Key Points
EGF promotes HSC DNA repair and hematopoietic regeneration via activation of DNA-PKcs and NHEJ.
EGF drives hematopoietic regeneration after chemotherapy and human HSC recovery after irradiation while increasing HSC intergenic mutations.

Abstract
Chemotherapy and irradiation cause DNA damage to hematopoietic stem cells (HSCs), leading to HSC depletion and dysfunction and the risk of malignant transformation over time. Extrinsic regulation of HSC DNA repair is not well understood, and therapies to augment HSC DNA repair following myelosuppression remain undeveloped. We report that epidermal growth factor receptor (EGFR) regulates DNA repair in HSCs following irradiation via activation of the DNA-dependent protein kinase–catalytic subunit (DNA-PKcs) and nonhomologous end joining (NHEJ). We show that hematopoietic regeneration in vivo following total body irradiation is dependent upon EGFR-mediated repair of DNA damage via activation of DNA-PKcs. Conditional deletion of EGFR in hematopoietic stem and progenitor cells (HSPCs) significantly decreased DNA-PKcs activity following irradiation, causing increased HSC DNA damage and depressed HSC recovery over time. Systemic administration of epidermal growth factor (EGF) promoted HSC DNA repair and rapid hematologic recovery in chemotherapy-treated mice and had no effect on acute myeloid leukemia growth in vivo. Further, EGF treatment drove the recovery of human HSCs capable of multilineage in vivo repopulation following radiation injury. Whole-genome sequencing analysis revealed no increase in coding region mutations in HSPCs from EGF-treated mice, but increased intergenic copy number variant mutations were detected. These studies demonstrate that EGF promotes HSC DNA repair and hematopoietic regeneration in vivo via augmentation of NHEJ. EGF has therapeutic potential to promote human hematopoietic regeneration, and further studies are warranted to assess long-term hematopoietic effects.
Introduction
Ionizing radiation (IR) and chemotherapy cause DNA damage in hematopoietic stem and progenitor cells (HSPCs), thereby contributing to a risk for hematopoietic stem cell (HSC) dysfunction, accelerated aging, and malignancy over time.1-5 Eukaryotic cells repair DNA damage primarily through homologous recombination (HR) and nonhomologous end-joining (NHEJ) repair mechanisms.1,2 HSCs, which are largely quiescent in the steady-state, primarily undergo NHEJ in response to IR, whereas proliferating HSCs and progenitor cells are able to undergo HR.1 NHEJ is considered a more error-prone mechanism than HR, potentially resulting in increased deletions, insertions, translocations, and genomic instability.2,3 Mohrin et al reported that NHEJ in quiescent HSCs was associated with increased genomic rearrangements that persisted in vivo.1
Because IR and chemotherapy induce genomic instability in HSCs and increase the risk of malignant transformation, the development of therapies capable of reducing DNA damage or increasing DNA repair in HSCs could be highly beneficial. Recently, de Laval et al demonstrated that thrombopoietin stimulated DNA repair in HSCs via augmentation of DNA-dependent protein kinase (DNA-PK)–dependent NHEJ, and this DNA-PK activation was dependent on Erk and NF-κB pathway activation.6,7 The broader role of extrinsic signals in regulating DNA repair in HSCs remains poorly understood.6,7 We previously showed that high-dose total body irradiation (TBI) depletes bone marrow (BM) HSCs and promotes myeloid skewing and immune cell depletion in mice.8 Systemic administration of epidermal growth factor (EGF), which is expressed by BM endothelial cells (ECs), mitigated these effects of TBI and promoted hematopoietic regeneration in vivo.8 However, the precise molecular mechanisms through which EGF promoted hematopoietic regeneration remained unclear. In tumor cells, epidermal growth factor receptor (EGFR) can promote DNA repair via activation of DNA-dependent protein kinase–catalytic subunit (DNA-PKcs).9-11 Here, we show that EGF treatment promotes HSC recovery and hematopoietic regeneration via augmentation of DNA-PKcs activity and NHEJ repair in HSCs. EGFR is essential for activation of NHEJ repair in HSPCs and hematopoietic regeneration in vivo following TBI. EGF treatment also increases NHEJ repair in human HSCs following irradiation and promotes the recovery of human HSCs with in vivo repopulating capacity.
Methods
Flow cytometry
BM cells from femurs and tibia were collected in Iscove modified Dulbecco medium, 10% fetal bovine serum, and 1% penicillin-streptomycin, following red blood cell lysis with ACK Buffer (MilliporeSigma, Burlington, MA). Cells were stained with V450 Mouse Lineage Antibody (BD Biosciences, San Jose, CA), c-kit (CD117) PE Rat Anti-Mouse (BD Biosciences), and Sca-1 APC-Cy7 Rat Anti-Mouse (BD Biosciences) to measure the percentage of ckit+sca-1+lin− (KSL) cells. Cells were also stained with Alexa Fluor 488 Anti-Mouse CD41 Antibody (BioLegend, San Diego, CA), FITC Hamster Anti-Mouse CD48 (BD Biosciences), and Alexa Fluor 647 Rat Anti-Mouse CD150 (BD Biosciences) to measure the percentage of CD150+CD48−CD41− KSL HSCs.12-13 For hematopoietic engraftment analysis, Brilliant Violet 605 Anti-Mouse CD45.1 Antibody (BioLegend), FITC-CD45.2, PE-Mac-1 (CD11b), PE-Gr-1 (Ly-6G and Ly-6C), V450-CD3, and APC-Cy7-B220 (CD45R) (BD Biosciences) were used. For analysis of phosphorylated (p)-EGFR, p-Akt, p-DNA-PKcs, and p-Artemis, cells were permeabilized with 0.5% Triton X-100 and 1% bovine serum albumin in phosphate-buffered saline (PBS) and fixed with methanol (all from Thermo Fisher Scientific, Waltham, MA) for 10 minutes. Cells were stained with 1:100 primary antibody for 60 minutes at 4°C, washed with PBS, and stained with 1:100 Alexa Fluor-488 Goat Anti-Rabbit IgG Secondary Antibody (Thermo Fisher Scientific) for 30 minutes at room temperature. 7-AAD (BD Biosciences) was used to exclude dead cells.
To measure the percentage of CD34+CD38− cells in cultures of human hematopoietic cells, cells were stained with FITC anti-human CD34 Antibody (BioLegend) and APC Mouse Anti-Human CD38 (BD Biosciences). Data acquisition was performed using a BD FACSCanto II system; BD FACSDiva software and FlowJo (Ashland, OR) were used for analysis.
Immunofluorescence microscopy
BM KSL cells (1 × 104) were plated on fibronectin-coated slides (Thermo Fisher Scientific), serum starved for 1 hour, and stimulated with 100 ng/mL EGF (R&D Systems, Minneapolis, MN), with and without 0.5 μM NU7441, a DNA-PKcs inhibitor (Selleckchem, Houston, TX),14 or 0.5 μM MK2206, a selective Akt inhibitor (Selleckchem).15 Cells were fixed with PBS + 4% paraformaldehyde for 10 minutes at room temperature, permeabilized with 0.5% Triton X-100 in PBS for 30 minutes, blocked for 30 minutes with 5% fetal bovine serum, and stained with antibodies. Cells were incubated at room temperature for 1 hour in buffer containing primary antibodies: Alexa Fluor 488 Anti-γ H2AX (phospho S139, 1:50; BioLegend), DNA-PKcs (p-Thr2609) Antibody (1:50; Novus Biological), and Phospho-Akt (Ser473) (1:200; Cell Signaling Technologies, Danvers, MA). Cells were washed twice with PBS, incubated with secondary antibodies (Alexa Fluor 488 Donkey Anti-Mouse [1:200; Thermo Fisher Scientific] and Alexa Fluor 647 Donkey Anti-Rabbit [1:200; Thermo Fisher Scientific]) for 1 hour at room temperature, washed twice with PBS, and mounted with Gold Antifade Reagent with DAPI (Cell Signaling Technologies). Images were captured with a Zeiss Axio Imager M2 microscope (63× objective) with ZEN software. Foci numbers and mean fluorescence intensity (MFI) were quantified by Fiji software (ImageJ; National Insitutes of Health, Bethesda, MD).
Comet assay
For in vitro analysis, BM KSL cells were cultured with EGF, with or without 0.5 μM NU7441 (Selleckchem),14 in Iscove modified Dulbecco medium supplemented 10% fetal bovine serum (VWR, Radnor, PA) plus 20 ng/ml thrombopoietin, 125 ng/ml stem cell factor and 50 ng/ml Flt-3 ligand (TSF media; R&D Systems) for 1 hour at 37°C. For in vivo studies, C57BL/6 mice were irradiated with 500 cGy and treated with 0.5 μg/g EGF subcutaneously, with or without 10 mg/kg NU7441. KSL cells were analyzed at +4 hours post-TBI. Comet Assay was performed using a Comet Assay Kit (Trevigen, Gaithersburg, MD). Cells were resuspended in CometAssay LMAgarose (Trevigen) and spread on CometSlides (Trevigen). Slides were immersed in Lysis Solution (Trevigen) overnight. Electrophoresis was performed with 1× Electrophoresis Buffer (100 mM Tris base, 300 mM sodium acetate). Slides were immersed in DNA Precipitation Solution (1 M ammonium acetate, 82% ethanol) for 30 minutes, and transferred to 70% ethanol for 30 minutes at RT. After drying, slides were stained with SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific). Comet tails were visualized using a Zeiss Axio Imager M2 microscope with ZEN software at 10× objective and analyzed with Comet Analysis Software (Trevigen).
Whole-genome sequencing analysis
C57BL/6 mice were irradiated with 500 cGy and treated subcutaneously with EGF or saline at 1 and 24 hours post-TBI. BM KSL cells were sorted from irradiated mice at 6 weeks post-TBI for whole-genome sequencing (WGS) analysis. Genomic DNA was extracted using a PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific). Quality control of genomic DNA was performed by electrophoresis using an Agilent 2200 TapeStation System (Agilent, Santa Clara, CA) and by measurement of DNA concentration with a Qubit 2.0 fluorometer (Thermo Fisher Scientific). A NEBNext DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA) was used for library construction. For library quality control, a Qubit 2.0 fluorometer was used to determine concentration, a 2100 Bioanalyzer Instrument (Agilent) was used to assess insert size, and quantitative polymerase chain reaction was performed to detect effective concentration of the library.
Paired sequencing reads were mapped to the GRCm38.p4 mouse genome assembly with the Burrows-Wheeler Aligner (BWA) Maximal Exact Matches algorithm in BWA version 0.7.17-r1188.16 Sequence alignment map files created by BWA Maximal Exact Matches were converted to binary alignment map (BAM) files with samtools version 1.9.17 The resulting BAM files were sorted by chromosome coordinate, polymerase chain reaction and optical duplicates were marked, and individual sample and sequencing lane read group information was added to each BAM file with Picard version 2.9.0-1-gf5b9f50-SNAPSHOT (http://broadinstitute.github.io/picard). Genomic variants were called with GATK v3.518 and subsequently split into files that contained only single nucleotide polymorphisms (SNPs) or insertions or deletions (InDels). Variants were hard filtered for quality using these criteria for SNPs (quality of depth < 2.0 || Fisher strand > 60.0 || mapping quality < 30.0 || HaplotypeScore > 13.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0) and InDels (quality of depth < 2.0 || Fisher strand > 200.0 || ReadPosRankSum < −20.0). To determine treatment-specific variants, variants found in individual-matched genomic samples (ear) were removed from variants discovered in treatment or control samples. The Program CNVnator19 was used to call copy number variants for each sample using the same BAM files as described above. We attempted to call structural variants using the program BreakDancer20 but found variation in library insert size that prohibited further analysis. All discovered variants were annotated using SnpEff.21
Statistics
Data are shown as mean ± standard error of the mean (SEM), unless otherwise indicated. We used an unpaired 2-tailed Student t test for simple comparisons. When making multiple comparisons using single data sets, 1-way analysis of variance (ANOVA) was used. Two-way ANOVA was used when comparing the mean differences between groups that were split regarding 2 independent variables. The log-rank (Mantel-Cox) test was used for survival analyses. Statistics were determined using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). All experiments were repeated to confirm findings. Supplemental Methods are available on the Blood Web site.
Results
EGF decreases DNA damage in irradiated HSCs via activation of DNA-PKcs
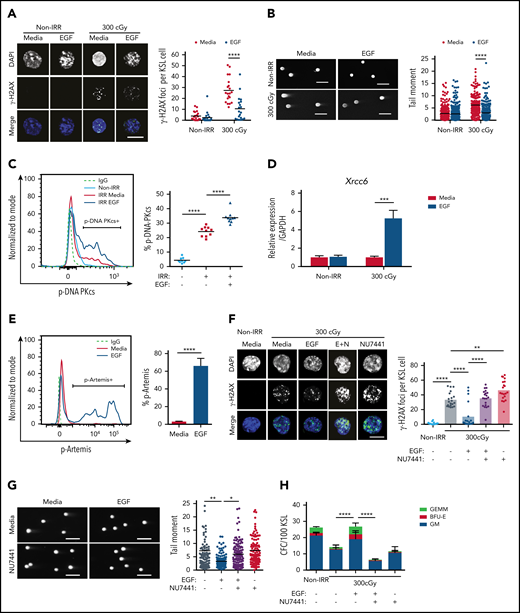
IR causes DNA strand breaks that lead to cellular apoptosis and necrosis.22 DNA damage responses are distinct between HSCs and committed progenitor cells.1,23-25 We sought to determine whether EGFR signaling regulates the DNA damage response in HSCs following exposure to IR. Irradiation with 300 cGy increased γ-H2AX foci, a marker of DNA double-strand breaks,1,26 in BM KSL HSPCs, whereas treatment with 100 ng/mL EGF decreased γ-H2AX foci in irradiated KSL cells (Figure 1A). Using the Comet assay, which detects DNA strand breaks as tail moments,27 300-cGy irradiation increased tail moment length in KSL cells, whereas EGF treatment decreased tail moment length (Figure 1B). EGF treatment had no effect on the numbers of γ-H2AX foci or tail moment lengths in nonirradiated KSL cells (Figure 1A-B).
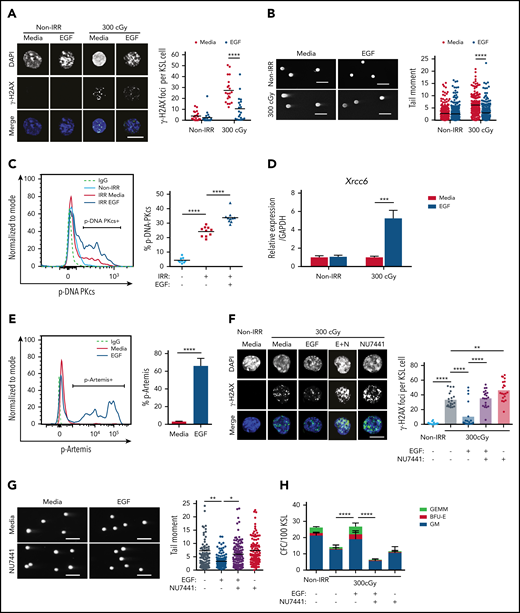
EGF decreases DNA damage in irradiated HSCs via DNA-PKcs. (A) Immunofluorescence microscopy of IR-induced foci of γ-H2AX in nonirradiated (Non-IRR) and 300-cGy–irradiated BM KSL cells cultured in complete media (Media), with or without 10 ng/mL EGF for 1 hour (scale bar, 10 µm) (left panel). Numbers of foci per KSL cell in Non-IRR KSL cells (n = 20 replicates per group, 2-way ANOVA, horizontal lines represent means). (B) Representative images of a Comet assay of BM KSL cells that were irradiated (300 cGy) or not (Non-IRR) and cultured in media, with or without EGF, for 1 hour (scale bars, 100 µm) (left panel). Quantification of tail moments in each condition (n = 179-256 cells per group, 2-way ANOVA) (right panel). (C) Flow cytometric analysis of p-DNA-PKcs levels in Non-IRR KSL cells and irradiated (IRR; 100 cGy) KSL cells treated with Media alone or Media plus EGF for 1 hour (left panel). Percentage of p-DNA-PKcs+ KSL cells in each condition (n = 9-10 per group, 1-way ANOVA) (right panel). (D) Gene expression of Xrcc6 in Non-IRR KSL cells and at 1 hour following 300 cGy, treated or not with EGF (n = 4 per group, mean ± SEM, 2-way ANOVA). (E) Representative line graph of p-Artemis (Ser516) in KSL cells irradiated with 300 cGy and then treated or not with EGF for 1 hour (left panel). Percentage of p-Artemis+ KSL cells (n = 4 per group, mean ± SEM, Student t test) (right panel). (F) Fluorescence microscopy images of γ-H2AX foci in Non-IRR KSL cells and irradiated KSL cells in Media with or without EGF and with or without NU7441 (E+N; scale bar, 10 µm) (left panel). Numbers of γ-H2AX foci per KSL cell in each condition (n = 20 per group, 2-way ANOVA) (right panel). (G) Representative images of Comet assay of 300-cGy–irradiated KSL cells cultured with Media with or without EGF and with or without NU7441 (scale bars, 100 µm) (left panel). Tail moments from KSL cells from each condition (n = 102-114 cells per group, 1-way ANOVA) (right panel). (H) Numbers of CFCs from Non-IRR BM KSL cells and 300-cGy–irradiated KSL cells cultured in Media with or without EGF and with or without NU7441 for 72 hours (n = 6 per group, mean ± SEM, 2-way ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001. IgG, immunoglobulin G.BFU-E, burst forming unit–erythroid; E+N, EGF + NU7441; GEMM, colony forming unit-granulocyte/erythroid/macrophage/megakaryocyte, GM, colony forming unit-granulocyte/macrophage.
EGF decreases DNA damage in irradiated HSCs via DNA-PKcs. (A) Immunofluorescence microscopy of IR-induced foci of γ-H2AX in nonirradiated (Non-IRR) and 300-cGy–irradiated BM KSL cells cultured in complete media (Media), with or without 10 ng/mL EGF for 1 hour (scale bar, 10 µm) (left panel). Numbers of foci per KSL cell in Non-IRR KSL cells (n = 20 replicates per group, 2-way ANOVA, horizontal lines represent means). (B) Representative images of a Comet assay of BM KSL cells that were irradiated (300 cGy) or not (Non-IRR) and cultured in media, with or without EGF, for 1 hour (scale bars, 100 µm) (left panel). Quantification of tail moments in each condition (n = 179-256 cells per group, 2-way ANOVA) (right panel). (C) Flow cytometric analysis of p-DNA-PKcs levels in Non-IRR KSL cells and irradiated (IRR; 100 cGy) KSL cells treated with Media alone or Media plus EGF for 1 hour (left panel). Percentage of p-DNA-PKcs+ KSL cells in each condition (n = 9-10 per group, 1-way ANOVA) (right panel). (D) Gene expression of Xrcc6 in Non-IRR KSL cells and at 1 hour following 300 cGy, treated or not with EGF (n = 4 per group, mean ± SEM, 2-way ANOVA). (E) Representative line graph of p-Artemis (Ser516) in KSL cells irradiated with 300 cGy and then treated or not with EGF for 1 hour (left panel). Percentage of p-Artemis+ KSL cells (n = 4 per group, mean ± SEM, Student t test) (right panel). (F) Fluorescence microscopy images of γ-H2AX foci in Non-IRR KSL cells and irradiated KSL cells in Media with or without EGF and with or without NU7441 (E+N; scale bar, 10 µm) (left panel). Numbers of γ-H2AX foci per KSL cell in each condition (n = 20 per group, 2-way ANOVA) (right panel). (G) Representative images of Comet assay of 300-cGy–irradiated KSL cells cultured with Media with or without EGF and with or without NU7441 (scale bars, 100 µm) (left panel). Tail moments from KSL cells from each condition (n = 102-114 cells per group, 1-way ANOVA) (right panel). (H) Numbers of CFCs from Non-IRR BM KSL cells and 300-cGy–irradiated KSL cells cultured in Media with or without EGF and with or without NU7441 for 72 hours (n = 6 per group, mean ± SEM, 2-way ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001. IgG, immunoglobulin G.BFU-E, burst forming unit–erythroid; E+N, EGF + NU7441; GEMM, colony forming unit-granulocyte/erythroid/macrophage/megakaryocyte, GM, colony forming unit-granulocyte/macrophage.
NHEJ is the predominant mechanism that regulates DNA repair in quiescent HSCs.1,28,29 DNA-PKcs is a principal enzyme effector of NHEJ in mammalian cells; together with its regulator subunits, Ku70 and Ku80, it stabilizes DNA breaks for repair.30-33 Following 300-cGy irradiation, BM KSL cells displayed increased levels of p-DNA-PKcs at +1 hour compared with nonirradiated KSL cells (Figure 1C). EGF treatment further increased p-DNA-PKcs levels in irradiated BM KSL cells. EGF treatment also increased expression of X-ray repair cross complementing 6 (Xrcc6), which encodes the Ku70 protein,34 and increased phosphorylation of Artemis, a nuclease involved in end-trimming during NHEJ (Figure 1D-E).35 Conversely, EGF treatment had no effect on the expression of genes encoding proteins involved in HR, including RPA1 and ATRIP, or foci of Rad51, a recombinase integral to HR, in irradiated KSL cells (supplemental Figure 1A-C).36,37 These results suggest that EGF treatment activates the NHEJ machinery in HSCs with less effect on HR. Because cell cycle status impacts DNA repair processes,1,27 we evaluated the effect of EGF treatment on BM KSL cell cycle status. We did not observe any difference in the percentages of KSL cells in the G0, G1, or G2/S/M phase between EGF-treated and saline-treated irradiated cells (supplemental Figure 1D).
To determine whether DNA-PKcs was necessary for EGF-mediated reduction in DNA damage in HSCs, we treated irradiated BM KSL cells with EGF, with or without the DNA-PKcs inhibitor NU7441.14 NU7441 treatment abrogated EGF-mediated DNA damage repair in irradiated KSL cells (Figure 1F-G). NU7441 treatment also suppressed EGF-mediated recovery of colony-forming cells (CFCs) from irradiated KSL cells in culture, suggesting that DNA-PKcs activation was necessary for EGF-mediated hematopoietic regenerative effects (Figure 1H).
EGF-mediated DNA repair in BM HSCs is dependent on Akt
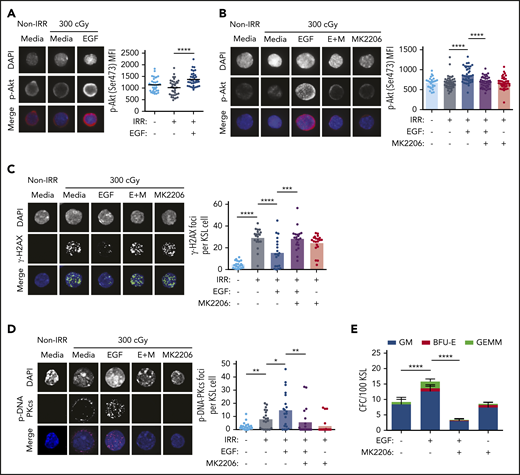
In tumor cells, EGFR has been shown to regulate DNA-PKcs activity via 2 distinct mechanisms following irradiation9-11 : EGFR can bind with DNA-PKcs and physically translocate DNA-PKcs from the cytoplasm into the nucleus, or it can increase DNA-PKcs activity via induction of phosphatidylinositol 3-kinase/Akt signaling.9-11,38 Irradiation with 300 cGy did not increase Akt phosphorylation in BM KSL cells (Figure 2A), whereas treatment of irradiated KSL cells with 100 ng/mL EGF increased phospho-Akt (Figure 2A; supplemental Figure 2A). Treatment with a selective Akt inhibitor, MK2206, blocked EGF-mediated phosphorylation of Akt (Figure 2B)15 and abrogated EGF-mediated reductions in γ-H2AX foci in irradiated KSL cells (Figure 2C). EGF-mediated phosphorylation of DNA-PKcs in irradiated KSL cells was also blocked by MK2206, as was EGF-mediated recovery of CFCs from irradiated BM KSL cells (Figure 2D-E).
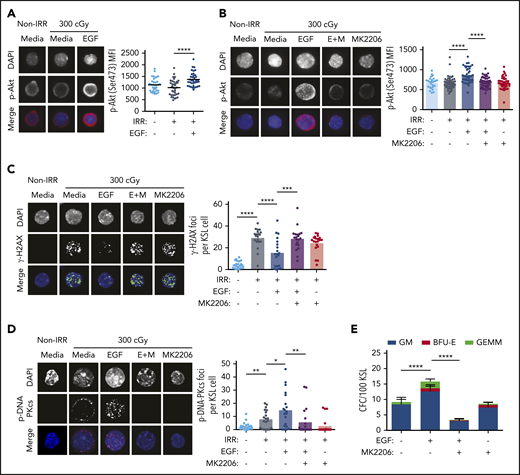
EGF-mediated activation of DNA-PKcs is dependent on Akt. (A) Representative microscopic images of p-Akt in nonirradiated (Non-IRR) and irradiated (IRR) KSL cells cultured in media, with or without EGF, for 5 minutes (scale bar, 10 µm) (left panel). p-Akt MFI in KSL cells for each condition (n = 34-37 cells per group, Student t test) (right panel). (B) Representative microscopic images of p-Akt in Non-IRR and irradiated KSL cells cultured with media with or without EGF and with or without MK2206 for 5 minutes (scale bar, 10 µm) (left panel). p-Akt MFI in KSL cells for each condition (n = 26-44 cells per group, 1-way ANOVA) (right panel). (C) Representative microscopic images of γ-H2AX foci in the conditions shown at 1 hour of culture (scale bar, 10 µm) (left panel). Numbers of foci per KSL cell for each condition (n = 20 fields of view per group, 1-way ANOVA) (right panel). (D) Representative microscopic images of p-DNA-PKcs foci in the conditions shown (scale bar, 10 µm). Numbers of p-DNA-PKcs foci per KSL cell for each condition (n = 20 fields of view per group, 1-way ANOVA) (right panel). (E) CFCs from KSL cells irradiated with 300 cGy and cultured in media with or without EGF and with or without MK2206 (n = 6 per group). Data are mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-way ANOVA. DAPI, 4′,6-diamidino-2-phenylindole. E+M, EGF + MK2206.
EGF-mediated activation of DNA-PKcs is dependent on Akt. (A) Representative microscopic images of p-Akt in nonirradiated (Non-IRR) and irradiated (IRR) KSL cells cultured in media, with or without EGF, for 5 minutes (scale bar, 10 µm) (left panel). p-Akt MFI in KSL cells for each condition (n = 34-37 cells per group, Student t test) (right panel). (B) Representative microscopic images of p-Akt in Non-IRR and irradiated KSL cells cultured with media with or without EGF and with or without MK2206 for 5 minutes (scale bar, 10 µm) (left panel). p-Akt MFI in KSL cells for each condition (n = 26-44 cells per group, 1-way ANOVA) (right panel). (C) Representative microscopic images of γ-H2AX foci in the conditions shown at 1 hour of culture (scale bar, 10 µm) (left panel). Numbers of foci per KSL cell for each condition (n = 20 fields of view per group, 1-way ANOVA) (right panel). (D) Representative microscopic images of p-DNA-PKcs foci in the conditions shown (scale bar, 10 µm). Numbers of p-DNA-PKcs foci per KSL cell for each condition (n = 20 fields of view per group, 1-way ANOVA) (right panel). (E) CFCs from KSL cells irradiated with 300 cGy and cultured in media with or without EGF and with or without MK2206 (n = 6 per group). Data are mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-way ANOVA. DAPI, 4′,6-diamidino-2-phenylindole. E+M, EGF + MK2206.
EGF-mediated hematopoietic regeneration in vivo is dependent on DNA-PKcs
To evaluate EGF effects in vivo, we irradiated mice with 500-cGy TBI, treated them with 0.5 μg/g EGF or saline subcutaneously one time, and measured DNA damage in BM KSL cells (Figure 3A). EGF-treated mice exhibited decreased DNA damage in BM KSL cells at +4 hours compared with control mice (Figure 3B). Systemic administration of NU7441 abrogated the EGF-mediated reduction in DNA damage in BM KSL cells (Figure 3B). Daily EGF treatment for 10 days decreased the percentages of annexin V+ BM KSL cells in irradiated C57BL/6 mice compared with controls, and NU7441 treatment blocked this effect (Figure 3C). Treatment of irradiated mice with NU7441 also blocked EGF-mediated recovery of BM CFCs (Figure 3D). EGF treatment increased the percentages of BM KSL cells and CD150+CD48−CD41− KSL HSCs39 at day +10 following 500-cGy irradiation, and NU7441 treatment suppressed these effects (Figure 3E). EGF-mediated recovery of peripheral blood (PB) white blood cells (WBCs) and lymphocytes was also blocked by NU7441 treatment (supplemental Figure 2B).
![EGF promotes HSC recovery in vivo in a DNA-PKcs–dependent manner. (A) Schematic design. C57BL/6 mice were irradiated with 500-cGy TBI, followed by daily injections of saline, EGF, NU7441, or EGF + NU7441 for 10 days. BM cells were collected at +4 hours for Comet assay and at day +10 for annexin V apoptosis assay and competitive repopulation assays. (B) Representative images of a Comet assay of BM KSL cells collected at +4 hours following 500-cGy TBI and treatment with EGF with or without NU7441 (scale bars, 100 µm). Measurements of tail moments in KSL cells in each condition (n = 6-7 fields per group [range 110-137 cells per field], mean ± SEM, Student t test) (right panel). (C) Representative flow cytometric analysis of annexin V and 7-AAD staining in BM KSL cells at day +10 from mice described in A (left panel). Percentage of annexin V+ KSL cells in each condition (n = 3 or 4 per group, 1-way ANOVA) (right panel). (D) Numbers of CFCs in BM at day +10 from the mice described in A (n = 6 per group, mean ± SEM, 2-way ANOVA). (E) Flow cytometric analysis of BM CD150+CD48−CD41− KSL cells at day +10 from mice described in A (left panel). Percentage of CD150+CD48−CD41− cells within the KSL population in each group (n = 7 or 8 per group, 1-way ANOVA). (F) Percentages of donor CD45.2+ cells in the BM of primary recipient mice at 16 weeks following transplant of BM cells collected at day +10 from the mice in A (n = 6 per group, 2-way ANOVA). (G) Percentages of donor CD45.2+ cells in the BM of secondary mice at 16 weeks posttransplant. Secondary mice were transplanted with 5 × 106 BM cells collected from primary mice at 16 weeks posttransplant, along with 2 × 105 competitor BM (CD45.1+) cells (n = 6 per group, 2-way ANOVA). (H) Percentages of CD45.2+Mac1/Gr1+ (myeloid) cells (left panel), CD45.2+B220+ B cells (middle panel), and CD45.2+CD3+ T cells (right panel) in the BM of secondary recipients at 16 weeks posttransplant (n = 6 per group, 2-way ANOVA). (I-J) C57BL/6 mice were injected with 1 dose of doxorubicin as chemotherapy, followed by EGF or saline treatment. (I) PB WBCs, neutrophils (NEU), lymphocytes (LYMPH), and platelets (PLT) in mice at +10 days postdoxorubicin (n = 4 or 5, Student t test). (J) Number of KSL cells per mouse at +10 days postdoxorubicin (n = 5 per group, Student t test) (left panel). Percentages of CD150+CD48−CD41− cells within the KSL cell population at day +10 postdoxorubicin (n = 5 per group, Student t test) (right panel). *P < .05, **P < .01, ***P < .001, ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/4/10.1182_blood.2020005895/1/m_bloodbld2020005895f3.png?Expires=1770650940&Signature=v4K02DlYAmFK11Dp8eIQDXFePDcgAduu95HEnKV3cueV066DZDne6Yqgq5XgjcWtVZ1tryFv6GQJO4WQSV4lCy0NNRNdHxD9-DqMwSb3C5MGMbB8O4BRbDU5Poef4n4fXoL~EBV7A0eEd2mimA1F4jw4l~2GYPWYb4m~Nm1SLBR-7UboBeB-qKH67IlsU26x8MwpILXcOoeoaSU5MfIfnFPPbAKltBTpTHKsCUqKMrrBW4X6TD5oC0rNRbt0KUt6uWGKekWnW4Yrw7bgOZ29Nj8Y40DEGQ3bMXcTlZwtpt5l8vFewDPMf6AiCgWBGaOUy5uV2uy2YeeqgAVzZbpIRg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
EGF promotes HSC recovery in vivo in a DNA-PKcs–dependent manner. (A) Schematic design. C57BL/6 mice were irradiated with 500-cGy TBI, followed by daily injections of saline, EGF, NU7441, or EGF + NU7441 for 10 days. BM cells were collected at +4 hours for Comet assay and at day +10 for annexin V apoptosis assay and competitive repopulation assays. (B) Representative images of a Comet assay of BM KSL cells collected at +4 hours following 500-cGy TBI and treatment with EGF with or without NU7441 (scale bars, 100 µm). Measurements of tail moments in KSL cells in each condition (n = 6-7 fields per group [range 110-137 cells per field], mean ± SEM, Student t test) (right panel). (C) Representative flow cytometric analysis of annexin V and 7-AAD staining in BM KSL cells at day +10 from mice described in A (left panel). Percentage of annexin V+ KSL cells in each condition (n = 3 or 4 per group, 1-way ANOVA) (right panel). (D) Numbers of CFCs in BM at day +10 from the mice described in A (n = 6 per group, mean ± SEM, 2-way ANOVA). (E) Flow cytometric analysis of BM CD150+CD48−CD41− KSL cells at day +10 from mice described in A (left panel). Percentage of CD150+CD48−CD41− cells within the KSL population in each group (n = 7 or 8 per group, 1-way ANOVA). (F) Percentages of donor CD45.2+ cells in the BM of primary recipient mice at 16 weeks following transplant of BM cells collected at day +10 from the mice in A (n = 6 per group, 2-way ANOVA). (G) Percentages of donor CD45.2+ cells in the BM of secondary mice at 16 weeks posttransplant. Secondary mice were transplanted with 5 × 106 BM cells collected from primary mice at 16 weeks posttransplant, along with 2 × 105 competitor BM (CD45.1+) cells (n = 6 per group, 2-way ANOVA). (H) Percentages of CD45.2+Mac1/Gr1+ (myeloid) cells (left panel), CD45.2+B220+ B cells (middle panel), and CD45.2+CD3+ T cells (right panel) in the BM of secondary recipients at 16 weeks posttransplant (n = 6 per group, 2-way ANOVA). (I-J) C57BL/6 mice were injected with 1 dose of doxorubicin as chemotherapy, followed by EGF or saline treatment. (I) PB WBCs, neutrophils (NEU), lymphocytes (LYMPH), and platelets (PLT) in mice at +10 days postdoxorubicin (n = 4 or 5, Student t test). (J) Number of KSL cells per mouse at +10 days postdoxorubicin (n = 5 per group, Student t test) (left panel). Percentages of CD150+CD48−CD41− cells within the KSL cell population at day +10 postdoxorubicin (n = 5 per group, Student t test) (right panel). *P < .05, **P < .01, ***P < .001, ****P < .0001.
EGF promotes HSC recovery in vivo in a DNA-PKcs–dependent manner. (A) Schematic design. C57BL/6 mice were irradiated with 500-cGy TBI, followed by daily injections of saline, EGF, NU7441, or EGF + NU7441 for 10 days. BM cells were collected at +4 hours for Comet assay and at day +10 for annexin V apoptosis assay and competitive repopulation assays. (B) Representative images of a Comet assay of BM KSL cells collected at +4 hours following 500-cGy TBI and treatment with EGF with or without NU7441 (scale bars, 100 µm). Measurements of tail moments in KSL cells in each condition (n = 6-7 fields per group [range 110-137 cells per field], mean ± SEM, Student t test) (right panel). (C) Representative flow cytometric analysis of annexin V and 7-AAD staining in BM KSL cells at day +10 from mice described in A (left panel). Percentage of annexin V+ KSL cells in each condition (n = 3 or 4 per group, 1-way ANOVA) (right panel). (D) Numbers of CFCs in BM at day +10 from the mice described in A (n = 6 per group, mean ± SEM, 2-way ANOVA). (E) Flow cytometric analysis of BM CD150+CD48−CD41− KSL cells at day +10 from mice described in A (left panel). Percentage of CD150+CD48−CD41− cells within the KSL population in each group (n = 7 or 8 per group, 1-way ANOVA). (F) Percentages of donor CD45.2+ cells in the BM of primary recipient mice at 16 weeks following transplant of BM cells collected at day +10 from the mice in A (n = 6 per group, 2-way ANOVA). (G) Percentages of donor CD45.2+ cells in the BM of secondary mice at 16 weeks posttransplant. Secondary mice were transplanted with 5 × 106 BM cells collected from primary mice at 16 weeks posttransplant, along with 2 × 105 competitor BM (CD45.1+) cells (n = 6 per group, 2-way ANOVA). (H) Percentages of CD45.2+Mac1/Gr1+ (myeloid) cells (left panel), CD45.2+B220+ B cells (middle panel), and CD45.2+CD3+ T cells (right panel) in the BM of secondary recipients at 16 weeks posttransplant (n = 6 per group, 2-way ANOVA). (I-J) C57BL/6 mice were injected with 1 dose of doxorubicin as chemotherapy, followed by EGF or saline treatment. (I) PB WBCs, neutrophils (NEU), lymphocytes (LYMPH), and platelets (PLT) in mice at +10 days postdoxorubicin (n = 4 or 5, Student t test). (J) Number of KSL cells per mouse at +10 days postdoxorubicin (n = 5 per group, Student t test) (left panel). Percentages of CD150+CD48−CD41− cells within the KSL cell population at day +10 postdoxorubicin (n = 5 per group, Student t test) (right panel). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Competitive repopulation assays were performed to measure functional HSC content at day +10 following 500-cGy TBI. Primary recipient mice transplanted with BM cells from irradiated EGF-treated mice displayed increased total donor CD45.2+ cell engraftment and multilineage engraftment in the BM at 16 weeks posttransplant compared with recipient mice transplanted with equal doses of BM from irradiated control mice (Figure 3F; supplemental Figure 2C). Recipient mice transplanted with BM cells from irradiated donor mice treated with EGF + NU7441 displayed no increase in the engraftment of total cells, myeloid cells, B cells, or T cells compared with irradiated controls. Secondary mice transplanted with BM cells from primary recipients in the irradiated EGF treatment group displayed increased total donor engraftment and multilineage engraftment at 16 weeks, but secondary recipient mice transplanted with equal doses of BM cells from irradiated mice treated with EGF + NU7441 demonstrated no difference in donor cell engraftment compared with the irradiated control group (Figure 3G-H). These results suggest that EGF-mediated HSC recovery in irradiated mice is dependent on DNA-PKcs activation. Of note, short-duration EGF treatment (2 doses at days +1 and +2 post-TBI) did not increase PB complete blood counts, BM cells, or HSCs in mice at day +10 compared with irradiated controls, suggesting that a longer duration of EGF treatment was necessary for hematopoietic regeneration after TBI (supplemental Figure 2D-G).
EGF promotes hematopoietic regeneration without leukemogenesis following chemotherapy
To determine whether EGF treatment could promote hematopoietic regeneration following chemotherapy, we treated mice with doxorubicin,40 followed by EGF or saline from day +1 to day +10. Doxorubicin treatment caused pancytopenia in control mice at day +10; conversely, mice treated with doxorubicin, followed by EGF, had increased PB WBCs, neutrophils, lymphocytes, and platelet counts compared with controls (Figure 3I). BM cell counts, KSL cells, and the percentages of CD150+CD48−41− KSL cells were also increased in EGF-treated mice compared with controls (Figure 3J; supplemental Figure 3A-B). BM KSL cells from doxorubicin-treated mice displayed increased γ-H2AX foci and tail moments at +12 hours; EGF treatment decreased doxorubicin-mediated DNA damage and increased p-DNA-PKcs in BM KSL cells at +12 hours following doxorubicin (supplemental Figure 3C-E).
To test whether EGF treatment might promote tumor growth in vivo, we transplanted CD45.1+B6.SJL mice with CD45.2+ BM lin− cells transduced with a retroviral HOXA9/MEIS1 vector that produces acute myeloid leukemia (AML) at 3 to 4 weeks posttransplantation.41 Recipient mice developed AML at 3 weeks posttransplant and were subsequently treated with cytarabine for 5 days to reduce tumor burden. Mice were then treated daily for 10 days with EGF or saline subcutaneously, and CD45.2+ AML cells and Mac1/Gr1+ myeloid cells were subsequently measured in the BM and PB. We did not observe any differences in the percentages of CD45.2+ AML cell numbers or Mac1/Gr1+ cells in the BM or PB of EGF-treated mice vs saline-treated mice (supplemental Figure 4).
Effects of EGF treatment on BM vascular ECs and stromal cells
Our prior study42 and a recent study by Tikhonova et al43 suggest that BM ECs and leptin receptor-positive (LepR+) stromal cells express EGFR; therefore, it is possible that systemic administration of EGF may promote hematopoietic regeneration via indirect effects on the BM microenvironment. At day +10 following 500-cGy TBI and treatment with EGF or saline, we detected increased percentages of VE-cad+ BM ECs in both groups compared with nonirradiated mice (supplemental Figure 5A). Conversely, the percentages of LepR+ BM stromal cells decreased following TBI in both groups of mice (supplemental Figure 5B). TBI increased the MFI of VE-cad+ BM ECs and BM vascular area at day +10 in EGF-treated mice and controls (supplemental Figure 5C-E). However, EGF treatment decreased BM vascular area in irradiated mice, suggesting a beneficial effect of EGF treatment on BM vascular recovery after TBI.44 We did not detect any effect of EGF treatment on the MFI of LepR+ stromal cells in the BM after TBI (supplemental Figure 5F).
EGFR is necessary for HSC DNA repair and hematopoietic regeneration in vivo
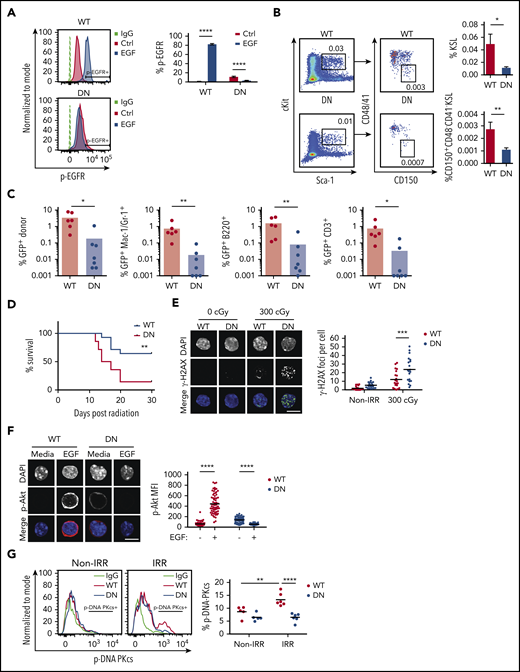
To determine whether EGFR signaling is necessary for HSC DNA repair and hematopoietic regeneration in vivo, we used EGFR-wild type (EGFR-WT) and EGFR-dominant negative (EGFR-DN) mice.45-47 EGFR-DN mice expressed high levels of the EGFR mutant extracellular domain (supplemental Figure 6A). In response to EGF treatment, BM KSL cells from EGFR-WT mice displayed increased p-EGFR, whereas KSL cells from EGFR-DN mice did not (Figure 4A). In steady-state, EGFR-WT mice and EGFR-DN mice did not exhibit any differences in hematopoietic parameters or HSC content (supplemental Figure 6B-G). At day +10 following 500 cGy TBI, EGFR-DN mice displayed decreased percentages of BM KSL cells and CD150+CD48−CD41− KSL HSCs, as well as decreased BM CFCs, compared with EGFR-WT controls, although PB complete blood counts were not substantially different (Figure 4B; supplemental Figure 7A-B). To compare long-term HSC content, we transplanted 2.5 × 104 BM GFP+ KSL cells, labeled via transduction with MSCV-IRES-GFP (Addgene #20672, gift from Tannishtha Reya) and isolated at day +10 from EGFR-DN mice or EGFR-WT mice that were irradiated with 500 cGy, into 950-cGy–irradiated F1 recipient mice, along with 2 × 105 competitor (F1) BM cells. At 16 weeks, recipient mice transplanted with KSL cells from EGFR-DN donors displayed decreased total donor cell and decreased donor myeloid, B-, and T-cell engraftment in the PB, as well as decreased donor KSL cells in the BM, compared with recipients transplanted with KSL cells from EGFR-WT mice (Figure 4C; supplemental Figure 7C). Separately, recipient F1 mice transplanted with BM cells collected at day +10 from 500-cGy–irradiated EGFR-DN mice displayed decreased survival compared with mice transplanted with BM cells from irradiated EGFR-WT mice, suggesting that EGFR deficiency in HSCs also depleted radioprotective HSPCs after TBI (Figure 4D).
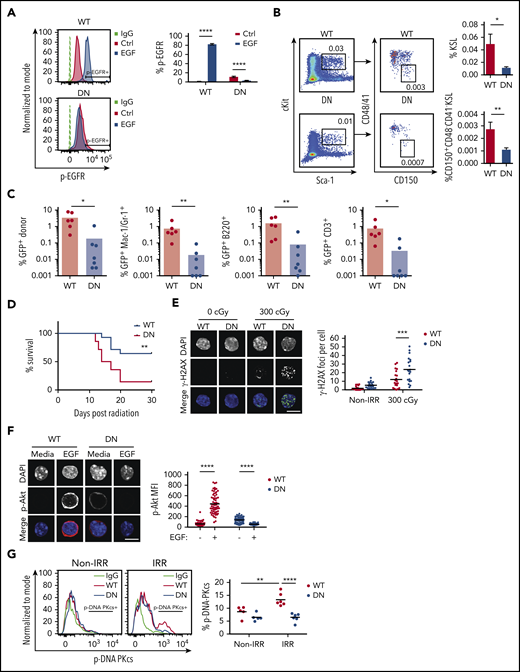
EGFR signaling is necessary for HSC DNA repair and hematopoietic regeneration following TBI. (A) Representative line graphs of p-EGFR levels in BM KSL cells from EGFR-WT (WT) mice and EGFR-DN (DN) mice at 45 minutes following EGF treatment (left panel). Percentage of p-EGFR+ KSL cells in each condition (n = 6 per group, mean ± SEM, 2-way ANOVA) (right panel). (B) Representative flow cytometric analysis of percentages of BM KSL cells and CD150+CD48−CD41− KSL HSCs in WT and DN mice at day +10 following 500-cGy TBI (left panels). Percentages of KSL cells and CD150+CD48−CD41− KSL cells in each group (n = 14 per group, mean ± SEM, Student t test) (right panels). (C) Percentages of total donor GFP+ cells, myeloid cells, B cells, and T cells in the PB of recipient mice at 16 weeks following transplantation of 2.5 × 104 GFP+ KSL cells collected from WT or DN mice at day +10 following 500-cGy TBI, coupled with 2 × 105 BM competitor cells (n = 6 or 7 per group, Student t test). (D) Percentage survival of recipient F1 mice over 30 days following 900-cGy TBI and transplantation with 5 × 105 BM cells collected from EGFR-DN or EGFR-WT mice at 24 hours post-500-cGy TBI (n = 14 mice per group, log-rank test). (E) Representative microscopic image of γ-H2AX foci in nonirradiated and 300-cGy–irradiated BM KSL cells from WT and DN mice cultured with TSF for 1 hour (scale bar, 10 µm) (left panel). Foci numbers per KSL cell (n = 20 fields of view per group, mean ± SEM, 2-way ANOVA) (right panel). (F) Microscopic images of p-Akt in KSL cells from WT and DN mice at 5 minutes following irradiation with 300 cGy and culture in media with or without EGF (scale bar, 10 µm) (left panel). p-Akt MFI in KSL cells from each condition (n = 56-69 cells per group, mean ± SEM, 2-way ANOVA) (right panel). (G) Representative line graphs of p-DNA-PKcs levels in KSL cells from WT and DN mice at 1 hour following 100 cGy (left panel). Percentage of p-DNA-PKcs+ KSL cells in each condition at 1 hour postirradiation (n = 5 or 6 per group, 2-way ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; IgG, immunoglobulin G; IRR, irradiated; Non-IRR, nonirradiated.
EGFR signaling is necessary for HSC DNA repair and hematopoietic regeneration following TBI. (A) Representative line graphs of p-EGFR levels in BM KSL cells from EGFR-WT (WT) mice and EGFR-DN (DN) mice at 45 minutes following EGF treatment (left panel). Percentage of p-EGFR+ KSL cells in each condition (n = 6 per group, mean ± SEM, 2-way ANOVA) (right panel). (B) Representative flow cytometric analysis of percentages of BM KSL cells and CD150+CD48−CD41− KSL HSCs in WT and DN mice at day +10 following 500-cGy TBI (left panels). Percentages of KSL cells and CD150+CD48−CD41− KSL cells in each group (n = 14 per group, mean ± SEM, Student t test) (right panels). (C) Percentages of total donor GFP+ cells, myeloid cells, B cells, and T cells in the PB of recipient mice at 16 weeks following transplantation of 2.5 × 104 GFP+ KSL cells collected from WT or DN mice at day +10 following 500-cGy TBI, coupled with 2 × 105 BM competitor cells (n = 6 or 7 per group, Student t test). (D) Percentage survival of recipient F1 mice over 30 days following 900-cGy TBI and transplantation with 5 × 105 BM cells collected from EGFR-DN or EGFR-WT mice at 24 hours post-500-cGy TBI (n = 14 mice per group, log-rank test). (E) Representative microscopic image of γ-H2AX foci in nonirradiated and 300-cGy–irradiated BM KSL cells from WT and DN mice cultured with TSF for 1 hour (scale bar, 10 µm) (left panel). Foci numbers per KSL cell (n = 20 fields of view per group, mean ± SEM, 2-way ANOVA) (right panel). (F) Microscopic images of p-Akt in KSL cells from WT and DN mice at 5 minutes following irradiation with 300 cGy and culture in media with or without EGF (scale bar, 10 µm) (left panel). p-Akt MFI in KSL cells from each condition (n = 56-69 cells per group, mean ± SEM, 2-way ANOVA) (right panel). (G) Representative line graphs of p-DNA-PKcs levels in KSL cells from WT and DN mice at 1 hour following 100 cGy (left panel). Percentage of p-DNA-PKcs+ KSL cells in each condition at 1 hour postirradiation (n = 5 or 6 per group, 2-way ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; IgG, immunoglobulin G; IRR, irradiated; Non-IRR, nonirradiated.
Following 300-cGy irradiation, BM KSL cells from EGFR-DN mice displayed increased γ-H2AX foci compared with KSL cells from EGFR-WT mice (Figure 4E). EGF treatment increased p-Akt in KSL cells from EGFR-WT mice, but no change was observed in EGFR-DN mice (Figure 4F). Similarly, p-DNA-PKcs levels increased in BM KSL cells from EGFR-WT mice, but not from EGFR-DN mice, following 300-cGy irradiation (Figure 4G).
EGF induces EGFR/Akt/DNA-PKcs pathway activation in HSPCs in irradiated mice
We detected increased activation of DNA-PKcs in BM KSL cells in irradiated EGFR-WT mice in the absence of EGF treatment, implying that endogenous EGF activates EGFR signaling and DNA-PKcs in HSPCs of EGFR-WT mice. Following 500-cGy TBI, we detected EGF levels in the BM of EGFR-WT and EGFR-DN mice over time (range, 22-5205 pg/mL; supplemental Figure 8A). In keeping with this, EGFR-WT mice demonstrated increased p-EGFR, p-Akt, p-Artemis, and p-DNA-PKcs in BM KSL cells at 5 minutes following 500-cGy TBI, whereas EGFR-DN mice did not show activation of these targets (supplemental Figure 8B-E). Treatment of BM HSCs with 100 pg/mL EGF in vitro also increased p-EGFR, p-Akt, p-Artemis, and p-DNA-PKcs in BM HSPCs compared with untreated BM HSPCs (supplemental Figure 8F-I). These data suggest that endogenous EGF promotes the activation of the EGFR/Akt/DNA-PKcs pathway in BM HSPCs in irradiated EGFR-WT mice, whereas EGFR-DN mice fail to activate this pathway. We did not detect any differences in nuclear localization of EGFR in response to irradiation or EGF treatment in EGFR-WT or EGFR-DN mice (supplemental Figure 8J).
In a complementary study, we compared p-EGFR, p-Akt, p-Artemis, and p-DNA PKcs levels in BM KSL cells from C57BL/6 mice irradiated with 500-cGy TBI and treated once with EGF vs irradiated C57BL/6 mice treated with saline. Both groups of mice displayed increased p-EGFR, p-Akt, p-Artemis, and p-DNA-PKcs in BM KSL cells compared with nonirradiated mice (supplemental Figure 9). However, irradiated mice treated with EGF demonstrated increased levels of p-EGFR, p-Akt, p-Artemis and, p-DNA-PKcs compared with irradiated controls (supplemental Figure 9).
EGF promotes human hematopoietic regeneration following irradiation
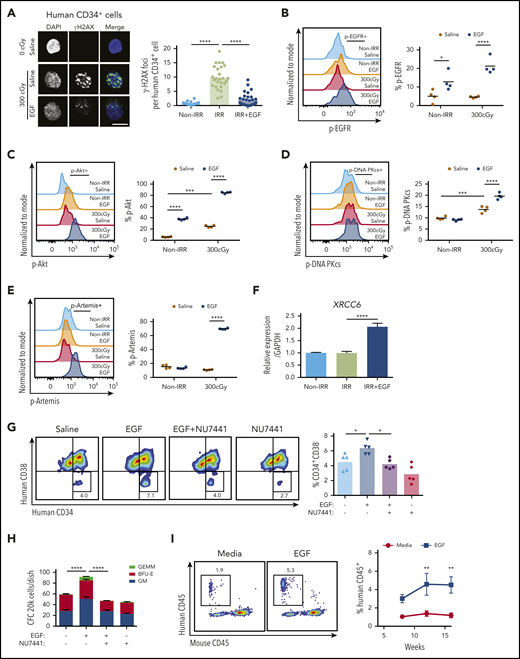
We also irradiated human BM CD34+ HSPCs with 300 cGy and cultured them in media, with or without 100 ng/mL EGF, for 36 hours. Irradiation increased γ-H2AX foci in CD34+ cells at +1 hour, and EGF treatment abrogated this effect (Figure 5A). EGF treatment significantly increased p-EGFR, p-Akt, p-Artemis, and p-DNA-PKcs, as well as increased the expression of XRCC6, in irradiated human CD34+ cells (Figure 5B-F). EGF treatment also increased the percentage of CD34+CD38− HSPCs and the numbers of CFCs in culture at +36 hours compared with control cultures (Figure 5G-H). Inhibition of DNA-PKcs with NU7441 blocked EGF’s effects on HSPC maintenance and CFC recovery after irradiation (Figure 5G-H).
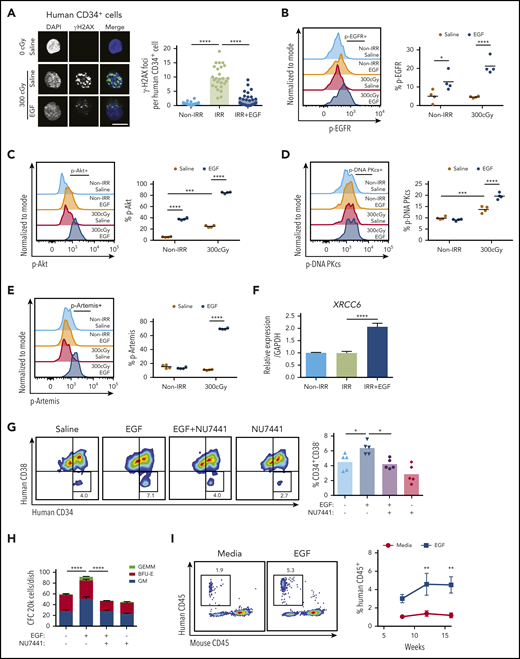
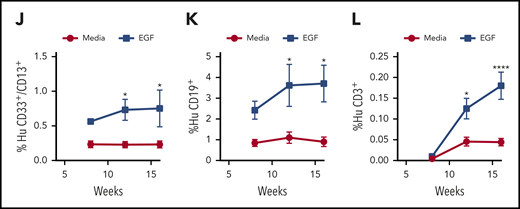
EGF treatment promotes recovery of human HSCs following irradiation. (A) Immunofluorescence microscopy of γ-H2AX foci in nonirradiated and 300-cGy–irradiated human BM CD34+ cells cultured in complete media (TSF) with and without 100 ng/mL EGF for 1 hour (scale bar, 10 µm). Numbers of foci per cell are quantified (n = 24-27 fields of view, 1-way ANOVA) (left panel). Nonirradiated (Non-IRR) or 300-cGy–irradiated human BM CD34+ cells were treated with EGF or saline for 5 minutes and analyzed for p-EGFR (B), p-Akt (C), p-DNA-PKcs (D), and p-Artemis (E) levels (n = 4 per group, 2-way ANOVA). (B) Representative flow cytometric analysis of p-EGFR in BM CD34+ cells (left panel). Quantification of the percentage of p-EGFR (right panel). (C) Representative analysis of p-Akt in BM CD34+ cells (left panel). Quantification of the percentage of p-Akt (right panel). (D) Representative analysis of p-DNA-PKcs in BM CD34+ cells (left panel). Quantification of the percentage of p-DNA PKcs (right panel). (E) Representative analysis of p-Artemis in BM CD34+ cells (left panel). Quantification of the percentage of p-Artemis (right panel). (F) XRCC6 gene expression in human BM CD34+ cells following 300-cGy irradiation and treatment with EGF or saline for 1 hour. Non-IRR BM CD34+ cells cultured for 1 hour were used as control (n = 3, mean ± SEM, 1-way ANOVA). (G) Representative flow cytometric analysis of human CD34+CD38− HSPCs in culture at 36 hours following 300-cGy irradiation of BM CD34+ cells and treatment with media with or without EGF and with or without NU7441 (left panel). Mean percentages of CD34+CD38− cells (n = 5 per group, 1-way ANOVA) (right panel). (H) Numbers of CFCs after 36 hours of culture of BM CD34+ cells following 300-cGy irradiation and treatment with or without EGF and with or without NU7441 (n = 6, mean ± SEM, 2-way ANOVA). (I) Representative human CD45+ cell engraftment in PB of NSG-S mice at 16 weeks following transplantation of irradiated BM CD34+ cells cultured in media + EGF for 36 hours or irradiated BM CD34+ cells cultured in media alone (left panel). Time course of human CD45+ hematopoietic cell engraftment in PB of NSG-S mice (n = 7 or 8, mean ± SEM, 2-way ANOVA) (right panel). Time course of human CD33+/CD13+ myeloid cell engraftment (J), human CD19+ B-cell engraftment (K), and human CD3+ T-cell engraftment (L) in NSG-S mice (n = 7 or 8, mean ± SEM, 2-way ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001.
EGF treatment promotes recovery of human HSCs following irradiation. (A) Immunofluorescence microscopy of γ-H2AX foci in nonirradiated and 300-cGy–irradiated human BM CD34+ cells cultured in complete media (TSF) with and without 100 ng/mL EGF for 1 hour (scale bar, 10 µm). Numbers of foci per cell are quantified (n = 24-27 fields of view, 1-way ANOVA) (left panel). Nonirradiated (Non-IRR) or 300-cGy–irradiated human BM CD34+ cells were treated with EGF or saline for 5 minutes and analyzed for p-EGFR (B), p-Akt (C), p-DNA-PKcs (D), and p-Artemis (E) levels (n = 4 per group, 2-way ANOVA). (B) Representative flow cytometric analysis of p-EGFR in BM CD34+ cells (left panel). Quantification of the percentage of p-EGFR (right panel). (C) Representative analysis of p-Akt in BM CD34+ cells (left panel). Quantification of the percentage of p-Akt (right panel). (D) Representative analysis of p-DNA-PKcs in BM CD34+ cells (left panel). Quantification of the percentage of p-DNA PKcs (right panel). (E) Representative analysis of p-Artemis in BM CD34+ cells (left panel). Quantification of the percentage of p-Artemis (right panel). (F) XRCC6 gene expression in human BM CD34+ cells following 300-cGy irradiation and treatment with EGF or saline for 1 hour. Non-IRR BM CD34+ cells cultured for 1 hour were used as control (n = 3, mean ± SEM, 1-way ANOVA). (G) Representative flow cytometric analysis of human CD34+CD38− HSPCs in culture at 36 hours following 300-cGy irradiation of BM CD34+ cells and treatment with media with or without EGF and with or without NU7441 (left panel). Mean percentages of CD34+CD38− cells (n = 5 per group, 1-way ANOVA) (right panel). (H) Numbers of CFCs after 36 hours of culture of BM CD34+ cells following 300-cGy irradiation and treatment with or without EGF and with or without NU7441 (n = 6, mean ± SEM, 2-way ANOVA). (I) Representative human CD45+ cell engraftment in PB of NSG-S mice at 16 weeks following transplantation of irradiated BM CD34+ cells cultured in media + EGF for 36 hours or irradiated BM CD34+ cells cultured in media alone (left panel). Time course of human CD45+ hematopoietic cell engraftment in PB of NSG-S mice (n = 7 or 8, mean ± SEM, 2-way ANOVA) (right panel). Time course of human CD33+/CD13+ myeloid cell engraftment (J), human CD19+ B-cell engraftment (K), and human CD3+ T-cell engraftment (L) in NSG-S mice (n = 7 or 8, mean ± SEM, 2-way ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001.
We next transplanted NOD/SCID–stem cell factor/granulocyte macrophage colony stimulating factor/interleukin-3 (NSG-S) mice48 with the progeny of 2 × 105 CD34+ cells at 36 hours following 300-cGy irradiation and culture with or without 100 ng/mL EGF. Mice transplanted with the progeny of irradiated human CD34+ cells treated with EGF displayed increased engraftment of total human donor cells, myeloid cells, and B and T cells at 12 and 16 weeks posttransplant compared with mice transplanted with the progeny of irradiated human CD34+ cells not treated with EGF (Figure 5I-L).
Effects of EGF treatment on HSC mutagenesis and gene expression following TBI
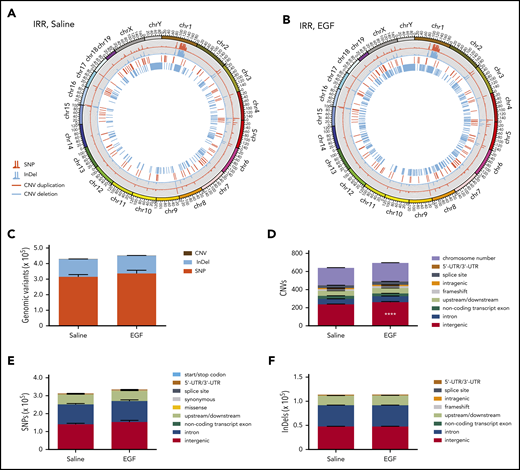
Because EGF treatment increases NHEJ in KSL cells, and NHEJ is considered error prone,1 we sought to measure whether EGF treatment affected the frequency of somatic mutations in BM KSL cells following TBI. We irradiated C57BL/6 mice with 500-cGy TBI and administered EGF or saline at +1 hour and +24 hours, and BM KSL cells were collected at 6 weeks for WGS. We did not detect any differences in the total numbers of InDels, copy number variations (CNVs), SNPs, coding region mutations, or intergenic SNPs and InDels in BM KSL cells from EGF-treated vs saline-treated mice (Figure 6); however, we detected increased numbers of intergenic CNVs in KSL cells from EGF-treated mice (Figure 6D; supplemental Table 3). Within a subset of AML oncogenes, we observed no differences in SNPs or InDels in BM KSL cells from EGF-treated vs saline-treated mice (supplemental Figure 10A). The WGS data set has been deposited in the Sequence Read Archive of the National Center for Biotechnology Information (accession number PRJNA612325).
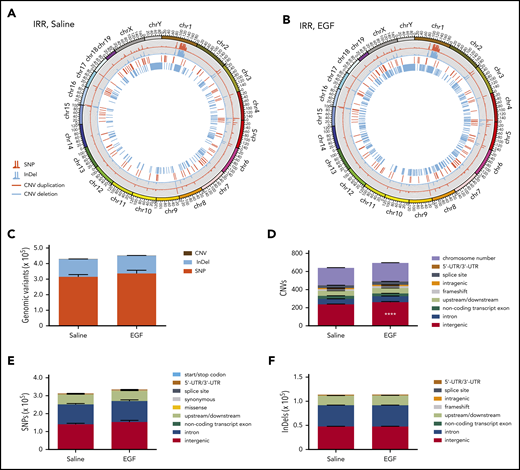
EGF treatment effects on HSC mutagenesis and gene expression following TBI. Representative Circos plots of WGS of BM KSL cells collected from C57BL/6 mice at 6 weeks following 500-cGy TBI and treatment with saline (A) or EGF (B). (C) Total genome variant numbers categorized by mutation type (n = 5 per group, mean ± SEM, 2-way ANOVA). (D) Numbers and locations of CNVs in BM KSL cells at 6 weeks following 500-cGy TBI and treatment with EGF or saline (n = 5 per group, mean ± SEM). ****P < .0001, 2-way ANOVA. (E) Numbers and locations of SNPs in BM KSL cells at 6 weeks following 500-cGy TBI and treatment with EGF or saline (n = 5 per group, mean ± SEM, 2-way ANOVA). (F) Numbers and locations of InDels in BM KSL cells at 6 weeks following 500-cGy TBI and treatment with EGF or saline (n = 5 per group, mean ± SEM, 2-way ANOVA). UTR, untranslated region.
EGF treatment effects on HSC mutagenesis and gene expression following TBI. Representative Circos plots of WGS of BM KSL cells collected from C57BL/6 mice at 6 weeks following 500-cGy TBI and treatment with saline (A) or EGF (B). (C) Total genome variant numbers categorized by mutation type (n = 5 per group, mean ± SEM, 2-way ANOVA). (D) Numbers and locations of CNVs in BM KSL cells at 6 weeks following 500-cGy TBI and treatment with EGF or saline (n = 5 per group, mean ± SEM). ****P < .0001, 2-way ANOVA. (E) Numbers and locations of SNPs in BM KSL cells at 6 weeks following 500-cGy TBI and treatment with EGF or saline (n = 5 per group, mean ± SEM, 2-way ANOVA). (F) Numbers and locations of InDels in BM KSL cells at 6 weeks following 500-cGy TBI and treatment with EGF or saline (n = 5 per group, mean ± SEM, 2-way ANOVA). UTR, untranslated region.
BM KSL cells from irradiated EGF-treated mice demonstrated a distinct pattern of gene expression compared with irradiated saline-treated mice (supplemental Figure 10B), as well as upregulation of inflammatory signaling pathways compared with saline-treated mice (supplemental Figure 10C). The complete RNA-sequencing analysis of KSL cells from this study has been deposited in the Gene Expression Omnibus of the National Center for Biotechnology Information (accession number GSE146371).
Discussion
DNA damage response mechanisms are essential for HSCs to maintain genomic integrity over time in response to intrinsic and extrinsic stresses.1,4,5,23,28,29,49,50 DNA repair is also critical for the HSC response to medically relevant stresses, such as chemotherapy and TBI.1,51,52 Recently, it was shown that administration of thrombopoietin just before irradiation protected HSCs from DNA damage by inducing DNA-PKcs activity.6,7 Here, we show that EGF treatment delivered after TBI suppresses DNA damage in HSCs in vivo via activation of EGFR and several components of the NHEJ machinery, including DNA-PKcs and Artemis, while increasing expression of Xrcc6, which encodes the Ku70 protein in the Ku70/Ku80 heterodimer.34 We hypothesize that EGF treatment mitigates DNA damage in HSCs after irradiation because amplification of the Ku70/Ku80 heterodimer can increase the affinity of DNA-PKcs for the Ku:DNA complex by up to 100-fold, thereby increasing DNA-PKcs function.34 Therefore, EGF treatment increases DNA-PKcs activity and augments DNA-PKcs localization to sites of DNA damage in HSCs.
Our studies suggest that EGF treatment decreased BM vascular area after TBI,44 implying that EGF may have direct action on EGFR-expressing BM ECs.42 We did not detect substantial effects of EGF treatment on the recovery of LepR+ stromal cells after TBI. Going forward, to define the role of EGFR-expressing BM niche cells in regulating EGF effects on hematopoietic regeneration and HSC recovery after myelosuppression, we will use Cre-loxP technology to control EGFR expression in BM hematopoietic cells and BM niche cells in mice treated with EGF in homeostasis and following myelosuppression.
Our studies demonstrate that EGF treatment accelerates hematologic recovery in mice treated with chemotherapy. One concern related to developing a growth factor, such as EGF, for clinical application is that EGF could promote the relapse or progression of residual tumor. Our studies of EGF treatment of mice bearing HOXA9/MEIS1+ AML did not demonstrate any significant effects on AML growth or progression in vivo. A rational strategy for the clinical application of EGF or EGF mimetics might be to include only patients in complete remission and with EGFR− malignancy.
To further highlight the translational potential of EGF, we show that EGF abrogates radiation-induced DNA damage in human HSCs and promotes human hematopoietic regeneration via activation of DNA-PKcs. Importantly, EGF treatment of irradiated human HSPCs caused a marked increase in the recovery of human HSCs capable of multilineage in vivo repopulation in immune-deficient mice. These results demonstrate the potential importance of EGF-EGFR signaling in regulating human hematopoietic regeneration and provide the basis for further studies to define the efficacy of EGF treatment of human hematopoietic cell engraftment and regeneration.
Because EGF treatment augments NHEJ repair in HSPCs, it is important to assess potential adverse effects on the HSC genome. Mohrin et al showed that >30% of quiescent HSCs that underwent NHEJ repair following 2-Gy irradiation displayed major genomic mutations.1 Systemic administration of EGF did not increase the overall frequency of mutations in BM KSL cells harvested from mice at 6 weeks post-TBI. This may be explained by the possibility that EGF treatment upregulated canonical NHEJ, which is dependent on DNA-PKcs and relatively accurate,53-55 rather than alternative NHEJ, which is DNA-PKcs independent and associated with error-prone end joining.53,56,57 However, it is noteworthy that BM KSL cells from irradiated EGF-treated mice displayed increased numbers of intergenic mutations, specifically CNVs, compared with control mice. The significance of noncoding intergenic mutations remains poorly understood, but such mutations occur in cancers and are the subject of intense ongoing research.58-60 In our model, long-term studies will be necessary to determine the impact of EGF treatment on clonal hematopoiesis or myelodysplasia over time. These studies will provide better understanding of the balance between near-term beneficial effects of hematopoietic regenerative factors and potentially deleterious late effects.
The data reported in this article have been deposited in the Sequence Read Archive (accession number PRJNA612325) and the Gene Expression Omnibus (accession number GSE146371) of the National Center for Biotechnology Information.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Matteo Pellegrini (Director, UCLA Institute for Quantitative and Computational Biosciences) for guidance on the whole genome sequencing analysis and Destiny Batton for assistance with the microscopy analysis. The authors also thank the UCLA Broad Stem Cell Research Center FACS Core Facility for assistance with hematopoietic cell sorting.
This work was supported, in part, by National Institutes of Health, National Institute of Allergy and Infectious Diseases grants AI107333 and AI067769, and California Institute for Regenerative Medicine Leadership Award (LA1-08014) (all J.P.C.).
Authorship
Contribution: J.P.C. conceptualized the study; T.F., Y.Z., V.Y.C., M.R., C.M.T., P.K.L., L.S., M.Q., A.P., J.K., X.Y., A.J., K.P., L.Z., P.S., and H.A.H. performed and analyzed experiments; C.M.T. designed the visual abstract; and T.F. and J.P.C. wrote the manuscript, with input from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John P. Chute, Division of Hematology/Oncology, Department of Medicine, University of California, Los Angeles, 615 Charles E. Young Dr South, Los Angeles, CA 90095; e-mail: jchute@mednet.ucla.edu.


![EGF promotes HSC recovery in vivo in a DNA-PKcs–dependent manner. (A) Schematic design. C57BL/6 mice were irradiated with 500-cGy TBI, followed by daily injections of saline, EGF, NU7441, or EGF + NU7441 for 10 days. BM cells were collected at +4 hours for Comet assay and at day +10 for annexin V apoptosis assay and competitive repopulation assays. (B) Representative images of a Comet assay of BM KSL cells collected at +4 hours following 500-cGy TBI and treatment with EGF with or without NU7441 (scale bars, 100 µm). Measurements of tail moments in KSL cells in each condition (n = 6-7 fields per group [range 110-137 cells per field], mean ± SEM, Student t test) (right panel). (C) Representative flow cytometric analysis of annexin V and 7-AAD staining in BM KSL cells at day +10 from mice described in A (left panel). Percentage of annexin V+ KSL cells in each condition (n = 3 or 4 per group, 1-way ANOVA) (right panel). (D) Numbers of CFCs in BM at day +10 from the mice described in A (n = 6 per group, mean ± SEM, 2-way ANOVA). (E) Flow cytometric analysis of BM CD150+CD48−CD41− KSL cells at day +10 from mice described in A (left panel). Percentage of CD150+CD48−CD41− cells within the KSL population in each group (n = 7 or 8 per group, 1-way ANOVA). (F) Percentages of donor CD45.2+ cells in the BM of primary recipient mice at 16 weeks following transplant of BM cells collected at day +10 from the mice in A (n = 6 per group, 2-way ANOVA). (G) Percentages of donor CD45.2+ cells in the BM of secondary mice at 16 weeks posttransplant. Secondary mice were transplanted with 5 × 106 BM cells collected from primary mice at 16 weeks posttransplant, along with 2 × 105 competitor BM (CD45.1+) cells (n = 6 per group, 2-way ANOVA). (H) Percentages of CD45.2+Mac1/Gr1+ (myeloid) cells (left panel), CD45.2+B220+ B cells (middle panel), and CD45.2+CD3+ T cells (right panel) in the BM of secondary recipients at 16 weeks posttransplant (n = 6 per group, 2-way ANOVA). (I-J) C57BL/6 mice were injected with 1 dose of doxorubicin as chemotherapy, followed by EGF or saline treatment. (I) PB WBCs, neutrophils (NEU), lymphocytes (LYMPH), and platelets (PLT) in mice at +10 days postdoxorubicin (n = 4 or 5, Student t test). (J) Number of KSL cells per mouse at +10 days postdoxorubicin (n = 5 per group, Student t test) (left panel). Percentages of CD150+CD48−CD41− cells within the KSL cell population at day +10 postdoxorubicin (n = 5 per group, Student t test) (right panel). *P < .05, **P < .01, ***P < .001, ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/4/10.1182_blood.2020005895/1/m_bloodbld2020005895f3.png?Expires=1770650941&Signature=Or2VmrzdzAjoKwUUbfS5ySpOK~HGyJGTuDvmz7b23ZehQX47Rnlleq7USnqPAF-okfMGvcr17rAnVjMfzP0vZBUMMee2bWlm1dDvx-4bP0SxXeZajIk62otLtYKKgp6blriErUF6llNbu3~MLVIYfnQCFjfva-56YmpHJ5rKsJ6WaBg~-L~qF6cVXhNk2H2BjVbVgo-lCgKZRQekZVIetAcjK7X1s1WiNIpcoxGTvNgks0Nsdq7B-udyIpQtEVzWfLF6kEtHubry07vfhWO8KiTrdX5-l7Ipi5oLODU45vInTv8uiQgAP1NONX~x6eTaV9cvEPZmMF3Cpo1D2Kzb0A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)