Key Points
Fully human BCMA-targeting CAR exerted safety and efficacy in patients with RRMM.
Patients who relapsed from prior murine BCMA CAR T-cell therapy may still benefit from CT103A.

Abstract
B-cell maturation antigen (BCMA)-specific chimeric antigen receptor (CAR) T-cell therapies have shown efficacy in relapsed/refractory multiple myeloma (RRMM). Because the non-human originated antigen-targeting domain may limit clinical efficacy, we developed a fully human BCMA-specific CAR, CT103A, and report its safety and efficacy in a phase 1 trial. Eighteen consecutive patients with RRMM, including 4 with prior murine BCMA CAR exposures, were enrolled. CT103A was administered at 1, 3, and 6 × 106 CAR-positive T cells/kg in the dose-escalation phase, and 1 × 106 CAR-positive T cells/kg in the expansion cohort. The overall response rate was 100%, with 72.2% of the patients achieving complete response or stringent complete response. For the 4 murine BCMA CAR–exposed patients, 3 achieved stringent complete response, and 1 achieved a very good partial response. At 1 year, the progression-free survival rate was 58.3% for all cohorts and 79.1% for the patients without extramedullary myeloma. Hematologic toxicities were the most common adverse events; 70.6% of the patients experienced grade 1 or 2 cytokine release syndromes. No immune effector cell–associated neurotoxicity syndrome was observed. To the cutoff date, CAR transgenes were detectable in 77.8% of the patients. The median CAR transgene persistence was 307.5 days. Only 1 patient was positive for the anti-drug antibody. Altogether, CT103A is safe and highly active in patients with RRMM and can be developed as a promising therapy for RRMM. Patients who relapsed from prior murine BCMA CAR T-cell therapy may still benefit from CT103A. This trial was registered at http://www.chictr.org.cn as #ChiCTR1800018137.
Introduction
Despite recent advances in multiple myeloma (MM) treatment strategies, particularly the emergence and clinical application of immunomodulatory drugs, proteasome inhibitors, and monoclonal antibodies that have improved the survival of MM,1-4 it remains an incurable plasma cell cancer, and relapse is almost inevitable in all patients. Results from previously published clinical trials showed that ∼33% to 88% of patients with relapsed/refractory MM (RRMM) had objective antimyeloma responses after treatment with anti–B-cell maturation antigen (BCMA) chimeric antigen receptor (CAR) T cells.5-11 However, it remains a great challenge to achieve durable responses, and relapse or disease progression is observed in ∼28% to 88% of patients at a median follow-up time of 2 to 15 months.5,7-11
The level of CAR T-cell proliferation and the duration of cellular persistence in the blood may be one determinant of the duration of response (DOR).11 Multiple mechanisms, including antigen escape, T-cell intrinsic mechanisms, tumor microenvironment–mediated suppression, and host anti-CAR immunity, may be responsible for the inability of certain CAR T cells to survive in vivo.12 Particularly, previous studies have suggested that CARs with humanized13,14 or fully human15-18 single-chain variable fragments (scFvs) may bypass the potential host anti-CAR immunogenicity and retain antitumor activity.
In the current study, we developed a novel BCMA-targeting CAR construct (CT103A) with a fully human scFv and conducted an open-label, single-arm phase 1 clinical trial to evaluate the safety and preliminary efficacy of CT103A for patients with RRMM.
Methods
Study design
This open-label, phase 1 study was conducted at the Department of Hematology of Tongji Hospital in Wuhan, China. The study consisted of 2 parts: a dose escalation phase using a traditional 3 + 3 protocol and a dose expansion phase (Figure 1). The study protocol was approved by the institutional review board of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and registered with the Chinese Clinical Trial Registry (http://www.chictr.org.cn; #ChiCTR1800018137). Written informed consent was obtained from each participant in compliance with the Declaration of Helsinki.

Flowchart of this trial. *Three patients were excluded because they did not meet inclusion criteria or because of rapid progression. †Three patients who received lymphodepletion were not dosed because of heart failure, severe liver function damage, and severe infection, respectively. ‡Two patients died of infection. §One patient refused follow-up in the study site but remained in a state of sCR until the cutoff date.
Flowchart of this trial. *Three patients were excluded because they did not meet inclusion criteria or because of rapid progression. †Three patients who received lymphodepletion were not dosed because of heart failure, severe liver function damage, and severe infection, respectively. ‡Two patients died of infection. §One patient refused follow-up in the study site but remained in a state of sCR until the cutoff date.
CAR T-cell preparation
The second-generation CAR construct was composed of a fully human scFv, a CD8a hinge, and transmembrane domain, 4-1BB costimulatory, and CD3ζ activation domains (Figure 2A). The procedures of CAR T-cell production, validation, and in vitro/in vivo antimyeloma effect evaluation are described in the supplemental Methods (available on the Blood Web site).
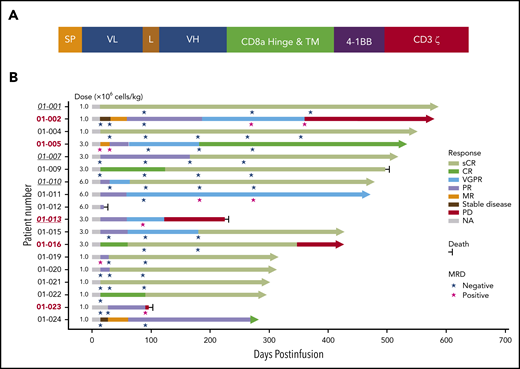
Schematic diagram of CT103A CAR construct and treatment response and outcome in patients with CT103A infusion. (A) The second-generation CAR consisted of a scFv from a fully human antibody against human BCMA, the hinge and transmembrane domain from CD8a, the costimulatory domain from 4-1BB (CD137), and the activation domain from CD3ζ. (B) The best responses of all 18 patients with different infusion doses (1 × 106 cells/kg to 6 × 106 cells/kg) are shown. The responses were assessed according to the criteria described in the Methods. Underlined italic numbers indicate patients who relapsed after a prior murine BCMA CAR T-cell treatment. Bold numbers in red indicate patients with EMM. Arrows indicate ongoing remission. L, linker; MR, minor response; NA, not applicable; PR, partial response; SP, signal peptide; VGPR, very good partial response; VL, variable light chain; VH, variable heavy chain.
Schematic diagram of CT103A CAR construct and treatment response and outcome in patients with CT103A infusion. (A) The second-generation CAR consisted of a scFv from a fully human antibody against human BCMA, the hinge and transmembrane domain from CD8a, the costimulatory domain from 4-1BB (CD137), and the activation domain from CD3ζ. (B) The best responses of all 18 patients with different infusion doses (1 × 106 cells/kg to 6 × 106 cells/kg) are shown. The responses were assessed according to the criteria described in the Methods. Underlined italic numbers indicate patients who relapsed after a prior murine BCMA CAR T-cell treatment. Bold numbers in red indicate patients with EMM. Arrows indicate ongoing remission. L, linker; MR, minor response; NA, not applicable; PR, partial response; SP, signal peptide; VGPR, very good partial response; VL, variable light chain; VH, variable heavy chain.
Patients and treatments
RRMM patients with positive BCMA expression (defined in the supplemental Methods) according to either immunohistochemistry or multiparametric flow cytometry were enrolled. Patients were diagnosed according to the updated criteria of the International Myeloma Working Group.19 The major inclusion criteria were as follows: age 18 to 70 years, an Eastern Cooperative Oncology Group performance status score of 0 or 1,20 adequate major organ function, a life expectancy of ≥12 weeks, and at least 3 lines of prior therapies that must include a proteasome inhibitor and an immunomodulatory agent. All eligible patients underwent leukapheresis for peripheral blood mononuclear cell (PBMC) collection and received lymphodepletion chemotherapy with a regimen of fludarabine at 25 mg/m2 and cyclophosphamide at 20 mg/kg daily for 3 consecutive days (days −4 to −2) before CT103A infusion. Bridging therapy was allowed between PBMC collection and lymphodepletion. CT103A was administered at 1, 3, and 6 × 106 CAR-positive (CAR+) T cells/kg in the dose escalation phase and 1 × 106 CAR+ T cells/kg in the expansion phase. Related laboratory and imaging evaluations were performed for toxicity and response assessment according to study procedures in the supplemental Trial Protocols.
Anti-drug antibody detection assay
The anti-drug antibody (ADA) of CT103A was evaluated by using an electrochemiluminescence bridging assay on the MSD-ECL platform (Meso Scale Discovery, Gaithersburg, MD).21,22 A multi-tiered ADA testing approach was used to detect ADA, including screening assay, confirmatory assay, and titration assay. The positive samples for screening assay underwent confirmatory assay to test the ADA specificity and the titration assay to determine the titer.
End point assessments
The primary end point was to evaluate the safety and tolerability, including the dose-limiting toxicity (DLT) and the maximum tolerated dose, and to determine the recommended phase 2 dose of CT103A. The secondary end points were efficacy and pharmacokinetic and pharmacodynamic data of CT103A. Toxicity was graded by using the National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0 (https://academy.myeloma.org.uk/wp-content/uploads/sites/2/2015/04/CTCAE_v5.pdf). All adverse events (AEs) and severe AEs (SAEs) were recorded throughout the follow-up; these included cytokine release syndrome (CRS) and symptoms of immune effector cell–associated neurotoxicity syndrome (ICANS), which were graded according to the criteria of Lee et al.23 The definition of parameters for end point assessment, including progression-free survival (PFS), overall survival (OS), time to response (TTR), and DOR, is provided in the supplemental Trial Protocols.
Clinical response and disease progression were evaluated according to the International Myeloma Working Group consensus criteria24 at serial time points after CT103A infusion. Bone marrow examination was applied to assess disease response for nonsecretory patients. Minimal residual disease (MRD) was assessed by using multiparametric flow cytometry and next-generation sequencing if experimentally feasible, and CAR transgene copies in the patient PBMCs were monitored by using digital droplet polymerase chain reaction,25 detailed in the supplemental Methods. The definition of extramedullary myeloma (EMM) is the presence of soft tissue masses in extraosseous locations resulting from hematogenous spread that are not contiguous to the involved bone.26 The cutoff date was 30 April 2020.
Statistical analysis
The analysis of categorical variables was performed by using Fisher’s exact test. For continuous variables, the Wilcoxon rank sum test was used, and the Kaplan-Meier method was used to estimate the probabilities of OS and PFS. Statistical analyses were performed by using SPSS version 22 (IBM SPSS Statistics, IBM Corporation, Armonk, NY) and GraphPad Prism 8 (GraphPad Software, La Jolla, CA). P values <.05 (two-tailed) were considered statistically significant.
Results
Production and validation of CT103A
The fully human anti-BCMA scFv of CT103A was selected from 42 leading scFvs from >5300 candidates discovered through yeast display and functional assays in T cells (supplemental Figure 1). The affinity between CT103A cell and BCMA-Fc fusion protein was determined (supplemental Figure 2; supplemental Table 1). In vitro validation of antimyeloma efficacy showed that CT103A had stable and specific degranulation activity and excellent specific cytotoxicity against U266 cells (supplemental Figures 3-5). In vivo study using a U266 human myeloma xenograft mice model showed the robust antimyeloma efficacy of CT103A (supplemental Figures 6-8).
Patient characteristics
Between 7 September 2018, and 8 July 2019, a total of 24 consecutive adult subjects with BCMA-positive RRMM were screened according to the study protocol (supplemental Trial Protocols), and 6 patients were excluded (Figure 1). The results presented are from 18 patients (10 male subjects and 8 female subjects) who received CT103A in the dose escalation and expansion cohorts. As shown in Table 1, the median age was 53.5 years (range, 38.0-66.0 years), and the median time since diagnosis was 31.9 months (range, 8.8-94.3 months). Seven patients had a high-risk cytogenetic profile. Five patients had EMM, one of whom (patient 01-013) also developed secondary plasma cell leukemia before infusion. The median number of prior therapies before enrollment was 4 (range, 3-6). All the patients had been treated and were refractory to both bortezomib and lenalidomide; 7 patients (38.9%) were also refractory to other novel agents, including carfilzomib, ixazomib, pomalidomide, and daratumumab. Six patients (33.3%) had previously undergone autologous hematopoietic stem cell transplantation. Four patients (22.2%) had received prior murine anti-BCMA CAR T-cell treatment (registered at Chinese Clinical Trial Registry, http://www.chictr.org.cn; #ChiCTR-OPC-16009113). Only one patient received bridging therapy. Detailed information is provided in supplemental Tables 2 and 3.
CT103A is highly active and induces rapid responses in patients with RRMM
All patients received the prescribed cell dose of CT103A. Patients were assessed for clinical responses and were observed for up to 587 days (median, 394 days) by the cutoff date. Each patient’s response and survival profile are visualized as a bar chart in Figure 2B. In the first 2 weeks after infusion, the overall response rate (ORR) was 77.8% (14 of 18). At 1 month postinfusion, the ORR was 88.9% (16 of 18), with a complete response (CR) or a stringent CR (sCR) rate of 44.4% (8 of 18). The ORR was 100% for all patients (18 of 18), with enhanced responses over time. A total of 72.2% (13 of 18) of the patients finally achieved a CR or sCR (supplemental Figure 9). All patients evaluated for MRD (17 of 17) in the bone marrow were MRD-negative at 10−4 nucleated cells by flow cytometry within 1 month; 9 of them were tested by next-generation sequencing, and 4 patients achieved the best responses of MRD negativity by the level of 10−6 nucleated cells over time (Figure 2B; supplemental Table 4). The median TTR was 15 days (range, 14-62 days) for all patients.
CT103A induces a persistent response in patients without EMM
The median DOR was 325 days (range, 7-573 days) for all 18 patients and 412 days (range, 213-573 days) for the 13 patients with CR/sCR (supplemental Figure 10). Four patients (22.2%) had relapsed or progressive disease (PD). Two of the patients feasible for re-evaluation of BCMA expression exhibited a reduction but not a complete loss of BCMA expression on myeloma cells post–CT103A infusion (mean fluorescence intensity and BCMA-positive percentage by flow cytometry of bone marrow samples, respectively, 1883 vs 583, 81% vs 29% for subject 01-002; 9053 vs 1825, 99% vs 39% for subject 01-013). Two patients (01-013 and 01-023) died during the follow-up due to PD. In addition, an early death on day 20 was recorded for patient 01-012, and another patient (01-009) with persistent sCR died of a sudden severe infection (both detailed in the supplemental Results). Survival analysis showed that the rates of PFS and OS at 1 year were 58.3% and 75%, respectively (Figure 3). In 5 patients with EMM, 4 with multiple extramedullary lesions had PD or relapse (supplemental Table 5). The other patient (01-005) with an isolated extramedullary lesion achieved CR and had an ongoing CR status (Figure 2B; Figure 4A). Kaplan-Meier analysis indicated that EMM was associated with a shortened PFS (79.1% vs 20.0%, P = .015) but not OS (79.1% vs 60.0%, P = .328) at 1 year (supplemental Figure 11), possibly due to the limited sample size and/or follow-up time.
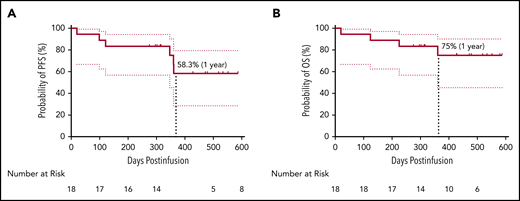
Survival analysis of patients. PFS (A) and OS (B) with a 95% confidence interval of all 18 patients. The median PFS and OS were not reached at the cutoff date. The rate of PFS and OS at 1 year was 58.3% and 75%, respectively.
Survival analysis of patients. PFS (A) and OS (B) with a 95% confidence interval of all 18 patients. The median PFS and OS were not reached at the cutoff date. The rate of PFS and OS at 1 year was 58.3% and 75%, respectively.
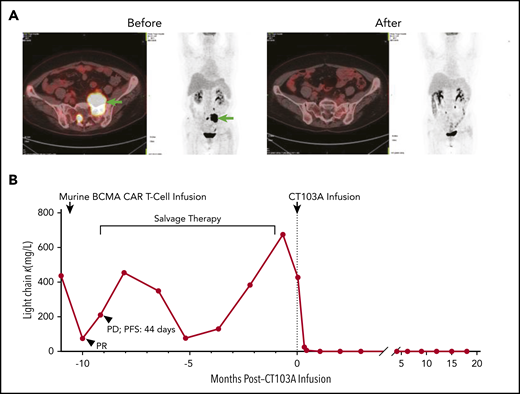
Elimination of myeloma cells by CT103A. (A) Comparison of an extramedullary lesion in the abdomen of patient 01-005 before and 98 days after CT103A infusion. The lesion was denoted by arrows in the left 2 images from the positron emission tomography/computed tomography scan before infusion. At day 98, after infusion, no lesion was observed in the 2 images on the right, which was nearly the same section as that on the positron emission tomography/computed tomography scan. (B) Change in the serum light chain levels in patient 01-001. After the murine BCMA CAR infusion, the serum light chain level decreased for a short time and soon recovered, and the PFS was 44 days. In comparison, after CT103A infusion, the light chain level quickly decreased and remained stable at a normal level with a normal κ/λ ratio, indicating continuous sCR. The PFS was 587 days at the cutoff date.
Elimination of myeloma cells by CT103A. (A) Comparison of an extramedullary lesion in the abdomen of patient 01-005 before and 98 days after CT103A infusion. The lesion was denoted by arrows in the left 2 images from the positron emission tomography/computed tomography scan before infusion. At day 98, after infusion, no lesion was observed in the 2 images on the right, which was nearly the same section as that on the positron emission tomography/computed tomography scan. (B) Change in the serum light chain levels in patient 01-001. After the murine BCMA CAR infusion, the serum light chain level decreased for a short time and soon recovered, and the PFS was 44 days. In comparison, after CT103A infusion, the light chain level quickly decreased and remained stable at a normal level with a normal κ/λ ratio, indicating continuous sCR. The PFS was 587 days at the cutoff date.
Patients treated with a prior murine BCMA CAR benefit from CT103A
Four patients who had previously received a murine BCMA CAR T-cell treatment and then relapsed or had PD were enrolled in this trial. After CT103A infusion, 3 patients achieved sCR and remained in the sCR state for at least 455 days, and patient 01-013, who had EMM and secondary plasma cell leukemia, achieved the best response of very good partial response and survived for 225 days (supplemental Table 6). The change in the serum light chain level of patient 01-001 is shown in Figure 4B. The best response ever achieved before his enrollment in this study was a partial response resulting from murine BCMA CAR T-cell therapy; the disease then quickly progressed, with a PFS of only 44 days. Impressively, the light chain level rapidly decreased and remained at a normal level with a normal κ/λ ratio post–CT103A infusion.
Humoral immunogenicity
ADA was tested in all patients before infusion and/or at serial follow-up visits postinfusion if possible. Four plasma samples from 3 patients were above the detection threshold in the screening assay, of which 2 samples from patient 01-016 were further validated as positive by confirmatory assay. The rest of the samples were all negative for ADA (Figure 5A). The titer of the 2 positive samples was 8.30 (day 90) and 126.64 (day 120), respectively. Notably, loss of CAR transgene occurred almost simultaneously with the emergence of ADA in this patient around day 90 (Figure 5B). However, the relapse was detected at day 347.
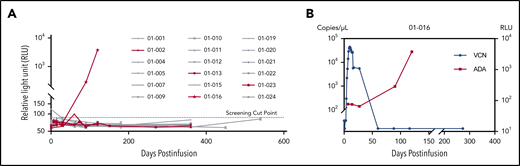
Detection of ADA. (A) Relative light unit (RLU) values of serial time points for all 18 patients in the screening assay. The four PD/relapse patients are shown in red, and others are in gray. The screening cut-point of RLU was derived by multiplying the median of negative control RLU values in each plate by the threshold factor 1.2, which was approximately 86.6, as shown in the dotted blue line. Four samples from 3 patients were putative positive and further validated in the confirmatory assay. (B) The relationship between ADA and CAR transgenes in patient 01-016. The left y-axis represented the viral copy number (VCN); the right y-axis represented the RLU.
Detection of ADA. (A) Relative light unit (RLU) values of serial time points for all 18 patients in the screening assay. The four PD/relapse patients are shown in red, and others are in gray. The screening cut-point of RLU was derived by multiplying the median of negative control RLU values in each plate by the threshold factor 1.2, which was approximately 86.6, as shown in the dotted blue line. Four samples from 3 patients were putative positive and further validated in the confirmatory assay. (B) The relationship between ADA and CAR transgenes in patient 01-016. The left y-axis represented the viral copy number (VCN); the right y-axis represented the RLU.
Adverse events
During the first 8 weeks’ postinfusion, a total of 73 different types of treatment-related AEs were recorded in all 18 patients, and those with an incidence ≥15% are presented in supplemental Table 7. All patients experienced grade 3 or higher AEs, most of which were hematologic toxicities, including leukopenia (18 of 18 [100%]), neutropenia (18 of 18 [100%]), lymphopenia (18 of 18 [100%]), anemia (16 of 18 [88.9%]), and thrombocytopenia (17 of 18 [94.4%]). These AEs are the expected toxic effects of lymphodepleting chemotherapy and CT103A infusions. Delayed recovery from cytopenia was observed. The median recovery times for the absolute neutrophil count (1.0 × 109 cells/L) and platelet count (50 × 109 cells/L) were 14.5 days (range, 8-58 days) and 38 days (range, 13-274 days) after infusion, respectively (supplemental Figure 12). No difference in cytopenia duration was observed between patients who had received previous murine BCMA CAR T-cell therapy and other patients (supplemental Figure 13). Other grade 3 or higher AEs were fibrinogenopenia, elevated aspartate aminotransferase, hyponatremia, fever, hypoxia, and hypotension. Recovery from all AEs except for hematologic toxicities was observed within 4 weeks.
The treatment-related SAEs recorded during the first 8 weeks’ postinfusion included CRS, coagulation disorder, hypoxemia, pleuritis, prolonged cytopenia, and pulmonary infection. Other SAEs that occurred during long-term follow-up (from 8 weeks’ postinfusion to cutoff date) included appendicitis, cellulitis, herpes zoster, pulmonary infection, and septic shock (Table 2; supplemental Table 8). In total, 94.4% (17 of 18) of all patients experienced CRS, of which 70.6% (12 of 17) were grades 1 and 2, 23.5% (4 of 17) were grade 3, and 5.9% (1 of 17) were grade 4. The median time to onset of CRS was 2 days (range, 0-7 days), and the median duration of CRS was 8 days (range, 1-19 days) (Figure 6A). The timing of tocilizumab and glucocorticoid treatment is also shown in Figure 6A. Subgroup analysis showed that severe CRS (grade 3 or higher) was only associated with the 6 × 106 CAR+ T cells/kg dose level (supplemental Figure 14). A significant increase in ferritin and interleukin-6 (IL-6) levels during the CRS phase was observed. The elevation of serum ferritin and IL-6 levels seemed parallel with the CRS grade, and the peak of IL-6 appeared slightly earlier than that of ferritin in most patients (Figure 6B; supplemental Figure 15). No ICANS was observed in any of the dose groups.

The timing and severity of CRS, CRS-related treatment, and increases in serum ferritin and IL-6 levels after CT103A infusion. (A) The timing of CRS onset, grades, and durations are shown in lines with different colors for each patient. Magenta and blue ticks indicate treatment of tocilizumab and steroids, respectively. (B) A heatmap of the serum ferritin and IL-6 levels in 18 patients in the first month after CT103A infusion. Each block is composed of cells that represent the serum ferritin or IL-6 level collected at baseline and serial time point after infusion. The color of each cell is determined by the value (log10) of the serum ferritin or IL-6 level from low (black) to high (red). The elevation of serum ferritin and IL-6 levels seem parallel with the CRS grade. The peak of IL-6 seems slightly earlier than that of ferritin in most patients.
The timing and severity of CRS, CRS-related treatment, and increases in serum ferritin and IL-6 levels after CT103A infusion. (A) The timing of CRS onset, grades, and durations are shown in lines with different colors for each patient. Magenta and blue ticks indicate treatment of tocilizumab and steroids, respectively. (B) A heatmap of the serum ferritin and IL-6 levels in 18 patients in the first month after CT103A infusion. Each block is composed of cells that represent the serum ferritin or IL-6 level collected at baseline and serial time point after infusion. The color of each cell is determined by the value (log10) of the serum ferritin or IL-6 level from low (black) to high (red). The elevation of serum ferritin and IL-6 levels seem parallel with the CRS grade. The peak of IL-6 seems slightly earlier than that of ferritin in most patients.
Toxicity among different dosage of CT103A
During dose escalation, all 3 patients had grade 3 or higher CRS at the 6 × 106 CAR+ T cells/kg dose level. The incidence of grade 3 or higher CRS was significantly higher in this dose group compared with the other 2 dose groups (supplemental Figure 16A). Specifically, patient 01-012 developed a grade 4 CRS, which was considered a DLT, at this dose level. Therefore, 3 additional patients were further enrolled in the second dose group (3.0 × 106 CAR+ T cells/kg), and 6 additional patients were enrolled in the first dose group (1.0 × 106 CAR+ T cells/kg) as the expansion cohort. No other DLT was observed, suggesting that a dose of ≤3.0 × 106 CAR+ T cells/kg was well tolerated in these subjects. Tocilizumab and glucocorticoids were applied in 61.1% (11 of 18) and 66.6% (12 of 18) of all patients, respectively (Figure 6A). The patients receiving a dose of ≤3.0 × 106 CAR+ T cells/kg required less treatment of CRS than the patients who received a dose of 6.0 × 106 CAR+ T cells/kg (supplemental Figure 16B). No dose-dependent effect could be observed among the 3 groups in terms of responses, PFS, or OS (supplemental Figure 9; supplemental Figure 17), possibly due to limited sample size. However, a lower dosage of CT103A seemed to have a better safety profile.
Pharmacokinetic parameters of CT103A
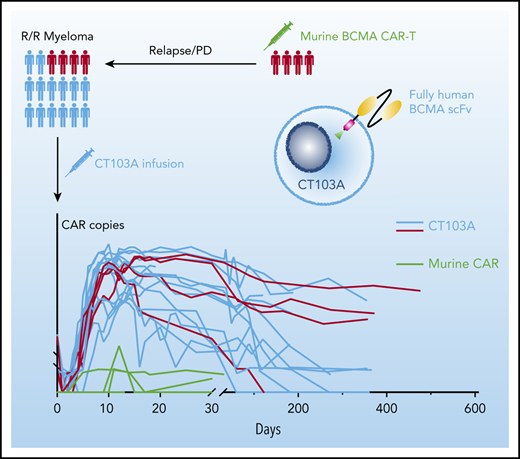
The CAR copies at serial points before and after infusion are shown in Figure 7A. After infusion, the median time to reach peak concentration was 12 days (range, 7-26 days), a strong indication of rapid CAR expansion. Values for the maximum plasma drug concentration (Cmax), time to reach peak concentration, and area under the curve from 0 to 28 days did not show significant differences among the 3 dosing groups (supplemental Figure 18). High expansion of CT103A, roughly represented by the Cmax and area under the curve from 0 to 28 days of the CAR transgene copies, was associated with the severity of CRS and CRS-related use of tocilizumab or glucocorticoid (supplemental Figure 19). CAR transgenes were not detectable only in 4 patients to the cutoff date. The median CAR T-cell persistence was 307.5 days (range, 20-587 days). No significant difference was observed in CAR T-cell expansion or durability of response based on baseline BCMA intensity on myeloma cells (supplemental Figure 20). For the 4 patients who had participated in prior murine BCMA CAR T-cell trials, 2 unique probes were designed to simultaneously monitor the expansion of CT103A and the previous murine CAR T cells. Low levels (<1000 copies/μg genomic DNA) of expansions of previously infused murine CAR T cells were observed (3 of 4 [patients 01-001, 01-007, and 01-013]) with short durations (<20 days) after CT103A infusion (Figure 7B).

Kinetics of CAR T cells after CT103A infusion. (A) Kinetics of the CAR T cells in the patients are shown by the CAR copies per microgram of genomic DNA (copies/μg gDNA) at serial time points post-infusion using digital droplet polymerase chain reaction. The data were obtained until the latest follow-up of each patient. The horizontal line denotes the lower limit of quantitation (LLQ: 20 CAR copies/μg gDNA). Red, light green, and blue lines represent patients receiving infusion doses of 1, 3, and 6 × 106 cells/kg, respectively. By the end of the last follow-up, expansion of CT103A was not detected in 4 patients only. (B) Four patients had relapsed from a prior murine BCMA CAR T-cell therapy. Copy numbers of CT103A and previous murine BCMA CARs were simultaneously monitored by digital droplet polymerase chain reaction in these patients. In each panel, the upper red curve is CT103A, and the lower light green curve is the murine BCMA CAR.
Kinetics of CAR T cells after CT103A infusion. (A) Kinetics of the CAR T cells in the patients are shown by the CAR copies per microgram of genomic DNA (copies/μg gDNA) at serial time points post-infusion using digital droplet polymerase chain reaction. The data were obtained until the latest follow-up of each patient. The horizontal line denotes the lower limit of quantitation (LLQ: 20 CAR copies/μg gDNA). Red, light green, and blue lines represent patients receiving infusion doses of 1, 3, and 6 × 106 cells/kg, respectively. By the end of the last follow-up, expansion of CT103A was not detected in 4 patients only. (B) Four patients had relapsed from a prior murine BCMA CAR T-cell therapy. Copy numbers of CT103A and previous murine BCMA CARs were simultaneously monitored by digital droplet polymerase chain reaction in these patients. In each panel, the upper red curve is CT103A, and the lower light green curve is the murine BCMA CAR.
Serum BCMA may serve as an indicator of response
The serum BCMA (sBCMA) levels were measured at screening, before infusion, and then regularly postinfusion. Compared with the baseline level, sBCMA levels showed a significant reduction at 1 month postinfusion, with a median clearance of 95.3% (range, 31.9%-98.5%). No significant difference was observed in the clearance of sBCMA 1 month postinfusion between patients who achieved very good partial response or better and patients who did not (median, 95.3% vs 95.1%; P = .297) (supplemental Figure 21A). Three patients with nonsecretory MM (patients 01-004, 01-021, and 01-022) (supplemental Table 2) had a similar clearance rate of sBCMA as the patients with secretory MM (median, 95.2% vs 95.3%; P = .912) (supplemental Figure 21B). For the patients with relapse or PD, either delayed clearance or an increase in sBCMA was observed (supplemental Figure 21C). These results indicate that sBCMA may be used as a biomarker for response monitoring, especially for patients with nonsecretory MM, who lack measurable M protein or light chain.
Discussion
Anti-BCMA CAR T-cell trials have already shown promising results in patients with RRMM.5-11 However, a relatively high incidence of relapse remains a significant challenge to anti-BCMA CAR T-cell therapies. The short persistence of CAR T cells in vivo may be one of the most important reasons for BCMA-positive relapse. Theoretically, fully human CAR might offer the advantage of reduced immunogenicity and therefore facilitate a better persistence of CAR T cells.27 The current study used a second-generation anti-BCMA CAR with a fully human component and reported the clinical trial of this product.
The impressive responses to CT103A include a median TTR of 15 days and 100% ORR, indicating highly efficient plasma cell elimination by CT103A. The median PFS was not reached at the median follow-up time of 394 days (∼13 months). The response time, rate, and durability seem to be comparable to the best results achieved from other published BCMA CAR T-cell trials using nonhuman CARs, in which the TTR was ∼1 month, the ORR was ∼33% to 88%,5,7-10 and the median PFS was ∼7 to 15 months.7-10 This was corroborated by swift and robust expansions of CT103A, as indicated by pharmacokinetic data from digital droplet polymerase chain reaction. In comparison, other studies reported a discrepancy in CAR transgenes between responders and nonresponders, in which the Cmax could differ by orders of magnitude.9-11 Given the relatively small variation of Cmax among subjects across all dose groups in the current study, a minimal dose of CT103A can be administered to obtain a better safety profile without compromising efficacy in future trials. Interestingly, slight expansions of previously infused murine CAR following lymphodepletion and CT103A infusion were observed, but the underlying mechanisms remain unclear. Given the minor expansions with short durations of murine CAR, it is unlikely that previously infused murine CAR would have significant antimyeloma activity to induce a sustained therapeutic response.
Several features of the trial, including scFv-binding properties, patient population, conditioning regimen, and immunogenicity, may contribute to the persistency of CT103A observed. Compared with the scFv used in another BCMA CAR T-cell trial,9 our fully human scFv may bind to BCMA slower, remain longer, and dissociate slower, as indicated by in vitro analysis (supplemental Table 1). Subtle differences in binder properties, in combination with other signals such as 4-1BB and CD3ζ incorporated in the CAR molecule, may lead to complex variations in antigenic signaling strength and duration.
Although all patients in the current study received at least 3 lines of prior therapies and were refractory to both bortezomib and lenalidomide, the cohort seems to be less heavily pretreated compared with those from other BCMA CAR T-cell trials,9,28 due to limited availability of certain drugs in China such as carfilzomib, pomalidomide, and daratumumab. The proportion of patients who previously received autologous hematopoietic stem cell transplantation was also relatively low but comparable to the average level in Chinese patients with MM.29,30 Such differences in patient populations may confer a favorable impact on the efficacies of CT103A.
The lymphodepletion regimen including a total of 60 mg/kg cyclophosphamide was adopted from previous trials of CD19-CAR T cells in patients with non-Hodgkin lymphoma and adult B-cell acute lymphoblastic leukemia.31,32 However, such a dose of cyclophosphamide was higher than that used in other BCMA CAR T-cell trials.7-11 To our knowledge, the optimal dose of cyclophosphamide for lymphodepletion prior to BCMA CAR T-cell therapy remains inconclusive. Speculatively, a higher dose of cyclophosphamide may offer potential benefits on response rate and CAR-T persistence by reduction of tumor burden and/or intensified lymphodepletion prior CAR-T infusion. To test this hypothesis, new clinical trials should be designed to directly compare lymphodepletion regimens with different doses of cyclophosphamide.
Several lines of evidence have indicated that CARs derived from murine scFvs can elicit cellular and humoral immune responses, which may cause elimination of certain CAR T cells.33,34 This potential immunogenicity against CAR T cells in vivo is marked by the very limited success in re-dosing of CAR-T products in various trials.35-37 Particularly, CAR-specific T-cell responses were detected in patients upon infusions of anti-CD19 CAR-T.31,38 However, the attempt to evaluate the levels of anti–CAR-T antibody yielded inconsistent results across different CAR-T trials. Xu et al10 reported that 85.7% (6 of 7) of PD/relapsed patients were ADA positive, which may be one of the major risk factors for PD/relapse of murine BCMA CAR T-cell therapies. In contrast, preexisting antimurine CD19-CAR antibodies detected in 84.8% of patients and treatment-related ADA did not affect the expansion or cellular kinetics of a murine anti-CD19 CAR-T, nor did preexisting antibodies affect response or relapse.39 Interestingly, in the current study, only 1 (5.6%) in 18 patients was ADA positive, and such emergence of ADA may be associated with the loss of CAR existence in the patient. Nevertheless, thorough evaluation including both cellular and humoral immunogenicity should be conducted in further studies to provide a rationale for re-dosing of CT103A. Notably, it was the first time that prior BCMA CAR-exposed patients were eligible to participate in an anti-BCMA CAR T-cell trial. The CT103A expansion did not seem to be influenced by prior murine BCMA CAR, indicating that CT103A may be able to bypass the potential immunogenicity induced by the previously infused CAR. In addition, because potential selection of escape mutations and alternative splicing of the target antigen have been reported in CD19-directed immunotherapy,40 whether the cognate BCMA epitope played a role in different responses for both BCMA-targeting CAR with different scFvs is yet to be determined.
Relapse after CAR T-cell therapy is common, including both antigen-negative and antigen-positive relapse. Tumor antigen loss usually results from clonal evolution/devolution41,42 and, in some cases, by other causes such as trogocytosis.43 The reason for antigen-positive relapse is more complicated, however, such as T-cell exhaustion and senescence, costimulatory domain selection, generation of ADA, and immune escape,31,44-47 which may cause poor persistency of CAR T cells. Notably, all PD/relapse cases in this study were EMM with multiple lesions, indicating that CT103A could only serve as a bridging rather than a definitive therapy in these patients. Other mechanisms may contribute to the short DOR in patients with EMM. First, extramedullary lesions are highly heterogenic, and thus tumor cells can more easily generate clones with escape mutations of BCMA.26 Second, the microenvironment of extramedullary lesions is relatively more “hostile” for the penetration and persistence of CAR T cells.14 Due to the limited sample size of the current trial, this phenomenon should be further validated in a larger cohort.
A relatively high rate of CRS was observed in the study. However, the majority of grade 3 or higher CRS cases (4 of 5) were rapidly relieved after conventional CRS treatment, including tocilizumab and steroids. Except for 1 patient (who required intensive care unit treatment and who died of possible pulmonary infection after mechanical ventilation to treat grade 4 CRS), there were no CRS-related fatal cases after CT103A infusion. The infection complications and prolonged cytopenia observed might be related to the higher dose of cyclophosphamide.
Notably, no ICANS was observed in the entire cohort. As a common AE of CAR T-cell therapy, ICANS was found in 1.8% to 42% of previous BCMA CAR T-cell trials.5,7-9,11 The incidence of ICANS in this study did not parallel that of CRS. The occurrence of ICANS was reported to be associated with specific cytokines, such as IL-6, IL-8, monocyte chemoattractant protein-1, and interferon inducible protein-10 in patients with acute lymphoblastic leukemia.48 The spectrum of cytokines activated by CT103A might be different from those in other CARs. The relatively high proportion of glucocorticoid usage to control CRS might be another reason for the low incidence of ICANS, as glucocorticoids are the most common treatment of ICANS.49 We also observed no association between glucocorticoid usage and the response to CT103A.
In conclusion, data from this phase 1 clinical study showed that CT103A is safe and highly active in patients with RRMM. At the lower dosage levels (1 and 3 × 106 CAR+ T cells/kg), CT103A remained active and effective, with minimal side effects. Notably, patients who relapsed after prior murine BCMA CAR T-cell therapy may still benefit from CT103A.
The data sets supporting the conclusions of this article are included within the article and additional files. Requests regarding original data may be submitted by contacting the corresponding authors via jfzhou@tjh.tjmu.edu.cn or cunrui5650@hust.edu.cn.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all the faculty and staff in the Clinical and Laboratory Unit of the Department of Hematology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, for their clinical and technical support.
This work was supported by funding from the National Natural Science Foundation of China (81873452, C.L.), the Key Program of the National Natural Science Foundation of China (81830008, J.Z.), and the Clinical Research Program of Huazhong University of Science and Technology affiliated Tongji Hospital (2020003, J.Z.).
Authorship
Contribution: J.Z., C.L., and L.Q. designed and supervised the clinical study; D.W., Jue Wang, G.H., W.W., and C.L. analyzed data, and wrote and revised the manuscript; G.H., W.W., Y.Y., and Z.Y. supervised the CAR T-cell production; G.H., W.W., and Y.Y. conducted preclinical validation and quality control; D.W., Jue Wang, H.C., L.J., Z.H., and X.Z. collected clinical data; D.W., Jue Wang, and Z.Y. performed statistical analyses; D.W., Jue Wang, Y.X., L.J., L.M., Z.H., and X.Z. enrolled patients and took care of the patients; and H.C., M.X., L.C., X.M., L.Z., and Jin Wang contributed to laboratory tests and response monitoring of the patients.
Conflict-of-interest disclosure: G.H., W.W., Y.Y., and Z.Y. are employees of Nanjing IASO Therapeutics Ltd. and held interests in the company. G.H., Y.Y., and J.Z. are among inventors of patent applications related to the CT103A. J.Z. is a nonpaid member of Scientific and Medical Advisory Board of Nanjing IASO Therapeutics Ltd. The remaining authors declare no competing financial interests.
Correspondence: Jianfeng Zhou, Department of Hematology, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, 1095 Jie-Fang Ave, Wuhan, Hubei, 430030, China; e-mail: jfzhou@tjh.tjmu.edu.cn; or Chunrui Li, Department of Hematology, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, 1095 Jie-Fang Ave, Wuhan, Hubei, 430030, China; e-mail: cunrui5650@hust.edu.cn; or Lugui Qiu, State Key Laboratory of Experimental Hematology, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Science & Peking Union Medical College, Tianjin 300020, China; e-mail: qiulg@ihacams.ac.cn.