

TO THE EDITOR:
Therapy-related myeloid neoplasms (TMNs) constitute one of the most challenging complications of cancer treatment.1 Although understanding of the pathogenesis of TMNs remains fragmentary, genomic studies in adults have thus far refuted the notion that TMNs simply result from cytotoxin-induced DNA damage.2-4 Analysis of the preclinical evolution of a limited number of adult TMNs have traced the majority of cases to clonal hematopoiesis (CH) that predates cytotoxic treatment and lacks the mutational footprint of genotoxic therapies.2-6 Balanced translocations, generally attributed to treatment with topoisomerase II inhibitors, are implicated in a minority of TMNs.1 TMNs are a leading cause of premature death in childhood cancer survivors and affect 7% to 11% of children treated for high-risk neuroblastoma and sarcoma.7,8 However, the origin of pediatric TMNs remains unclear. Targeted sequencing of known cancer genes detects CH in ∼4% of children after cytotoxic treatment,6,9 whereas CH is vanishingly rare in young individuals in the general population.10,11 Moreover, to our knowledge, no cases of childhood TMNs have been traced to pretreatment CH. In light of these observations, we asked whether a broader driver landscape had eluded targeted CH screens in pediatric cancer patients and/or whether therapy-induced mutagenesis may be an under-recognized catalyst of CH and TMN in this patient group.
As proof of concept, we first applied whole-genome sequencing and deep targeted sequencing of serial bone marrow and blood samples to investigate the pathogenesis of TMNs arising in 2 children after treatment for high-risk neuroblastoma. This study was approved by the National Health Service Research Ethics Service (reference 16/EE/0394). Patients’ guardians provided written informed consent. DNA extracted from blood, bone marrow, and tumors (supplemental Table 1, available on the Blood Web site) underwent whole-genome sequencing and/or targeted sequencing of hematologic cancer genes (supplemental Table 2). Mapping to the human reference genome GRCh37 and somatic variant calling were performed using an extensively validated pipeline.12 From somatic mutations, we reconstructed phylogenetic relationships between samples by using methods described previously.12 We assessed the signatures of base substitutions, as defined by their trinucleotide context, to search for evidence of therapy-related mutagenesis. Sequencing data are accessible at the European Genome Archive (EGAD00001006423, EGAD00001006424).
Patient 1 (PD31013), a 7-year-old girl, developed therapy-related myelodysplastic syndrome (t-MDS) 10 months after completing treatment for high-risk neuroblastoma (Figure 1A), which included induction (7 agents, including cisplatin and carboplatin) followed by myeloablative chemotherapy and autologous hematopoietic stem cell transplantation (HSCT).13 Persistent thrombocytopenia developed 6 months after treatment was completed. Bone marrow examination revealed t-MDS with del(7q) and monosomy 7 in 4 of 20 and 11 of 20 metaphases, respectively, and leukemogenic PTPN11 G503E and SETBP1 D868G variants. Repeat bone marrow assessment 4 months later identified a stable blast percentage alongside neuroblastoma relapse, to which the child succumbed.
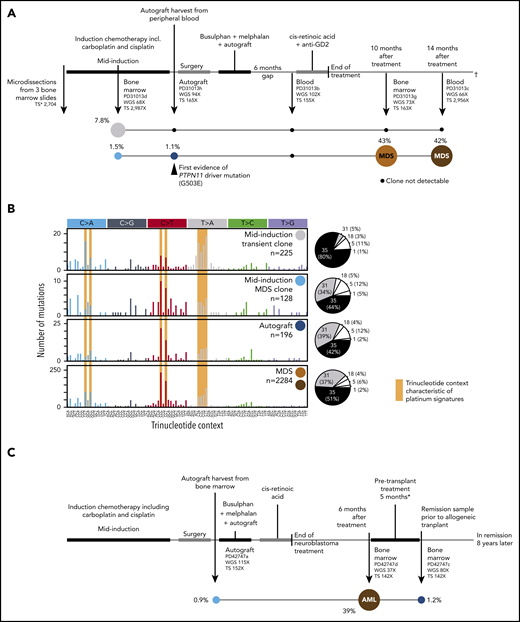
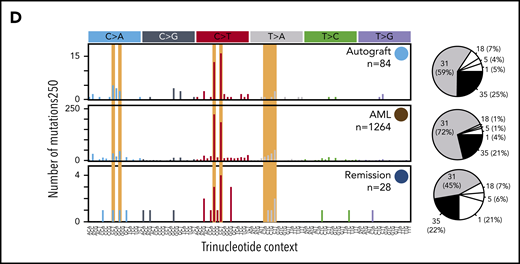
Development of CH and TMNs in patients 1 and 2. (A) The neuroblastoma treatment course for patient 1 in parallel with the emergence of CH (clone 1 in blue and clone 2 in gray) and the progression of clone 1 to t-MDS denoted in orange and red. Sequencing coverage (X) is indicated for whole-genome sequencing (WGS) and targeted sequencing (TS). For patient 1, targeted sequencing coverage is reported at the PTPN11 G503E locus. The median VAFs of mutations defining each clone are indicated as a percentage value next to their respective circle. Black circles indicate that no evidence (ie, no significant enrichment of mutant reads) of the clone in question was found at that time point. (B) The mutational spectra defining clone 2 for patient 1 (light blue and dark blue circles), clone 2 (gray circle) and t-MDS (red and orange circles) are defined by the number of single-base substitutions (SBSs, y-axis) per trinucleotide context (x-axis). SBS spectra characteristic of platinum agent-induced mutagenesis are shaded yellow. The pie charts to the right of each mutational spectra plot indicate that the majority of the SBSs at all time points are accounted for by platinum agent–associated mutational signatures (SBS31 and SBS35), with the small remainder of mutations attributed to clock-like mutational processes associated with ageing (SBS1 and SBS5) and oxidative stress (SBS18). (C) The neuroblastoma treatment timeline for patient 2 in parallel with the progression of CH (light blue circle) to AML (red circle), with persistence of residual CH after t-AML remission (dark blue circle). As in panel A, the median VAF of mutations defining each clone is indicated as a percentage value. *Treatment for t-AML before allogeneic transplant comprised cytarabine, daunorubicin, and etoposide (ADE) and fludarabine, high-dose cytarabine, idarubicin, and granulocyte colony-stimulating factor (FLAG-IDA) chemotherapy. (D) The mutational spectra defining clonal hematopoiesis and t-AML for patient 2 is shown in the same manner as that for patient 1 in panel B.
Development of CH and TMNs in patients 1 and 2. (A) The neuroblastoma treatment course for patient 1 in parallel with the emergence of CH (clone 1 in blue and clone 2 in gray) and the progression of clone 1 to t-MDS denoted in orange and red. Sequencing coverage (X) is indicated for whole-genome sequencing (WGS) and targeted sequencing (TS). For patient 1, targeted sequencing coverage is reported at the PTPN11 G503E locus. The median VAFs of mutations defining each clone are indicated as a percentage value next to their respective circle. Black circles indicate that no evidence (ie, no significant enrichment of mutant reads) of the clone in question was found at that time point. (B) The mutational spectra defining clone 2 for patient 1 (light blue and dark blue circles), clone 2 (gray circle) and t-MDS (red and orange circles) are defined by the number of single-base substitutions (SBSs, y-axis) per trinucleotide context (x-axis). SBS spectra characteristic of platinum agent-induced mutagenesis are shaded yellow. The pie charts to the right of each mutational spectra plot indicate that the majority of the SBSs at all time points are accounted for by platinum agent–associated mutational signatures (SBS31 and SBS35), with the small remainder of mutations attributed to clock-like mutational processes associated with ageing (SBS1 and SBS5) and oxidative stress (SBS18). (C) The neuroblastoma treatment timeline for patient 2 in parallel with the progression of CH (light blue circle) to AML (red circle), with persistence of residual CH after t-AML remission (dark blue circle). As in panel A, the median VAF of mutations defining each clone is indicated as a percentage value. *Treatment for t-AML before allogeneic transplant comprised cytarabine, daunorubicin, and etoposide (ADE) and fludarabine, high-dose cytarabine, idarubicin, and granulocyte colony-stimulating factor (FLAG-IDA) chemotherapy. (D) The mutational spectra defining clonal hematopoiesis and t-AML for patient 2 is shown in the same manner as that for patient 1 in panel B.
To reconstruct TMN development, we interrogated blood or bone marrow samples, including neuroblastoma-infiltrated marrow taken before treatment, from 7 time points from which we microdissected hematopoietic islands. The TMNs were characterized by an increased burden of point mutations (2284) compared with de novo pediatric acute myeloid leukemia (AML; median, 600) (supplemental Figure 1).14 Most TMN mutations (88%) were attributed to single-base substitution signatures 31 and 35, which have been closely linked to platinum chemotherapy exposure (Figure 1B).15-17 Similarly, doublet-base substitutions clearly exhibited the imprint of platinum agents (DBS5; supplemental Figure 2A).
We found the first evidence of a premalignant expansion during induction chemotherapy: a clone sharing 128 mutations with the TMN at a median variant allele frequency (VAF) of 1.5%. The majority (78%) of these mutations exhibited platinum signatures (Figure 1B). Targeted sequencing first detected the PTPN11 G503E variant in the autograft, but there was no evidence of the SETBP1 D868G mutation until t-MDS was diagnosed (supplemental Figure 3A). Of note, the sequence context of the founding PTPN11 driver mutation confers a 99% probability of this lesion arising as a result of platinum mutagenesis (supplemental Methods). However, because of the sensitivity limits of copy number variant calling (supplemental Methods), we cannot conclusively rule out the presence of del(7q) or monosomy 7 in the autograft.18 In addition to interrogating the loci of TMN mutations, we called somatic mutations independently in each sample. This analysis revealed a second clone, separate from the TMN lineage, within the mid-induction bone marrow (Figure 1A). The second clone comprised 225 substitutions with a remarkable median VAF of 7.8%, again predominantly attributed to exposure to a platinum agent (Figure 1B). This clone regressed after induction treatment.
Patient 2 (PD42747), a girl almost 4 years old, developed therapy-related AML (t-AML) 6 months after completing treatment for metastatic neuroblastoma (with a protocol similar to that used for patient 1; Figure 1C). The t-AML harbored a balanced KMT2A-MLLT1 translocation (supplemental Figure 3B), commonly attributed to topoisomerase II inhibitors such as etoposide, which she had received during induction.1 She remains in remission 8 years after allogeneic HSCT.
Samples from 3 time points were available for patient 2: autograft harvest, t-AML diagnosis, and t-AML remission. The t-AML harbored an elevated number of substitutions (1264) compared with de novo childhood AML,14 93% of which exhibited platinum signatures (Figure 1D; supplemental Figure 2B). We detected t-AML variants, although not the KMT2A fusion, in both autograft and remission bone marrow at median VAFs of 0.9% and 1.2%, respectively. There were no other clones in the autograft or t-AML remission sample.
As mentioned earlier, targeted sequencing of known cancer genes identified CH in ∼4% of children after cytotoxic treatment.6,9 The finding that both patients harbored at least 1 clone without a recognized driver event prompted us to extend our unbiased sequencing approach to 18 further pediatric patients with solid tumors for whom blood, parental blood (to assess inherited and de novo germline variants), and detailed clinical information were available. For 17 patients, we were also able to sequence tumor samples (supplemental Table 1). Included was the sister (PD31012) of patient 1. She had undergone treatment of low-risk infant neuroblastoma (3 cycles of etoposide and carboplatin only) and did not have CH. Germline analysis revealed that the sisters’ predisposition to cancer may have been attributable to an inherited germline pathogenic variant in BARD1 (c.1935_1954dup, p.Glu652Valfs*69),19 although neither had an exceptional burden of de novo germline mutations (Figure 2A).
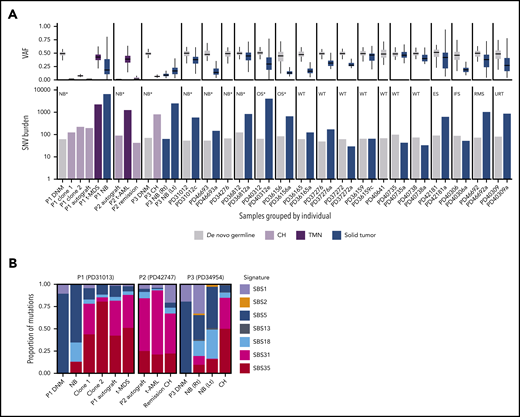
De novo germline and somatic mutations in CH, TMNs, and solid tumors in childhood cancer patients. (A) The VAF distribution (upper plot) and single nucleotide variant (SNV) burden (lower plot) of de novo germline mutations, solid tumors, CH, and TMNs in 20 pediatric oncology patients. Samples are grouped by individual. The patient’s solid tumor diagnosis is indicated in the upper left corner of the respective bar plot. Asterisks denote patients who had been exposed to platinum chemotherapy at the time of sampling. (B) Mutational signature profile of de novo germline, CH, TMN, and solid tumor mutation for the 3 individuals with clonal hematopoiesis, which demonstrates the preponderance of platinum-agent mutational signatures SBS31 and SBS35 in CH and TMN. DNM, de novo germline mutation; ES, Ewing sarcoma; IFS, infantile fibrosarcoma; Lt, left; NB, neuroblastoma; OS, osteosarcoma; RMS, rhabdomyosarcoma; Rt, right; URT, unclassifiable renal tumor; WT, Wilms tumor. Asterisk indicates previous exposure to platinum chemotherapy at the time of sampling.
De novo germline and somatic mutations in CH, TMNs, and solid tumors in childhood cancer patients. (A) The VAF distribution (upper plot) and single nucleotide variant (SNV) burden (lower plot) of de novo germline mutations, solid tumors, CH, and TMNs in 20 pediatric oncology patients. Samples are grouped by individual. The patient’s solid tumor diagnosis is indicated in the upper left corner of the respective bar plot. Asterisks denote patients who had been exposed to platinum chemotherapy at the time of sampling. (B) Mutational signature profile of de novo germline, CH, TMN, and solid tumor mutation for the 3 individuals with clonal hematopoiesis, which demonstrates the preponderance of platinum-agent mutational signatures SBS31 and SBS35 in CH and TMN. DNM, de novo germline mutation; ES, Ewing sarcoma; IFS, infantile fibrosarcoma; Lt, left; NB, neuroblastoma; OS, osteosarcoma; RMS, rhabdomyosarcoma; Rt, right; URT, unclassifiable renal tumor; WT, Wilms tumor. Asterisk indicates previous exposure to platinum chemotherapy at the time of sampling.
Analysis of the extension cohort revealed 1 instance of CH, again lacking a recognized oncogenic mutation, in a 4-year-old girl (patient 3, PD34954) who was treated for relapsed bilateral neuroblastoma (which ultimately proved fatal). This clone was defined by 810 substitutions (median VAF, 6.5%), 85% of which exhibited platinum signatures. Neither the 2 tumors from this child, nor the tumor from patient 1 bore evidence of platinum mutagenesis, despite previous exposure (Figure 2B). We did not detect any further cases of CH in the 6 other children exposed to platinum chemotherapy.
Collectively, our results reveal that the imprint of platinum-agent mutagenesis dominated all clones in the 3 children with detectable CH in marked contrast to CH observed in adults treated with these drugs.5,6 The reasons for this disparity are unclear. It is conceivable that the age of hematopoietic stem cells has an impact on susceptibility to mutagenesis or capacity to survive it and continue replicating. Furthermore, all 3 patients with CH harbored at least 1 clone without a known driver mutation, which corroborates evidence that knowledge of the somatic events under selective pressure is incomplete and that driverless CH cannot be accounted for by neutral drift alone.11,20,21
In the 2 patients with TMN, the preponderance of platinum signatures in nascent premalignant clones, high TMN mutation burdens, and 99% probability that the founding driver mutation in the tumor of patient 1 arose to therapy-related mutagenesis contrast sharply with findings in adult TMN.2-6 Together, these results suggest that the role of therapy-related mutagenesis in pediatric TMN may extend beyond the rare generation of balanced translocations linked to topoisomerase II inhibitors.1,22,23
The overall survival benefit of high-dose chemotherapy with autologous HSCT is unclear for several pediatric cancers, including neuroblastoma,24 and TMN is a leading cause of nonrelapse mortality for these patients.7,8 The only factor clearly associated with improved survival in childhood TMN is a shorter time between diagnosis and allogeneic HSCT.25 In both of our patients with TMN, preleukemic clones predated myeloablative treatment and pervaded the autograft. Pretransplant CH is emerging as a biomarker of the risk of TMN and nonrelapse mortality in adult autograft patients.26 This raises the possibility that early detection of pediatric CH, with or without known leukemogenic drivers, may inform personalized autograft decisions, enable earlier TMN diagnosis, and improve outcomes. Unbiased, systematic evaluation of the true frequency and prognostic implications of pediatric therapy-related CH is needed to determine any role for screening in clinical practice.
Sequencing data is accessible at the European Genome Archive (EGAD00001006423, EGAD00001006424). Code will be deposited on Github along with complete lists of variant calls.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the research nursing team and laboratory staff at Cambridge University Hospitals and Great Ormond Street Hospital (London, United Kingdom) and are indebted to the patients and their families for participating in this research.
This work was supported by fellowships from Sanger Institute core funding, Wellcome Trust (S.B., T.H.H.C., G.C., and E.M.), by the Robert J. Arceci International Award from St Baldrick’s Foundation (S.B.), by fellowships from the Biomedical Research Centre Great Ormond Street, Cambridge Human Research Tissue Bank, Oxford Biomedical Research Centre, National Institute for Health Research (T.R.W.O. and G.C.), and Great Ormond Street Hospital Children’s Charity (J.A.).
Authorship
Contribution: S.B., M.J.M., and G.C. designed the experiment; T.H.H.C., S.B., G.C., W.L., J.I., and E.M. analyzed the data; T.R., A.L.G., M.J.M., J.A., T.R.W.O., G.A.A.B., E.D., M.G., and J.N. curated samples, provided pathology input, and/or performed experiments; M.T. provided clinical genetics expertise; A.S. provided statistical input; S.B. and G.C. wrote the manuscript with input from all authors; and S.B. and M.J.M. directed the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sam Behjati, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridgeshire, CB10 1SA, United Kingdom; e-mail: sb31@sanger.ac.uk.

