed in technical triplicate. Bar graph in panel H showing Npm1CA/+Flt3ITD/+ cells represents 2 independent experiments performed in technical triplicate. Error bars represent standard deviation.
Gene and protein expression changes upon menin-MLL inhibition in NPM1mut and MLL-r AML. (A) Human (left) and murine (right) AML cells were treated for 11 days with MI-503. Viable (4′,6-diamidino-2-phenylindole [DAPI]–negative) cells were assessed by flow cytometry, and IC50 values were calculated with GraphPad Prism software. (B) Summary of IC50 values (MI-503), MLL-rearrangement, and NPM1 and FLT3 mutation status in the AML cells assessed. (C) Venn diagram showing downregulated genes identified by RNA-seq (more than twofold decrease; adjusted P < .05), in NPM1mut OCI-AML3, MLL-r MOLM13, and MV411 cells after MI-503 treatment (2.5 µM) compared with the DMSO control. (D) Volcano plots of RNA-seq data obtained from OCI-AML3, MOLM13, and MV411 cells treated with MI-503 (2.5 µM). FLT3 and selected MLL-fusion targets are labeled. (E) FLT3 and MEIS1 mRNA expression in human and murine leukemia cells after 4 days of MI-503 treatment (2.5 µM), as assessed by qRT-PCR. ChIP was performed with antibodies against menin (F) or MLL1 (G) and IgG as the negative control, followed by qPCR to detect a sequence within the MEIS1 gene body or SOX2 as negative control. Cells were treated with MI-503 (2.5 µM) or vehicle control for 4 days. (H) FLT3 protein (cell surface) expression assessed by flow cytometry in human and murine NPM1mut and MLL-rearranged AML cells after MI-503 treatment (2.5 µM for 4 or 7 days, as indicated). Representative histograms of 3 independent experiments are shown. Bar graphs in panels A and E-G represent the mean of 3 independent experiments, each performed in technical triplicate. Bar graph in panel H showing Npm1CA/+Flt3ITD/+ cells represents 2 independent experiments performed in technical triplicate. Error bars represent standard deviation.
To assess global transcriptional changes after menin-MLL inhibition in NPM1mut and MLL-r leukemia cells, we performed RNA-seq analysis after MI-503 treatment in OCI-AML3, MOLM13, and MV411 cells. After 4 days of treatment, gene expression was assessed in OCI-AML3 and MOLM13 cells and after 3 days in MV411 as these exhibited an earlier antiproliferative response to the treatment. In the MLL-r FLT3-ITD+ leukemia cells, we found that 1051 (MOLM13) and 1386 (MV411) genes were downregulated by at least twofold (adjusted P < .05; Figure 1C; supplemental Table 1). Many of the most profoundly downregulated genes were known MLL-target genes, including MEIS1, PBX3, JMJD1C, and MEF2C. As hypothesized, we also found FLT3 expression to be dramatically suppressed (Figure 1D). In the NPM1mut OCI-AML3 cells, MI-503 led to downregulation of 578 genes, including MEIS1, PBX3, and FLT3, when using the same thresholds (Figure 1C-D; supplemental Table 1). It is interesting to note that, in contrast with the previously published menin-MLL inhibitor MI-2-2, HOXA/B genes were not substantially repressed in these cells (supplemental Figure 2A). Forty genes were significantly downregulated in both NPM1mut and MLL-r leukemia cells and included MEIS1 and FLT3 (Figure 1C). Using qPCR, we confirmed that MEIS1 and FLT3 were uniformly downregulated by menin-MLL inhibition in all other NPM1mut and MLL-r leukemia cells that we assessed in this study (Figure 1E). Dramatic suppression of FLT3 in MV411 cells known to harbor a hemizygous FLT3-ITD mutation (with no remaining FLT3 wild-type [WT] copy) indicated that menin-MLL inhibition also suppresses the mutated FLT3-ITD transcript. qPCR, with individually designed ITD-specific primers, confirmed this finding in MOLM13 and MV411 cells (supplemental Figure 2B).
ChIP, followed by qPCR, revealed that transcriptional downregulation of MEIS1 by MI-503 treatment was accompanied by abrogation of menin and MLL protein binding to the MEIS1 gene locus (Figure 1F-G; supplemental Figure 2C-D). These data support the view that binding of the menin-MLL complex is necessary for MEIS1 gene expression in these AML-subtypes.
Next, to assess FLT3 protein expression after 7 days of MI-503 treatment (except MV411, 4-day treatment), we performed flow cytometry with antibodies that recognize an extracellular FLT3 epitope. In fact, FLT3 protein expression was also significantly reduced in all NPM1mut and MLL-r leukemias (Figure 1H).
These data showed that pharmacological menin-MLL inhibition causes uniform downregulation of MEIS1 and of WT and mutant FLT3 in NPM1mut and MLL-r leukemias.
Combined menin-MLL and FLT3 inhibition exerts a synergistic effect against MLL-r or NPM1mut leukemias that harbor a concurrent FLT3-ITD
The data presented thus far indicate that mutant FLT3 expression can be targeted via menin-MLL inhibition in MLL-r and NPM1mut leukemias. We therefore sought to assess the effects of combining menin-MLL inhibition with direct FLT3 kinase inhibitors that have been shown to be highly active against FLT3 mutant AML. First, we determined the IC50 concentrations of the specific and potent FLT3 inhibitors quizartinib, crenolanib, gilteritinib, and ponatinib in human MLL-r FLT3-ITD+ leukemia cell lines after 48 hours of treatment. Both cell lines were highly sensitive to the FLT3 inhibitors (Figure 2A; supplemental Figure 3A). Consistent with published data, quizartinib was the most potent inhibitor and was highly selective against FLT3-ITD+ leukemias, with IC50 values within the subnanomolar range, whereas AML cells without FLT3 mutation were unaffected (Figure 2A-B; supplemental Figure 3B-C).
![Synergistic effects of combined menin-MLL and FLT3 inhibition. (A) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in FLT3-ITD+ MOLM13 and MV411 cells. (B) FLT3-ITD+ and FLT3 WT human leukemia cell lines were treated with quizartinib for 48 hours. IC50 values were graphically determined by GraphPad Prism. (C) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in murine Npm1CA/+Flt3ITD/+ cells. (D) Ponatinib IC50 concentrations in murine Npm1CA/+Flt3ITD/+ and Hoxa9-Meis1–transformed cells after 48 hours of treatment. (E) Summary of FLT3 inhibitor IC50 concentrations in the human and murine leukemia cell lines assessed in this study. (F) Dose-response curves from cell-viability assays of MV411 and MOLM13 cells, comparing MI-503 (MI; 3 days for MV411 cells and 4 days for MOLM13 cells), quizartinib (Qz; 24 hours), and combinatorial MI-503 (3 or 4 days) and quizartinib (24 hours) treatment. Dashed lines indicate IC50 values. (G) Dose-response curves from cell-viability assays of Npm1CA/+Flt3ITD/+ cells comparing MI-503 (MI, 4 days), ponatinib (Po, 24 hours, left), and gilteritinib (Gil, 24 hours; right), or their combination (4 days, MI-503; 24 hours, ponatinib and gilteritinib). Dashed lines indicate IC50 values. (H) Effect of MI-503 (2.5 µM), ponatinib (100 nM), and combinatorial treatment (2.5 µM and 100 nM) on the number of total and blast-like colonies in murine Npm1CA/+Flt3ITD/+ cells, normalized to DMSO. Micrographs were taken at ×20 amplification. (I-J) Percentage of apoptotic (annexin V) and dead (4′,6-diamidino-2-phenylindole [DAPI]-stained) cells after single and combinatorial treatment with MI-503 (2.5 µM) and quizartinib (3 nM) in human cell lines (I) or MI-503 (2.5 µM) and ponatinib (100 nM) or gilteritinib (400 nM) in murine cells (J). (K) Giemsa-stained cytospins showing human MV411 and MOLM13 cells and murine Npm1CA/+Flt3ITD/+ cells after single and combinatorial treatment with MI-503 (2.5 µM; 4 days and 3 days for MV411) and FLT3 inhibitor (quizartinib, 3 nM, 24 hours; ponatinib, 100 nM, 24 hours) or their combination (day 4/24 hours and day 3/24 hours for MV411, respectively). Micrographs were taken at ×100 amplification. Error bars represent SD of 3 independent experiments, each performed in 3 technical replicates.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f2.png?Expires=1771105444&Signature=Al~B~G8Ug9o0zh6clFKEoi3k1oQLwt~ZbE-PV0U1o~QGvGRVrr7-sC3kAOEkJYQizHFLxhiAuVOj2KU1Eop6J3v3eqkELvOb672x6qjm6Kn0uYe6HaRNQETuGX5w90BgDxmWT1C1ltVTSHSWwCQawHLl5hHRIQmSYs7en1AfbAgZpOdui~YSPzs8ZoGb4GbmueUVldcbfXkKE2LsOHeF-o-WT33xPUSckNoED57GoarvQpGyyKQQDTIJPH67RxUaZ9qeBoLLZPFlQuCtl1RdJ5szTyTx6orXWnRyr0hvoEbvcRli1BoF62UlL5Oi3Ij2KCuYedKsUjUExNzALJ2c2Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Synergistic effects of combined menin-MLL and FLT3 inhibition. (A) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in FLT3-ITD+ MOLM13 and MV411 cells. (B) FLT3-ITD+ and FLT3 WT human leukemia cell lines were treated with quizartinib for 48 hours. IC50 values were graphically determined by GraphPad Prism. (C) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in murine Npm1CA/+Flt3ITD/+ cells. (D) Ponatinib IC50 concentrations in murine Npm1CA/+Flt3ITD/+ and Hoxa9-Meis1–transformed cells after 48 hours of treatment. (E) Summary of FLT3 inhibitor IC50 concentrations in the human and murine leukemia cell lines assessed in this study. (F) Dose-response curves from cell-viability assays of MV411 and MOLM13 cells, comparing MI-503 (MI; 3 days for MV411 cells and 4 days for MOLM13 cells), quizartinib (Qz; 24 hours), and combinatorial MI-503 (3 or 4 days) and quizartinib (24 hours) treatment. Dashed lines indicate IC50 values. (G) Dose-response curves from cell-viability assays of Npm1CA/+Flt3ITD/+ cells comparing MI-503 (MI, 4 days), ponatinib (Po, 24 hours, left), and gilteritinib (Gil, 24 hours; right), or their combination (4 days, MI-503; 24 hours, ponatinib and gilteritinib). Dashed lines indicate IC50 values. (H) Effect of MI-503 (2.5 µM), ponatinib (100 nM), and combinatorial treatment (2.5 µM and 100 nM) on the number of total and blast-like colonies in murine Npm1CA/+Flt3ITD/+ cells, normalized to DMSO. Micrographs were taken at ×20 amplification. (I-J) Percentage of apoptotic (annexin V) and dead (4′,6-diamidino-2-phenylindole [DAPI]-stained) cells after single and combinatorial treatment with MI-503 (2.5 µM) and quizartinib (3 nM) in human cell lines (I) or MI-503 (2.5 µM) and ponatinib (100 nM) or gilteritinib (400 nM) in murine cells (J). (K) Giemsa-stained cytospins showing human MV411 and MOLM13 cells and murine Npm1CA/+Flt3ITD/+ cells after single and combinatorial treatment with MI-503 (2.5 µM; 4 days and 3 days for MV411) and FLT3 inhibitor (quizartinib, 3 nM, 24 hours; ponatinib, 100 nM, 24 hours) or their combination (day 4/24 hours and day 3/24 hours for MV411, respectively). Micrographs were taken at ×100 amplification. Error bars represent SD of 3 independent experiments, each performed in 3 technical replicates.
Synergistic effects of combined menin-MLL and FLT3 inhibition. (A) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in FLT3-ITD+ MOLM13 and MV411 cells. (B) FLT3-ITD+ and FLT3 WT human leukemia cell lines were treated with quizartinib for 48 hours. IC50 values were graphically determined by GraphPad Prism. (C) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in murine Npm1CA/+Flt3ITD/+ cells. (D) Ponatinib IC50 concentrations in murine Npm1CA/+Flt3ITD/+ and Hoxa9-Meis1–transformed cells after 48 hours of treatment. (E) Summary of FLT3 inhibitor IC50 concentrations in the human and murine leukemia cell lines assessed in this study. (F) Dose-response curves from cell-viability assays of MV411 and MOLM13 cells, comparing MI-503 (MI; 3 days for MV411 cells and 4 days for MOLM13 cells), quizartinib (Qz; 24 hours), and combinatorial MI-503 (3 or 4 days) and quizartinib (24 hours) treatment. Dashed lines indicate IC50 values. (G) Dose-response curves from cell-viability assays of Npm1CA/+Flt3ITD/+ cells comparing MI-503 (MI, 4 days), ponatinib (Po, 24 hours, left), and gilteritinib (Gil, 24 hours; right), or their combination (4 days, MI-503; 24 hours, ponatinib and gilteritinib). Dashed lines indicate IC50 values. (H) Effect of MI-503 (2.5 µM), ponatinib (100 nM), and combinatorial treatment (2.5 µM and 100 nM) on the number of total and blast-like colonies in murine Npm1CA/+Flt3ITD/+ cells, normalized to DMSO. Micrographs were taken at ×20 amplification. (I-J) Percentage of apoptotic (annexin V) and dead (4′,6-diamidino-2-phenylindole [DAPI]-stained) cells after single and combinatorial treatment with MI-503 (2.5 µM) and quizartinib (3 nM) in human cell lines (I) or MI-503 (2.5 µM) and ponatinib (100 nM) or gilteritinib (400 nM) in murine cells (J). (K) Giemsa-stained cytospins showing human MV411 and MOLM13 cells and murine Npm1CA/+Flt3ITD/+ cells after single and combinatorial treatment with MI-503 (2.5 µM; 4 days and 3 days for MV411) and FLT3 inhibitor (quizartinib, 3 nM, 24 hours; ponatinib, 100 nM, 24 hours) or their combination (day 4/24 hours and day 3/24 hours for MV411, respectively). Micrographs were taken at ×100 amplification. Error bars represent SD of 3 independent experiments, each performed in 3 technical replicates.
We next assessed FLT3 inhibition in the murine Npm1CA/+Flt3ITD/+ leukemia cells known to harbor the F692L point (“gatekeeper”) mutation that mediates resistance to most FLT3 inhibitors.48 In accordance with published data, the cells showed hardly any response to quizartinib, and the IC50 values of ponatinib, crenolanib, and gilteritinib, were shifted to higher concentrations compared with the values determined from the MOLM13 and MV411 cells that lacked the F692L mutation. Murine Hoxa9-Meis1–transformed cells without an FLT3 mutation were not affected by FLT3 inhibition (Figure 2C-E; supplemental Figure 3D-F). Because the multityrosine kinase inhibitor ponatinib showed the lowest IC50 (48 nM; 48 hours) among the different compounds, we used this inhibitor for the treatment of the murine Npm1CA/+Flt3ITD/+ leukemias and validated the results with higher doses of the more specific drug gilteritinib. Quizartinib was chosen for experiments in the human MLL-r FLT3-ITD+ cells.
Next, we sought to assess combined menin-MLL and FLT3 inhibition in the MLL-r FLT3-ITD human and the murine Npm1CA/+Flt3ITD/+ leukemia cells (Figure 2F-G). Because single-drug treatment with menin-MLL inhibitors inhibits proliferation with a latency of several days20,26 and single-drug FLT3 inhibition induces a more rapid cytotoxic response in vitro, we pretreated the leukemia cells for 2 (MV411) or 3 days (MOLM13, murine Npm1CA/+Flt3ITD/+ cells) with MI-503 and then added the FLT3 inhibitor for an additional 24 hours (combination treatment). Menin-MLL and FLT3 inhibition resulted in dramatically enhanced inhibition of proliferation compared with single-drug treatment in all 3 of the assessed leukemias (Figure 2F-G; supplemental Figure 3G). Notably, for all of those leukemias we found dramatic drug synergism when using the Chou-Talalay algorithm (supplemental Figure 3H-I). Human and murine AML cells without MLL-r, NPM1mut, or FLT3 mutation (HL-60, NB4, and Hoxa9-Meis1–transformed cells), which served as negative controls, were not affected by single-drug or combinatorial treatment (supplemental Figure 3J-K). Murine Npm1CA/+Flt3ITD/+ leukemia cells treated with both drugs in methylcellulose for 7 days showed significantly better suppression of total and blast colony formation compared with single-drug treatment or vehicle control (Figure 2H; supplemental Figure 4A).
The enhanced killing of MLL-r FLT3-ITD+ and Npm1CA/+Flt3ITD/+ leukemia cells was associated with a significantly enhanced induction of apoptosis compared with single-drug or vehicle treatment (Figure 2I-J; supplemental Figure 4B). Menin-MLL inhibitors are known to inhibit proliferation by apoptosis and induction of differentiation in MLL-r and NPM1mut leukemias with a latency of several days, and we wondered whether early-onset induction of differentiation also contributes to the quick-killing effect of the combination treatment. Therefore, we assessed cell morphology in all 3 cell types at the time when efficient inhibition of proliferation was noted with combination treatment (3 and 4 days of menin-MLL inhibition and 24 hours of FLT3 inhibition). The mild-to-moderate myelomonocytic differentiation observed at this early time point (Figure 2K) indicates that apoptosis induction may be the main mechanism of the antileukemic activity of the combination treatment.
These data indicate that combined menin-MLL and FLT3 inhibition enhances apoptosis induction compared with single-drug treatment and synergistically inhibits proliferation in NPM1mut and MLL-r leukemias with FLT3-ITD.
Menin-MLL inhibition enhances FLT3-inhibitor–mediated abrogation of phosphorylated FLT3
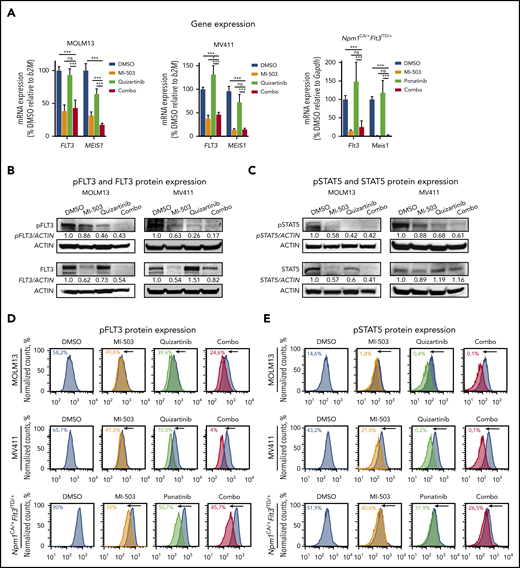
To characterize the effects of combined menin-MLL and FLT3 inhibition in more detail, we first assessed MEIS1 and FLT3 gene expression after combinatorial drug treatment. As described, menin-MLL inhibition alone or in combination with an FLT3 inhibitor suppressed MEIS1 and FLT3 transcription in MLL-r FLT3-ITD+ and Npm1CA/+Flt3ITD/+ leukemias. As expected, we did not find significant downregulation of MEIS1 and FLT3 gene expression with single inhibition of FLT3-phosphorylation. In fact, FLT3 transcription was even upregulated in MV411 and Npm1CA/+Flt3ITD/+ cells with single FLT3 inhibition, which is consistent with previous reports49-51 (Figure 3A).

Effects of single and combined menin-MLL and FLT3 inhibition on FLT3 and phosphorylated FLT3 protein levels. (A) FLT3 and MEIS1 mRNA expression in human MOLM13 (left) and MV411 (middle) cells and murine Npm1CA/+Flt3ITD/+ (right) leukemia cells after single or combinatorial treatment with MI-503 (2.5 µM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells and 3 days for MV411 cells) and FLT3 inhibitors (quizartinib, 3 nM; ponatinib, 100 nM, 24 hours) as assessed by qRT-PCR. Bar graphs represent the mean with SD of 3 independent experiments, each performed in technical triplicate. (B) Immunoblot analysis of FLT3 and phosphorylated (p)FLT3 in MOLM13 cells (left) and MV411 cells (right) upon treatment with 2.5 µM MI-503 (for 3 and 4 days in MV411 and MOLM13 cells), quizartinib (3 nM, 24 hours), or the 2 combined. One representative blot of 3 independent experiments is shown. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry with ImageJ software. (C) Immunoblot analysis of STAT5 and phosphorylated (p)STAT5 in MOLM13 (left) and MV411 (right) cells after treatment as described in panel B. One representative blot of 3 independent experiments is shown. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry with ImageJ software. pFLT3 (D) and pSTAT5 (E) protein expression in human MOLM13 and MV411 and murine Npm1CA/+Flt3ITD/+ cells after treatment with MI-503 (2.5 µM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells; 3 days for MV411 cells), FLT3 inhibitor (quizartinib, 3 nM, and ponatinib, 100 nM; 24 hours) or their combination, as assessed by flow cytometry. One representative histogram of 3 independent experiments is shown. The colored numbers in the flow histograms indicate the percentage of pFLT3+ and pSTAT5+ cells, respectively.
Effects of single and combined menin-MLL and FLT3 inhibition on FLT3 and phosphorylated FLT3 protein levels. (A) FLT3 and MEIS1 mRNA expression in human MOLM13 (left) and MV411 (middle) cells and murine Npm1CA/+Flt3ITD/+ (right) leukemia cells after single or combinatorial treatment with MI-503 (2.5 µM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells and 3 days for MV411 cells) and FLT3 inhibitors (quizartinib, 3 nM; ponatinib, 100 nM, 24 hours) as assessed by qRT-PCR. Bar graphs represent the mean with SD of 3 independent experiments, each performed in technical triplicate. (B) Immunoblot analysis of FLT3 and phosphorylated (p)FLT3 in MOLM13 cells (left) and MV411 cells (right) upon treatment with 2.5 µM MI-503 (for 3 and 4 days in MV411 and MOLM13 cells), quizartinib (3 nM, 24 hours), or the 2 combined. One representative blot of 3 independent experiments is shown. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry with ImageJ software. (C) Immunoblot analysis of STAT5 and phosphorylated (p)STAT5 in MOLM13 (left) and MV411 (right) cells after treatment as described in panel B. One representative blot of 3 independent experiments is shown. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry with ImageJ software. pFLT3 (D) and pSTAT5 (E) protein expression in human MOLM13 and MV411 and murine Npm1CA/+Flt3ITD/+ cells after treatment with MI-503 (2.5 µM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells; 3 days for MV411 cells), FLT3 inhibitor (quizartinib, 3 nM, and ponatinib, 100 nM; 24 hours) or their combination, as assessed by flow cytometry. One representative histogram of 3 independent experiments is shown. The colored numbers in the flow histograms indicate the percentage of pFLT3+ and pSTAT5+ cells, respectively.
These transcriptional changes translated into similar changes on the protein level, as assessed by immunoblot analysis. Although total FLT3 protein expression was reduced with menin-MLL or combination treatment, single-drug FLT3 inhibition caused no change (MOLM13) or upregulation (MV411) of total FLT3 protein levels (Figure 3B; supplemental Figure 5A).
Assessment of FLT3-receptor phosphorylation (pFLT3; activated FLT3) in these cell lines showed a strong reduction in pFLT3 after its direct inhibition by quizartinib and was also mildly reduced after menin-MLL inhibition, the latter most likely reflecting the decreased total FLT3 protein level. Combined-drug treatment caused even more pronounced reduction of pFLT3 compared with single-drug treatment, as assessed by immunoblot analysis (Figure 3B; supplemental Figure 5A). The observed reduction of pFLT3 was also associated with reduced phosphorylation of the downstream signaling proteins STAT5 (pSTAT5) and ERK (pERK). Similar to pFLT3, we observed reduced phosphorylation of both proteins with single-drug treatment that was even more pronounced with combinatorial treatment for pERK in MV411 and MOLM13 cells and for pSTAT5 in MV411 cells, whereas the dramatic reduction of pSTAT5 observed with quizartinib in MOLM13 was not enhanced (Figure 3C; supplemental Figure 5B).
Similar results were obtained when we assessed pFLT3 by flow cytometry in the human MLL-r FLT3-ITD and the Npm1CA/+Flt3ITD/+ leukemia cells, in which combined treatment abrogated pFLT3 levels significantly more than single-drug or vehicle treatment (Figure 3D; supplemental Figure 5C-D). The specificity of the antibody for pFLT3 is indicated by FLT3 ligand-stimulation experiments (supplemental Figure 5E). Again, the decrease in the FLT3 downstream signaling proteins pSTAT5 and pERK was more pronounced after combinatorial treatment than after single-drug treatment in MV411 cells and murine leukemia cells examined by flow cytometry, whereas quizartinib and combinatorial treatment showed a similar reduction of pSTAT5 and pERK in MOLM13 cells (Figure 3E; supplemental Figure 5F-J).
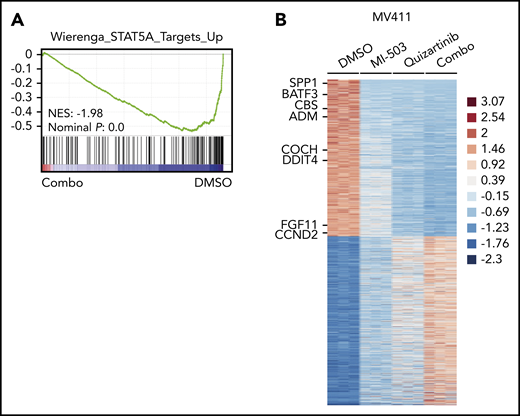
To explore the global transcriptional consequences of the enhanced reduction of pFLT3, we performed RNA-seq analysis in the MV411 cells and compared combinatorial vs single-drug or vehicle treatment. Gene set enrichment analysis revealed that the genes downregulated by combinatorial treatment were significantly enriched for target genes of the FLT3-activated transcription factor STAT5A (Figure 4A). When comparing the expression levels of STAT5A target genes between the different treatment groups in more detail, we found most of these genes to be moderately suppressed by single-drug treatment with either menin-MLL or FLT3 inhibitors. The combinatorial treatment further reduced the expression levels of these genes more efficiently (Figure 4B; supplemental Figure 6A-B; supplemental Table 2).
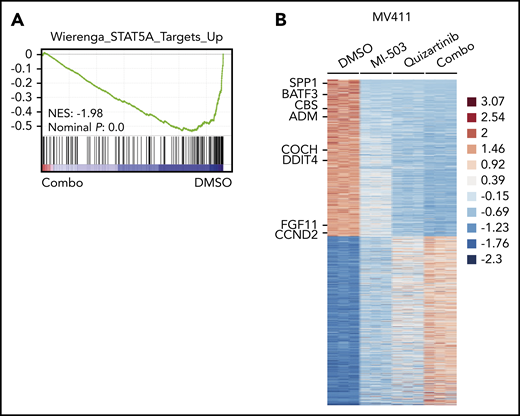
Downregulated genes after combined menin-MLL and FLT3 inhibition are enriched for STAT5A target genes. (A) Gene set enrichment analysis of gene expression changes in MV411 cells treated with combined MI-503 (2.5 µM; 3 days) and quizartinib (3 nM; 24 hours) compared with STAT5A targets. (B) Heatmap of differentially expressed genes (log2 change >1 and < −1, and adjusted P < .05) in MV411 cells after single and combined treatment with MI-503 (2.5 µM, 3 days) and quizartinib (3 nM, 24 hours). Target genes of STAT5A are indicated.
Downregulated genes after combined menin-MLL and FLT3 inhibition are enriched for STAT5A target genes. (A) Gene set enrichment analysis of gene expression changes in MV411 cells treated with combined MI-503 (2.5 µM; 3 days) and quizartinib (3 nM; 24 hours) compared with STAT5A targets. (B) Heatmap of differentially expressed genes (log2 change >1 and < −1, and adjusted P < .05) in MV411 cells after single and combined treatment with MI-503 (2.5 µM, 3 days) and quizartinib (3 nM, 24 hours). Target genes of STAT5A are indicated.
As FLT3 is a reported MEIS1 and HOXA9 transcriptional and potential binding target,52-54 we next assessed the effect of retrovirally induced ectopic expression of Meis1 and Hoxa9-Meis1 on Flt3 expression in Npm1CA/+Flt3ITD/+ murine leukemia cells. Both scenarios increased the expression of endogenous Flt3 by 1.5- and 2.8-fold, respectively (Figure 5A; supplemental Figure 7A). Of interest, ectopic Meis1 and Hoxa9-Meis1 expression partially rescued the Npm1CA/+Flt3ITD/+ cells from the antiproliferative activity of the menin-MLL inhibitor and of the combinatorial treatment (Figure 5B-C; supplemental Figure 7B-C). Similar rescue effects on colony-forming potential were observed when the cells were treated in methylcellulose (supplemental Figure 8A-B).
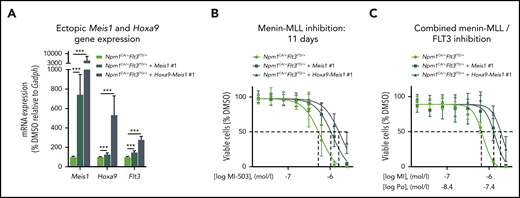
Effects of ectopic Meis1 expression in murine Npm1mut Flt3-ITD+ leukemias. (A) Relative mRNA expression of Meis1, Hoxa9, and Flt3 in murine Npm1CA/+Flt3ITD/+ cells ectopically expressing Meis1 or both Meis1 and Hoxa9, normalized to cells with just endogenous Meis1 and Hoxa9 expression. (B) Dose-response curves from cell viability assays after 11 days of MI-503 treatment comparing Npm1CA/+Flt3ITD/+ cells vs Npm1CA/+Flt3ITD/+ cells overexpressing Meis1 or Meis1-Hoxa9. Dashed lines indicate the shift of IC50 values. (C) Dose-response curves from cell viability assays after combinatorial treatment with MI-503 (MI, 6 days) and ponatinib (Po; 72 hours) comparing Npm1CA/+Flt3ITD/+ cells vs Npm1CA/+Flt3ITD/+ cells overexpressing Meis1 or Meis1-Hoxa9. Dashed lines indicate the shift of IC50 values. Cells with ectopic Meis1 and Hoxa9 expression in panels A-C were each obtained from 2 different clones after retroviral transduction. Shown are the average results of 3 different experiments with clone #1, all performed in triplicate. Error bars, standard deviation.
Effects of ectopic Meis1 expression in murine Npm1mut Flt3-ITD+ leukemias. (A) Relative mRNA expression of Meis1, Hoxa9, and Flt3 in murine Npm1CA/+Flt3ITD/+ cells ectopically expressing Meis1 or both Meis1 and Hoxa9, normalized to cells with just endogenous Meis1 and Hoxa9 expression. (B) Dose-response curves from cell viability assays after 11 days of MI-503 treatment comparing Npm1CA/+Flt3ITD/+ cells vs Npm1CA/+Flt3ITD/+ cells overexpressing Meis1 or Meis1-Hoxa9. Dashed lines indicate the shift of IC50 values. (C) Dose-response curves from cell viability assays after combinatorial treatment with MI-503 (MI, 6 days) and ponatinib (Po; 72 hours) comparing Npm1CA/+Flt3ITD/+ cells vs Npm1CA/+Flt3ITD/+ cells overexpressing Meis1 or Meis1-Hoxa9. Dashed lines indicate the shift of IC50 values. Cells with ectopic Meis1 and Hoxa9 expression in panels A-C were each obtained from 2 different clones after retroviral transduction. Shown are the average results of 3 different experiments with clone #1, all performed in triplicate. Error bars, standard deviation.
These data support the concept that FLT3 expression is driven by the MEIS1 transcription factor in these leukemias and that transcriptional suppression of FLT3 via menin-MLL inhibition contributes to a more efficient reduction of phosphorylated FLT3.
The novel menin-MLL inhibitor VTP-50469, in combination with FLT3 inhibitors, exerts a highly synergistic effect against NPM1mut and MLL-r leukemias
To confirm the effects of pharmacological menin-MLL inhibition by MI-503, we used 2 validated small-hairpin RNAs to perform knockdown of MEN1 in MOLM13 and MV411 cells. At 48 hours after lentiviral transduction, we observed downregulation of MEIS1 and FLT3 gene expression in a MEN1 suppression–dependent manner. Also, we found that MEN1 knockdown consistently sensitized the MLL-r AML cells to pharmacological FLT3 inhibition, thereby phenocopying the effects observed with MI-503 (Figure 6A-B).
![Synergistic inhibition of proliferation and FLT3 activation after combined treatment with next-generation menin-MLL and FLT3 inhibitors. (A) Dose-response curves of MOLM13 and MV411 cells treated with quizartinib for 24 hours, comparing cells transduced with short hairpin RNAs against MEN1 with control-transduced cells (shLUC). (B) mRNA expression levels of MEN1, MEIS1, and FLT3 in MOLM13 and MV411 cells with MEN1 knock down or control-transduced cells, assessed 48 hours after transduction. (C) Dose-response curves from cell-viability assays after 7 days of treatment with VTP-50469 in human (left) and murine (right) leukemia cells. Viable (4′,6-diamidino-2-phenylindole [DAPI]-negative) cells were assessed by flow cytometry. (D-E) Dose-response curves from cell viability assays of MV411 and MOLM13 cells comparing VTP-50469 (VTP; 3 days for MV411 and 4 days for MOLM13 cells), quizartinib (Qz, 24 hours; D), gilteritinib (Gil, 24 hours; E), and combinatorial VTP-50469 (3 or 4 days) and FLT3 inhibition (24 hours) treatment. Dashed lines indicate IC50 values. (F) Dose-response curves from cell viability assays of Npm1CA/+Flt3ITD/+ cells comparing VTP-50469 (VTP; 6 days), ponatinib (Po, 24 hours, [left]), or gilteritinib (Gil, 24 hours; [right]) with their combination (6 days VTP50469, 24 hours FLT3 inhibition). Dashed lines indicate IC50 values. (G-H) FLT3 and MEIS1 mRNA expression in murine Npm1CA/+Flt3ITD/+ (G), human MV411 (H; left), and MOLM13 (H; right) leukemia cells after single or combined treatment with VTP-50469 (100 nM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells and 3 days for MV411 cells) and FLT3 inhibitors (quizartinib, 3 nM and ponatinib, 100 nM; 24 hours) as assessed by qRT-PCR. Bar graphs represent the mean with standard deviation of 3 independent experiments, each performed in technical triplicate. (I) Immunoblot analysis of FLT3 and phosphorylated (p)FLT3 in MV411 cells (left) and MOLM13 cells (right) after treatment with VTP-50469 (100 nM; 3 and 4 days in MV411 and MOLM-13, respectively) and quizartinib (3 nM, 24 hours), or their combination. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry using the ImageJ software tool.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f6.png?Expires=1771105444&Signature=uFHwr9gwapcevSlcKF7NLW4wAhaY~UOI0bPYTyD0t~UGAlkb~AIr1FaehZ8aK3bJ0S58FczxzTVYFmGLXE3nCxQ20Jo~~CJMnniNgGZSfnxdjODJRA0PviLHhl9e3P-jrHI9MAxLvw2bLL-WgPHGkgdag7zwPlAR2uRrHcP0CycehB~SRgEHCeyyDE7w7nqIk5tlGgcgJtvyZE2F~IB7s2xGni17PbRKkWkuHAMqpyQBBiMtkaG0JATwob7zPwIQ-FblacLP2p~gAtH~LqVww8e8uk~RN0ah3ml6qeDQuddxZydJWWUmLULLtDhBIuVeW5YwsTIU-BBAJ-dyEvLBvw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Synergistic inhibition of proliferation and FLT3 activation after combined treatment with next-generation menin-MLL and FLT3 inhibitors. (A) Dose-response curves of MOLM13 and MV411 cells treated with quizartinib for 24 hours, comparing cells transduced with short hairpin RNAs against MEN1 with control-transduced cells (shLUC). (B) mRNA expression levels of MEN1, MEIS1, and FLT3 in MOLM13 and MV411 cells with MEN1 knock down or control-transduced cells, assessed 48 hours after transduction. (C) Dose-response curves from cell-viability assays after 7 days of treatment with VTP-50469 in human (left) and murine (right) leukemia cells. Viable (4′,6-diamidino-2-phenylindole [DAPI]-negative) cells were assessed by flow cytometry. (D-E) Dose-response curves from cell viability assays of MV411 and MOLM13 cells comparing VTP-50469 (VTP; 3 days for MV411 and 4 days for MOLM13 cells), quizartinib (Qz, 24 hours; D), gilteritinib (Gil, 24 hours; E), and combinatorial VTP-50469 (3 or 4 days) and FLT3 inhibition (24 hours) treatment. Dashed lines indicate IC50 values. (F) Dose-response curves from cell viability assays of Npm1CA/+Flt3ITD/+ cells comparing VTP-50469 (VTP; 6 days), ponatinib (Po, 24 hours, [left]), or gilteritinib (Gil, 24 hours; [right]) with their combination (6 days VTP50469, 24 hours FLT3 inhibition). Dashed lines indicate IC50 values. (G-H) FLT3 and MEIS1 mRNA expression in murine Npm1CA/+Flt3ITD/+ (G), human MV411 (H; left), and MOLM13 (H; right) leukemia cells after single or combined treatment with VTP-50469 (100 nM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells and 3 days for MV411 cells) and FLT3 inhibitors (quizartinib, 3 nM and ponatinib, 100 nM; 24 hours) as assessed by qRT-PCR. Bar graphs represent the mean with standard deviation of 3 independent experiments, each performed in technical triplicate. (I) Immunoblot analysis of FLT3 and phosphorylated (p)FLT3 in MV411 cells (left) and MOLM13 cells (right) after treatment with VTP-50469 (100 nM; 3 and 4 days in MV411 and MOLM-13, respectively) and quizartinib (3 nM, 24 hours), or their combination. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry using the ImageJ software tool.
Synergistic inhibition of proliferation and FLT3 activation after combined treatment with next-generation menin-MLL and FLT3 inhibitors. (A) Dose-response curves of MOLM13 and MV411 cells treated with quizartinib for 24 hours, comparing cells transduced with short hairpin RNAs against MEN1 with control-transduced cells (shLUC). (B) mRNA expression levels of MEN1, MEIS1, and FLT3 in MOLM13 and MV411 cells with MEN1 knock down or control-transduced cells, assessed 48 hours after transduction. (C) Dose-response curves from cell-viability assays after 7 days of treatment with VTP-50469 in human (left) and murine (right) leukemia cells. Viable (4′,6-diamidino-2-phenylindole [DAPI]-negative) cells were assessed by flow cytometry. (D-E) Dose-response curves from cell viability assays of MV411 and MOLM13 cells comparing VTP-50469 (VTP; 3 days for MV411 and 4 days for MOLM13 cells), quizartinib (Qz, 24 hours; D), gilteritinib (Gil, 24 hours; E), and combinatorial VTP-50469 (3 or 4 days) and FLT3 inhibition (24 hours) treatment. Dashed lines indicate IC50 values. (F) Dose-response curves from cell viability assays of Npm1CA/+Flt3ITD/+ cells comparing VTP-50469 (VTP; 6 days), ponatinib (Po, 24 hours, [left]), or gilteritinib (Gil, 24 hours; [right]) with their combination (6 days VTP50469, 24 hours FLT3 inhibition). Dashed lines indicate IC50 values. (G-H) FLT3 and MEIS1 mRNA expression in murine Npm1CA/+Flt3ITD/+ (G), human MV411 (H; left), and MOLM13 (H; right) leukemia cells after single or combined treatment with VTP-50469 (100 nM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells and 3 days for MV411 cells) and FLT3 inhibitors (quizartinib, 3 nM and ponatinib, 100 nM; 24 hours) as assessed by qRT-PCR. Bar graphs represent the mean with standard deviation of 3 independent experiments, each performed in technical triplicate. (I) Immunoblot analysis of FLT3 and phosphorylated (p)FLT3 in MV411 cells (left) and MOLM13 cells (right) after treatment with VTP-50469 (100 nM; 3 and 4 days in MV411 and MOLM-13, respectively) and quizartinib (3 nM, 24 hours), or their combination. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry using the ImageJ software tool.
During the work on the revision of this study, VTP-50469, a novel and more selective menin-MLL inhibitor with clinical utility was described and became available to us. We used VTP-50469 to independently validate the effects observed with MI-503. First, we confirmed selective growth inhibition of VTP-50469 on MLL-r and NPM1mut leukemia cells (Figure 6C; supplemental Figure 9A-B). Next, we assessed its combination with the FLT3 inhibitors quizartinib and gilteritinib in the human MLL-r and with ponatinib and gilteritinib in the murine Npm1CA/+Flt3ITD/+ AML cells. Similar to MI-503, all combinations resulted in synergistic inhibition of cell proliferation compared with single-drug treatment (Figure 6D-F; supplemental Figure 9C-G). VTP-50469 also suppressed MEIS1 and FLT3 expression dramatically and when combined with FLT3 inhibitors resulted in more pronounced abrogation of pFLT3 protein levels than each of the single-drug treatments alone (Figure 6G-I). These data independently confirm the synergistic antileukemic effects of combined menin-MLL and FLT3 targeting.
Combined menin-MLL and FLT3 inhibition suppresses primary NPM1mutFLT3ITD AML cells in vitro
To investigate combinatorial menin-MLL and FLT3 inhibition in samples from patients with primary AML, we used a previously described coculture assay that allowed us to maintain and treat these leukemia cells in serum-free medium with cytokines on a (HS27) stromal cell layer in vitro (Figure 7A). Five de novo NPM1mutFLT3-ITD samples from patients with AML were treated for 7 days with DMSO, MI-503, quizartinib, or the combination of the drugs. All 5 samples showed significantly enhanced reduction in the number of cells with combinatorial treatment compared with single-drug treatment. In all samples, MI-503 and quizartinib also reduced the number of viable cells significantly compared with the cells exposed to the drug vehicle (Figure 7B-C). To control for potential nonspecific drug toxicity, we also treated 2 primary AML samples lacking NPM1mut, MLL-r, and FLT3-ITD. In both samples the number of viable cells was not significantly affected by either single-drug or combinatorial treatment (Figure 7D). Four of the 5 primary NPM1mutFLT3-ITD samples were also treated in methylcellulose. Similar to the proliferation assays, blast-colony formation was significantly reduced by the drug combination vs all other treatment groups (Figure 7E).
![Effects of single and combined menin-MLL and FLT3 inhibition on primary NPM1mut FLT3ITD AML patient samples and on survival of in vivo–treated MLL-r FLT3-ITD leukemic xenograft mice. (A) The human stromal cell coculture assay, performed to maintain and treat patients’ primary AML blasts. (B) Summary of characteristics of patients providing the samples used in panels C-E. (C-D) Number of viable cells inde novoAML samples treated in coculture for 7 days with DMSO, MI-503 (2 µM), quizartinib (6 nM), or combinatorial MI-503 and quizartinib treatment. (C) Five independent samples of de novo NPM1mutFLT3ITD AML. (D) Two independent samples of de novo AML, WT for NPM1, FLT3, and MLL. Depicted are 4′,6-diamidino-2-phenylindole [DAPI]−, human CD45+ cell numbers as assessed by flow cytometry. (E) Effect of MI-503 (2.5 µM), quizartinib (3 nM), and combinatorial treatment (2.5 µM and 3 nM) on total and blast-like CFUs in primary patient sample cells. (F) Experimental setup for the treatment of MV411-derived leukemic xenograft mice (left); percentage of human CD45+ cells in the bone marrow of leukemic mice (right) after treatment with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combined MI-503 and quizartinib. (G) Kaplan-Meier survival analysis of MV411-derived leukemic xenograft mice treated with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combinatorial MI-503 and quizartinib (n = 5 mice/group). The treatment period is displayed in blue. The log-rank (Mantel-Cox) test was used to calculate the P-values.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f7.png?Expires=1771105444&Signature=USvavSVEysa-xgldO7HSpUWORU0UC-kk04q7ef6byCl~9h7sNhx4jHiyrGj~ZCehbr29BQsp3Nb~c1~LGAzigV33t6iy9KrIbEOZe5NjETth~k1ALpCn2h2mMOExn2Fh-HoUfmB87UedVipUSMPPjT4fH68XAIzvptq6fiKLt5F8-j5otr9cs~yQlIvJbylwLszIEc3s7A7xlckS7~ACh~K2N6rdb8yw8caHJmVnbtKbLRI8wxVM~1yEtEX0cFARA4vtyPHSc29VaeMPQJMe~sPs6K05nu10rBeU3dSg9w-q6nLygF0ajgKGBWlqLDDrw3ECeS5lDIr-xMfeOyxjAQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of single and combined menin-MLL and FLT3 inhibition on primary NPM1mut FLT3ITD AML patient samples and on survival of in vivo–treated MLL-r FLT3-ITD leukemic xenograft mice. (A) The human stromal cell coculture assay, performed to maintain and treat patients’ primary AML blasts. (B) Summary of characteristics of patients providing the samples used in panels C-E. (C-D) Number of viable cells inde novoAML samples treated in coculture for 7 days with DMSO, MI-503 (2 µM), quizartinib (6 nM), or combinatorial MI-503 and quizartinib treatment. (C) Five independent samples of de novo NPM1mutFLT3ITD AML. (D) Two independent samples of de novo AML, WT for NPM1, FLT3, and MLL. Depicted are 4′,6-diamidino-2-phenylindole [DAPI]−, human CD45+ cell numbers as assessed by flow cytometry. (E) Effect of MI-503 (2.5 µM), quizartinib (3 nM), and combinatorial treatment (2.5 µM and 3 nM) on total and blast-like CFUs in primary patient sample cells. (F) Experimental setup for the treatment of MV411-derived leukemic xenograft mice (left); percentage of human CD45+ cells in the bone marrow of leukemic mice (right) after treatment with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combined MI-503 and quizartinib. (G) Kaplan-Meier survival analysis of MV411-derived leukemic xenograft mice treated with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combinatorial MI-503 and quizartinib (n = 5 mice/group). The treatment period is displayed in blue. The log-rank (Mantel-Cox) test was used to calculate the P-values.
Effects of single and combined menin-MLL and FLT3 inhibition on primary NPM1mut FLT3ITD AML patient samples and on survival of in vivo–treated MLL-r FLT3-ITD leukemic xenograft mice. (A) The human stromal cell coculture assay, performed to maintain and treat patients’ primary AML blasts. (B) Summary of characteristics of patients providing the samples used in panels C-E. (C-D) Number of viable cells inde novoAML samples treated in coculture for 7 days with DMSO, MI-503 (2 µM), quizartinib (6 nM), or combinatorial MI-503 and quizartinib treatment. (C) Five independent samples of de novo NPM1mutFLT3ITD AML. (D) Two independent samples of de novo AML, WT for NPM1, FLT3, and MLL. Depicted are 4′,6-diamidino-2-phenylindole [DAPI]−, human CD45+ cell numbers as assessed by flow cytometry. (E) Effect of MI-503 (2.5 µM), quizartinib (3 nM), and combinatorial treatment (2.5 µM and 3 nM) on total and blast-like CFUs in primary patient sample cells. (F) Experimental setup for the treatment of MV411-derived leukemic xenograft mice (left); percentage of human CD45+ cells in the bone marrow of leukemic mice (right) after treatment with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combined MI-503 and quizartinib. (G) Kaplan-Meier survival analysis of MV411-derived leukemic xenograft mice treated with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combinatorial MI-503 and quizartinib (n = 5 mice/group). The treatment period is displayed in blue. The log-rank (Mantel-Cox) test was used to calculate the P-values.
These data from primary NPM1mutFLT3-ITD+ AML patient samples further support single and combinatorial menin-MLL and FLT3 inhibition as a potentially efficacious therapeutic concept for FLT3-mutated NPM1mut leukemia.
Combined in vivo treatment significantly prolongs survival of MLL-r FLT3-ITD+ leukemic mice
We next sought to explore the therapeutic potential of combined menin-MLL and FLT3 inhibition in vivo. First, we assessed the effects of these drugs on leukemic burden in a disseminated human MV411 xenotransplantation model. MV411 cells were transplanted into NSG mice via tail-vein injection, and the animals were randomly divided into 4 groups receiving MI-503, quizartinib, a combination of the 2 drugs, or vehicle control treatment. The treatment was started 1 week after transplantation (day 7) and the animals were euthanized after 14 days of treatment. Leukemia burden, as defined by the percentage of bone marrow cells expressing human CD45, was significantly reduced within the animal group treated with the drug combination vs all other groups (vehicle, 15%; MI-503, 8%; quizartinib, 3%; combination [combo], 1.5%; P < .05 for combo vs other groups; Figure 7F).
In a separate experiment, we assessed survival in the disseminated MV411 xenograft leukemia model. Drug treatment was initiated on day 12 after transplantation and continued until day 45. Combinatorial MI-503 and quizartinib treatment resulted in a significant survival advantage compared with single-drug– or vehicle-treated animals (hazard ratios for death, 0.15 and 0.35; 95% confidence intervals, 0.02845-0.8061 and 0.0285-0.8783; P = .0269 and P = .0350 for combo vs MI-503 and combo vs quizartinib, respectively; Figure 7G).
In summary, these data confirm that the combination of menin-MLL and FLT3 inhibition substantially improves survival of mice engrafted with MLL-r FLT3-ITD+ leukemia compared with single-drug– or vehicle-treated animals and supports this therapeutic concept as a synergistic approach to combat these leukemias.
Discussion
Intensive chemotherapy remains the backbone of curative treatment of AML.6 This aggressive therapy can induce long-term remissions in only about half of the patients with NPM1mut AML and one-third of the patients with MLL-r AML.3 Explanations for these relatively unsatisfactory survival rates is older age at diagnosis and the presence of concurrent poor prognostic disease markers, such as the FLT3-ITD mutations.33,34,55 These mutations are associated with treatment failure, relapse, and death56 and also represent important therapeutic targets. FLT3 inhibitors have been shown to increase survival rates in de novo and in relapsed or refractory AML,38,39,57,58 and potent, highly selective second-generation FLT3 inhibitors can induce high response rates as single agents. However, without further consolidating treatment, almost all patients relapse and ultimately succumb to their disease.38,39 Emerging resistance mutations have been reported as one reason for resistance to FLT3 inhibition,58,59 but there are other potential explanations. First, AML is commonly driven by multiple oncogenic mechanisms and not by a single FLT3 mutation. Recent deep-sequencing efforts found a median number of 5 and at least 2 genetic driver events to be present per AML case.5 Second, FLT3 mutations are considered late events during leukemogenesis.60-62 Therefore, FLT3 inhibitors target the dominant FLT3-mutant AML blast population, but potentially, not the AML founding clone from which relapse may originate. Because of its biologic heterogeneity, AML is generally not believed to be cured by single-drug treatment.63
In our study, we developed a synergistic treatment regimen that combines menin-MLL and FLT3 inhibitors to target leukemogenic gene expression in NPM1mut or MLL-r leukemia and activating FLT3 mutations. The development of this approach was based on the previous findings that inhibition of the menin-MLL interaction reverses leukemogenic gene expression in NPM1mut AML, including the most pronounced MEIS1 transcription factor and its putative transcriptional target gene FLT3.26 In a detailed assessment, we also demonstrated that both WT and mutant FLT3 transcript levels were consistently downregulated in response to MI-503 in NPM1mut and MLL-r AML models.
As FLT3 is a transcriptional target gene of MEIS1,53,54 FLT3 downregulation after menin-MLL inhibition is therefore a likely result of the dramatically suppressed MEIS1 expression. Reduced total FLT3 levels were also associated with the lower level of pFLT3 that we observed after menin-MLL inhibition. This may explain the superior reduction of pFLT3 that we observed with the combination treatment, compared with direct inhibition of pFLT3 or menin-MLL inhibition alone.
It is of interest to note that the pronounced reduction of pFLT3 after combinatorial menin-MLL and FLT3 inhibition also resulted in significantly enhanced suppression of STAT5A target genes in MV411 cells. Because STAT5A contributes to leukemia maintenance and is an important downstream mediator of activating FLT3 mutations,64-66 the synergistic antileukemic activity of combined menin-MLL and FLT3 inhibition is probably caused by its enhanced on-target activity in preventing FLT3 signaling.
We have also shown that upregulation of FLT3, which is commonly observed after FLT3 inhibition,49-51 can be reversed with the addition of the menin-MLL inhibitor. Although this mechanism may contribute to enhanced activity of FLT3 inhibitors, future studies will determine whether this concept may help to prevent or overcome resistance to FLT3 inhibition.
Menin-MLL inhibitors are particularly attractive partners in a combined treatment, as they allow the effective targeting of a core leukemogenic gene expression program driven by NPM1 mutations or MLL fusions including the transcription factors MEIS1 and PBX3.15,53,67,68 Our assessment of blast colony formation upon drug treatment is consistent with the view that menin-MLL inhibition targets immature leukemia-initiating cells, most likely via suppression of self-renewal–associated MEIS1- and PBX3-driven gene expression. Blast colony formation can be further inhibited by the addition of a FLT3 inhibitor, whereas FLT3 inhibition alone has only minor effects. The mechanisms behind this observation remain to be determined in detailed studies assessing single and combined drug effects on the leukemia-initiating fraction in these leukemia subtypes.
In contrast to previous menin-MLL inhibitors such as MI-2-2,25 many other MLL target genes including the HOX transcription factors were only moderately suppressed (MLL-r and murine Npm1CA/+Flt3ITD/+) or unchanged (human NPM1mut OCI-AML3) with MI-503. This is consistent with findings from recent studies assessing novel and more selective menin-MLL inhibitors, such as VTP-50469 and MI-3454, that also did not lead to broad downregulation of HOX genes and other MLL-fusion targets.22,28 These findings are in line with the observation that MLL fusion occupancy is lost only on a subset of genes in response to menin-MLL inhibition.22 Although we do not know the exact mechanism behind the different responses at specific loci, the 2 novel menin-MLL inhibitors also dramatically suppress a core transcriptional program that includes PBX3, MEIS1, and FLT3. In accordance with these observations, we present clear evidence that the novel menin-MLL inhibitor VTP-50469 has dramatic synergistic antileukemic activity in combination with various FLT3 inhibitors.
In summary, our data show that menin-MLL inhibition targets FLT3 transcriptionally via suppression of the leukemogenic transcription factor MEIS1, which is driven by NPM1 mutations or MLL fusions in AML and is not amenable to direct pharmacological inhibition. We further demonstrate that the combination of menin-MLL and FLT3 inhibition results in enhanced on-target activity against pFLT3, resulting in synergistic antileukemic effects against NPM1mut or MLL-r leukemias harboring the prognostically adverse FLT3-ITD mutation. This drug combination therefore represents a promising opportunity for the treatment of these leukemia subtypes that is already available for clinical testing.
The RNA-seq data reported in this article have been deposited Gene Expression Omnibus database (accession number GSE144759).
Original data are available by e-mail request to the corresponding author Michael W. M. Kühn (michael.kuehn@unimedizin-mainz.de).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Viral Shah, Jakub Szybinski, Birgit Enders, and Ashley Westerback for technical help; Maria Mendez-Lago, Clara Werner, and Hanna Lukas from the Institute of Molecular Biology (IMB) Genomics Core Facility for assisting with sequencing with NextSeq500 (INST 247/870-1 FUGG); the Translational Animal Research Center (TARC) of the University Medical Center, Mainz, for providing the animals; the Cytology Laboratory within the Department of Hematology and Medical Oncology of the University Medical Center, Mainz, for performing cytospins and Giemsa staining; and Basepair for bioinformatics support.
This work was supported by a grant from the German Research Foundation (DFG, KU 2688/2-1).
Authorship
Contribution: M.W.M.K. designed the research, analyzed and interpreted the data, wrote and revised the manuscript, and supervised the study; M.M.D. performed the experiments, analyzed and interpreted the data, wrote the original draft of the manuscript and edited the manuscript; M.S. performed experiments, analyzed and interpreted the data, and edited the manuscript; J.R. performed experiments and analyzed and interpreted the data; K.K., J.S., and M.C.T. performed experiments and analyzed the data; D.S. and R.P.K. analyzed and interpreted the RNA-seq data; A.M. provided Npm1CA/+Flt3ITD/+ murine leukemia cells and revised the manuscript; G.M.M. provided the menin-MLL inhibitor VTP-50469 and revised the manuscript; P.H. and T.K. provided administrative support and patient samples and revised the manuscript; and C.-W.C., M.T., G.S.V., and S.A.A. analyzed and interpreted the data and revised the manuscript.
Conflict-of-interest disclosure: M.W.M.K, is a consultant for Pfizer and Abbvie and receives travel support from Celgene and Daiichi Sankyo. S.A.A. is a consultant and/or shareholder for Epizyme Inc, Vitae/Allergan Pharmaceuticals, Imago Biosciences, Cyteir Therapeutics, C4 Therapeutics, Syros Pharmaceuticals, OxStem Oncology, Accent Therapeutics, and Mana Therapeutics and has received research support from Janssen, Novartis, and AstraZeneca. G.S.V. is a consultant for Kymab and Oxstem. G.M.M. is a shareholder of Syndax Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Michael W. M. Kühn, University Medical Center, Johannes Gutenberg-University Mainz, Langenbeckstr 1, 55131 Mainz, Germany; e-mail: michael.kuehn@unimedizin-mainz.de.

![Gene and protein expression changes upon menin-MLL inhibition in NPM1mut and MLL-r AML. (A) Human (left) and murine (right) AML cells were treated for 11 days with MI-503. Viable (4′,6-diamidino-2-phenylindole [DAPI]–negative) cells were assessed by flow cytometry, and IC50 values were calculated with GraphPad Prism software. (B) Summary of IC50 values (MI-503), MLL-rearrangement, and NPM1 and FLT3 mutation status in the AML cells assessed. (C) Venn diagram showing downregulated genes identified by RNA-seq (more than twofold decrease; adjusted P < .05), in NPM1mut OCI-AML3, MLL-r MOLM13, and MV411 cells after MI-503 treatment (2.5 µM) compared with the DMSO control. (D) Volcano plots of RNA-seq data obtained from OCI-AML3, MOLM13, and MV411 cells treated with MI-503 (2.5 µM). FLT3 and selected MLL-fusion targets are labeled. (E) FLT3 and MEIS1 mRNA expression in human and murine leukemia cells after 4 days of MI-503 treatment (2.5 µM), as assessed by qRT-PCR. ChIP was performed with antibodies against menin (F) or MLL1 (G) and IgG as the negative control, followed by qPCR to detect a sequence within the MEIS1 gene body or SOX2 as negative control. Cells were treated with MI-503 (2.5 µM) or vehicle control for 4 days. (H) FLT3 protein (cell surface) expression assessed by flow cytometry in human and murine NPM1mut and MLL-rearranged AML cells after MI-503 treatment (2.5 µM for 4 or 7 days, as indicated). Representative histograms of 3 independent experiments are shown. Bar graphs in panels A and E-G represent the mean of 3 independent experiments, each performed in technical triplicate. Bar graph in panel H showing Npm1CA/+Flt3ITD/+ cells represents 2 independent experiments performed in technical triplicate. Error bars represent standard deviation.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f1.png?Expires=1771105445&Signature=UuoirLRERqQ9Bfl0t-ftXHnODQ06QnIgSZaA2U9ia3MiyID7dJ-u3LBEy6W3IrRYTawKefbZ98NIao2SWJLbtJcWVtlLgZGrg2XO3y6nRrefKpg3RPB2yez0dEAUtCTk0wd9t-j3Lrlroafvueg7p8qt1BLVEbrITTfUW7JOip1IZzEpVshoiY8T49Ctioe7XvL9gJW-IGsH-n0azmEfvk24brdaxWux07MkApeJvJJ25cpeS5dXGjS~cGot0-dm2H~1Btj6TiGgIL1gXeTFKb62qo8VCu8e3Mf6IFRSMuV3hG8sLe-0qizY1YwreZ6ucc~0BCllIV17wz2MpWDZqw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Synergistic effects of combined menin-MLL and FLT3 inhibition. (A) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in FLT3-ITD+ MOLM13 and MV411 cells. (B) FLT3-ITD+ and FLT3 WT human leukemia cell lines were treated with quizartinib for 48 hours. IC50 values were graphically determined by GraphPad Prism. (C) Dose-response curves from cell-viability assays after 48 hours of treatment with various FLT3 inhibitors in murine Npm1CA/+Flt3ITD/+ cells. (D) Ponatinib IC50 concentrations in murine Npm1CA/+Flt3ITD/+ and Hoxa9-Meis1–transformed cells after 48 hours of treatment. (E) Summary of FLT3 inhibitor IC50 concentrations in the human and murine leukemia cell lines assessed in this study. (F) Dose-response curves from cell-viability assays of MV411 and MOLM13 cells, comparing MI-503 (MI; 3 days for MV411 cells and 4 days for MOLM13 cells), quizartinib (Qz; 24 hours), and combinatorial MI-503 (3 or 4 days) and quizartinib (24 hours) treatment. Dashed lines indicate IC50 values. (G) Dose-response curves from cell-viability assays of Npm1CA/+Flt3ITD/+ cells comparing MI-503 (MI, 4 days), ponatinib (Po, 24 hours, left), and gilteritinib (Gil, 24 hours; right), or their combination (4 days, MI-503; 24 hours, ponatinib and gilteritinib). Dashed lines indicate IC50 values. (H) Effect of MI-503 (2.5 µM), ponatinib (100 nM), and combinatorial treatment (2.5 µM and 100 nM) on the number of total and blast-like colonies in murine Npm1CA/+Flt3ITD/+ cells, normalized to DMSO. Micrographs were taken at ×20 amplification. (I-J) Percentage of apoptotic (annexin V) and dead (4′,6-diamidino-2-phenylindole [DAPI]-stained) cells after single and combinatorial treatment with MI-503 (2.5 µM) and quizartinib (3 nM) in human cell lines (I) or MI-503 (2.5 µM) and ponatinib (100 nM) or gilteritinib (400 nM) in murine cells (J). (K) Giemsa-stained cytospins showing human MV411 and MOLM13 cells and murine Npm1CA/+Flt3ITD/+ cells after single and combinatorial treatment with MI-503 (2.5 µM; 4 days and 3 days for MV411) and FLT3 inhibitor (quizartinib, 3 nM, 24 hours; ponatinib, 100 nM, 24 hours) or their combination (day 4/24 hours and day 3/24 hours for MV411, respectively). Micrographs were taken at ×100 amplification. Error bars represent SD of 3 independent experiments, each performed in 3 technical replicates.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f2.png?Expires=1771105445&Signature=t9DszE~21cxrA-Bs2hcpDeWDLV9x0B2AaAvmwpuEJba9LKhDh~CZF3~zuDOpNTcG7uj~Rqx3GHpoyfuKd6y5YKeUIk1f5W5Vf2ovG8c1ibWWnTkOyraN8dACn0qfxB4iX0XUoME4nm-DKA1zMmDXAGDZXF8INz0V4IONBSnpBG9olvz1zKIbxVBAI8WOuYBssU~rXOozM3KW0mSBhUTaSHruQoAQdtTRDUksYKGgCILU5tgFIHhIhC4paAUzOz7nD1wmydpIyklIPZMOWLECgnzx8gMJ-afFCE7jodqDb1f59La-ur01CgzmpQArELgkpVdWnw~ovOXofUB0Tg9pBQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)

![Synergistic inhibition of proliferation and FLT3 activation after combined treatment with next-generation menin-MLL and FLT3 inhibitors. (A) Dose-response curves of MOLM13 and MV411 cells treated with quizartinib for 24 hours, comparing cells transduced with short hairpin RNAs against MEN1 with control-transduced cells (shLUC). (B) mRNA expression levels of MEN1, MEIS1, and FLT3 in MOLM13 and MV411 cells with MEN1 knock down or control-transduced cells, assessed 48 hours after transduction. (C) Dose-response curves from cell-viability assays after 7 days of treatment with VTP-50469 in human (left) and murine (right) leukemia cells. Viable (4′,6-diamidino-2-phenylindole [DAPI]-negative) cells were assessed by flow cytometry. (D-E) Dose-response curves from cell viability assays of MV411 and MOLM13 cells comparing VTP-50469 (VTP; 3 days for MV411 and 4 days for MOLM13 cells), quizartinib (Qz, 24 hours; D), gilteritinib (Gil, 24 hours; E), and combinatorial VTP-50469 (3 or 4 days) and FLT3 inhibition (24 hours) treatment. Dashed lines indicate IC50 values. (F) Dose-response curves from cell viability assays of Npm1CA/+Flt3ITD/+ cells comparing VTP-50469 (VTP; 6 days), ponatinib (Po, 24 hours, [left]), or gilteritinib (Gil, 24 hours; [right]) with their combination (6 days VTP50469, 24 hours FLT3 inhibition). Dashed lines indicate IC50 values. (G-H) FLT3 and MEIS1 mRNA expression in murine Npm1CA/+Flt3ITD/+ (G), human MV411 (H; left), and MOLM13 (H; right) leukemia cells after single or combined treatment with VTP-50469 (100 nM; 4 days for MOLM13 and Npm1CA/+Flt3ITD/+ cells and 3 days for MV411 cells) and FLT3 inhibitors (quizartinib, 3 nM and ponatinib, 100 nM; 24 hours) as assessed by qRT-PCR. Bar graphs represent the mean with standard deviation of 3 independent experiments, each performed in technical triplicate. (I) Immunoblot analysis of FLT3 and phosphorylated (p)FLT3 in MV411 cells (left) and MOLM13 cells (right) after treatment with VTP-50469 (100 nM; 3 and 4 days in MV411 and MOLM-13, respectively) and quizartinib (3 nM, 24 hours), or their combination. Numbers indicate the DMSO-normalized quantification of western blot signals, relative to the loading control, performed by densitometry using the ImageJ software tool.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f6.png?Expires=1771105445&Signature=v0K9IUm7n1okIuVwWKfP4vjduFrLNgr1zMPdb4BqlfIbxcFP83aSXT~jL7dE7LW9IBqJyVPELPJj9DhuWoeaUrc0KpmU6EOwHevunLWuX0YP6LNE-wsjW9cdJCqbHRVSKHw7vlfxCtEmo~Io59-giuHHGfb9Hbd5rQMJcYcyCwK9HbELA2NqhSuKUjApMJiYwDpN4wbyYyy2A~INHxu3tJkftxmjRxZBW0xm~M467k3Ppj2MsZg1~0TDXfzhyCM57yKU1lqW4lz2qjmSgeYQ2UqjUVD99O8jB7xydOXmhtwf2QpWfD9-ZCryjsv5w4iPGz6SYjyJRiq6oCWudFCZ8g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Effects of single and combined menin-MLL and FLT3 inhibition on primary NPM1mut FLT3ITD AML patient samples and on survival of in vivo–treated MLL-r FLT3-ITD leukemic xenograft mice. (A) The human stromal cell coculture assay, performed to maintain and treat patients’ primary AML blasts. (B) Summary of characteristics of patients providing the samples used in panels C-E. (C-D) Number of viable cells inde novoAML samples treated in coculture for 7 days with DMSO, MI-503 (2 µM), quizartinib (6 nM), or combinatorial MI-503 and quizartinib treatment. (C) Five independent samples of de novo NPM1mutFLT3ITD AML. (D) Two independent samples of de novo AML, WT for NPM1, FLT3, and MLL. Depicted are 4′,6-diamidino-2-phenylindole [DAPI]−, human CD45+ cell numbers as assessed by flow cytometry. (E) Effect of MI-503 (2.5 µM), quizartinib (3 nM), and combinatorial treatment (2.5 µM and 3 nM) on total and blast-like CFUs in primary patient sample cells. (F) Experimental setup for the treatment of MV411-derived leukemic xenograft mice (left); percentage of human CD45+ cells in the bone marrow of leukemic mice (right) after treatment with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combined MI-503 and quizartinib. (G) Kaplan-Meier survival analysis of MV411-derived leukemic xenograft mice treated with drug vehicles, MI-503 (50 mg/kg; twice daily IP), quizartinib (10 mg/kg; PO; once daily), or combinatorial MI-503 and quizartinib (n = 5 mice/group). The treatment period is displayed in blue. The log-rank (Mantel-Cox) test was used to calculate the P-values.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005037/1/m_bloodbld2020005037f7.png?Expires=1771105445&Signature=ygwPTJD4v2-I5kC7LC~ohPef~wR3AoPDLxulPUTvNUpBsA~PBHNJSGuEeCNRlc95Unu~~-vBohtkTWXIkA-xKraKnOl4WrI~QFTlMX8hbjaTWGH8vpX706xpz25m~D7mlfKhXTOhxVsLgKysyror49BLtrs6Sc-9FVOGTkG9YMcSG2F3VHQJXW1FyUfstLolXJ7N4iDzl3B4D42GbCMNWRR1UEEjpjQxioHStfMvxy6Zg7gq-G2gOcunSnTjX2ZzYxBhArjdVPoSoQ~FLelMo3btxMJPPokVvXaCFSjDtjvk~-KVqcOHncBexU1bYoMavKMyLCN6enRmDd~~4D7J9g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
