Key Points
TET2 mutations, present in 34% of chronic lymphoproliferative disorders of NK cells, are associated with CD16low phenotype.
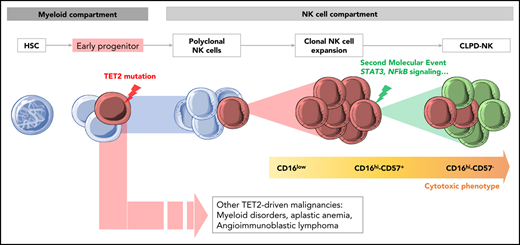
TET2 mutations can be an early event in the pathogenesis of CLPD-NK, explaining its association with other lymphoid or myeloid malignancies.

Abstract
Distinguishing chronic lymphoproliferative disorders of NK cells (CLPD-NK) from reactive NK-cell expansion is challenging. We assessed the value of killer immunoglobulin-like receptor(KIR) phenotyping and targeted high-throughput sequencing in a cohort of 114 consecutive patients with NK cell proliferation, retrospectively assigned to a CLPD-NK group (n = 46) and a reactive NK group (n = 68). We then developed an NK-cell clonality score combining flow cytometry and molecular profiling with a positive predictive value of 93%. STAT3 and TET2 mutations were respectively identified in 27% and 34% of the patients with CLPD-NK, constituting a new diagnostic hallmark for this disease. TET2-mutated CLPD-NK preferentially exhibited a CD16low phenotype, more frequently displayed a lower platelet count, and was associated with other hematologic malignancies such as myelodysplasia. To explore the mutational clonal hierarchy of CLPD-NK, we performed whole-exome sequencing of sorted, myeloid, T, and NK cells and found that TET2 mutations were shared by myeloid and NK cells in 3 of 4 cases. Thus, we hypothesized that TET2 alterations occur in early hematopoietic progenitors which could explain a potential link between CLPD-NK and myeloid malignancies. Finally, we analyzed the transcriptome by RNA sequencing of 7 CLPD-NK and evidenced 2 groups of patients. The first group displayed STAT3 mutations or SOCS3 methylation and overexpressed STAT3 target genes. The second group, including 2 TET2-mutated cases, significantly underexpressed genes known to be downregulated in angioimmunoblastic T-cell lymphoma. Our results provide new insights into the pathogenesis of NK-cell proliferative disorders and, potentially, new therapeutic opportunities.
Introduction
Within the spectrum of large granular lymphocyte (LGL) leukemia, chronic lymphoproliferative disorder of NK cells (CLPD-NK) was listed provisionally as an individual clinical entity in the latest (2016) World Health Organization classification.1 This disorder is characterized by the persistent expansion of mature NK cells with a typical LGL morphology and a restricted killer immunoglobulin-like receptor (KIR) pattern.2,,-5 Unlike T-LGL leukemias, which are characterized by a clonal T-cell receptor rearrangement, NK cell proliferations do not display T-cell receptor clonality, making the differential diagnosis between clonal CLPD-NK and reactive NK-cell expansion challenging.6,7 Indeed, after viral infection, autoimmune disease, or organ transplantation, some patients have an elevated circulating LGL count, which raises the question of a reactive or neoplastic origin and highlights the need for an unambiguous diagnosis.
Flow cytometry (FCM) analysis of NK receptors is the most appropriate tool for diagnosing malignant NK-cell proliferation.8 However, this technique is not widely available, and its results are sometimes difficult to interpret. Currently, STAT3 and STAT5B mutations, detected in ∼30% and 5% of cases of LGL leukemia, respectively, are considered to be the sole molecular diagnostic markers.9,-11 The exhaustive mutational profiling of CLPD-NK recently added new insights to the knowledge of this disease.12 Notably, genes involved in (1) other signaling pathways (MAPK and PI3K-AKT), (2) viral infections (DDX3X), (3) DNA repair (PAXIP1), or (4) epigenetic regulation (TET2, SETD1B), have been found to be altered in a cohort of 13 patients with CLPD-NK who were negative for STAT3 mutations.12 Mutations of TET2 have also been reported to be associated with myeloid malignancies in T-cell LGL.13 Thus, the diagnostic value and clinical impacts of TET2 mutations should be validated in a larger cohort of patients with CLPD-NK.
In this study, we retrospectively analyzed 114 consecutive patients with suspected CLPD-NK. We phenotyped NK receptors by FCM and performed targeted high-throughput sequencing (HTS) of a 76-gene panel designed for the diagnosis of hematologic malignancies.14 STAT3 and TET2 mutations were identified, respectively, in 27% and 34% of patients with CLPD-NK, constituting a new diagnostic hallmark for this disease.
To describe the involvement of TET2 alterations in the pathogenesis of CLPD-NK, we performed whole-exome sequencing (WES) on sorted myeloid and T- and NK-cell populations and compared transcriptomic profile and gene methylation status on tumor NK cells. TET2 mutations were always detected in the NK cell compartment, with a high allelic frequency, although some patients exhibited alterations in myeloid cells (probably as a result of clonal hematopoiesis). Finally, we proposed a practical algorithm to classify NK-cell proliferation.
Materials and methods
Patients
We included all consecutive patients with NK receptors phenotyped from January 2011 through January 2019 in our institution because of suspected CLPD-NK. Collected samples from 26 hospitals throughout France were obtained after approval by the local institutional review board. All patients gave their written consent to participate in the study, which was conducted in accordance with the Declaration of Helsinki. Samples were centralized in a single center for analyses. We extracted the patients’ clinical and biological data at the time of diagnosis. We retrospectively classified the patients, either in the CLPD-NK or reactive NK-cell proliferation group, based on the standard criteria for LGL leukemia.15 CLPD-NK was defined by a persistent level of >0.5 × 109/L of circulating LGLs with (1) an evocative clinical presentation (neutropenia, anemia, autoimmune disorder, and infections); (2) a proven clonality by flow cytometry; (3) a STAT3 mutation (Sanger sequencing when available); or (4) histological evidence of LGL involvement.
Flow cytometry analysis
FCM was performed prospectively as part of our routine workflow. All FCM data generated during the study were retrospectively reanalyzed, and gating strategies were cross-validated by 2 local expert reviewers. Ficoll-purified mononuclear cells were stained with the labeled antibodies listed in supplemental Table 1 (available on the Blood Web site). The NK cell count was calculated from the absolute lymphocyte count (determined with a routine hematologic analyzer) and the FCM-defined percentage of CD3−/CD56+ or CD16+ cells. Next, NK-cell receptor expression on NK cells was evaluated by using consecutive phycoerythrin-labeled CD158A, CD158B, NKB1, CD94, NKG2D, CD161, NKP44, NKP46, and NKP30 antibodies. Analyses were performed consecutively on the FC500 Navios (Beckman Coulter, Brea, CA), or Lyric (Becton Dickinson, Franklin Lake, NJ) flow cytometer. The proportion (percentage) of positive NK cells and the mean fluorescence intensity of each marker were calculated with Kaluza software (Beckman Coulter).
Targeted HTS
Using cell samples that had been frozen at diagnosis, we sequenced a panel dedicated to hematologic neoplasms and constituting 76 genes (supplemental Table 2), including a subset of genes recommended for T-/NK-lymphoid malignancies: STAT3, STAT5B, RHOA, PLCG1, JAK3, CD28, CARD11, DNMT3A, TET2, IDH1, and IDH2.14 Genomic DNA (gDNA) was extracted from Ficoll-isolated mononuclear cells with a Maxwell Rapid Sample Concentrator (Promega, Madison, WI) and quantified with a Quantus fluorimeter (Promega). Libraries were generated in duplicate by using the amplicon-based Access Array strategy (Fluidigm, San Francisco, CA) from 125 ng of DNA with Advanta NGS Library Prep reagents on an Access Array 48.48 Integrated Fluidic Circuit and sequenced on a NextSeq550 system (Illumina, San Diego, CA). Data were analyzed via an in-house bioinformatic pipeline (supplemental Method 1). For the whole cohort, the median depth of sequencing was 2735 reads by amplicon, ranging from 1362 to 6432 reads, ensuring a reliable identification of mutations with a variant allele frequency >2%. We evaluated copy number variations that affected the targeted genes with the CovCopCan tool.16
Sanger sequencing
To validate STAT3 and TET2 mutations, 100 ng of DNA was amplified by polymerase chain reaction (PCR) with AmpliTaq Gold Polymerase (Applied Biosystems, Foster City, CA) and the specific primers listed in supplemental Table 3. PCR products were purified and sequenced using the Big Dye Terminator kit on a 3130xl analyzer (Applied Biosystems).
Cell sorting of myeloid, T-cell, and NK-cell populations
Subsequent WES, transcriptomic and methylation analyses were performed on sorted myeloid, T, and NK cells. Ten million cells frozen in dimethyl sulfoxide were stained with 4′,6-diamidino-2-phenylindole, CD3-BUV395, CD33 and CD36-PerCP-Cy5.5, and CD56 and CD16-FITC. From live 4′,6-diamidino-2-phenylindole–negative cells, myeloid (SSChigh, CD33+or CD36+), T (SSClow, CD3+), and NK cells (CD16+ or CD56+ and CD3−, CD33−, and CD36−) were sorted on a BD FASCAria Fusion platform (Becton Dickinson; supplemental Figure 1). The purity of each cell subpopulation was measured with the same gating strategy. DNA and RNA extractions were performed with the AllPrep DNA/RNA Micro Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions.
WES
The genomic DNA (gDNA) was captured with the Twist Human Core Exome Enrichment System (Twist Bioscience, San Francisco, CA) without modification, except for library preparation, which was performed with NEBNext Ultra II kit (New England Biolabs, Ipswich, MA).17 Libraries were sequenced on an Illumina NovaSeq platform as paired-end 100-bp reads. Mutational analysis was performed with Mercury software (IntegraGen SA, Evry, France). Detailed methods and bioinformatic analyses are available in supplemental Method 2.
RNA-sequencing and bioinformatic analysis of tumor NK cells
Libraries were prepared with NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs) for the Illumina protocol, according to the supplier’s recommendations. RNA-sequencing (RNA-seq) was performed on paired-end 100-bp reads on an Illumina NovaSeq platform. Library preparation, sequencing, and bioinformatics (Galileo software) were performed with IntegraGen SA (details in supplemental Method 3).
Sodium bisulfite treatment and methylation-specific PCR
To detect methylation of selected genes, we used the quantitative multiplex methylation-specific PCR (MSP) method, as described previously.18 In brief, 500 ng of gDNA was sodium bisulfite–modified using EZ methylation-Gold Kit (Zymo Research, Irvine, CA). Then, 10 ng of converted gDNA was amplified using ZymoTaq PreMix (Zymo Research) and methylation- or nonmethylation-specific primer pairs (supplemental Table 3). After denaturation in a MasterCycler (Eppendorf, Hamburg, Germany), amplification was performed for 45 cycles before final extension. To test conversion efficiency and specificity of primers, universal methylated and nonmethylated human DNA standards were used to generate positive and negative controls. The analysis of the PCR products was performed on 2% agarose gels after staining with ClearSight DNA Stain (Euromedex, Souffelweyersheim, France).
5hmC dosage
The levels of gDNA-5hmC were measured with the Global 5-hmC DNA ELISA Kit (Active Motif, Carlsbad, CA). In brief, 20 ng of gDNA was fragmented by enzymatic digestion and then denatured to create single-stranded DNA (ssDNA). Ninety-six–well plates (Stripwell; Corning) were coated with a DNA-binding agent. After the addition of ssDNA to the coated wells, unbound DNA fragments were washed away, and the wells were treated with a primary antibody specific to 5-hydroxymethylcytosine (5hmC) and a secondary antibody conjugated to horseradish peroxidase. Bound hydroxymethylated fragments were quantified by spectrophotometry, and an absolute quantification was performed with concentrations of positive controls by the standard curve method. The amount of 5hmC (percentage) was calculated from 20 ng ssDNA samples.
Statistical analysis
Statistical analyses were performed with Prism software (version 7, GraphPad Inc, San Diego, CA). We used the Mann-Whitney U test to compare quantitative variables and Fisher’s exact test or the χ2 test to compare categorical variables. We divided the whole cohort into 2 groups: the training set, to build the NK-cell clonality score, and the validation set, to confirm the diagnostic value on individual patients. The optimal threshold for quantitative variables was defined with a receiver operating characteristic (ROC) curve. Parameters showing a significant difference between reactive NK and CLPD-NK cells were included as qualitative variables in a logistic regression model. We assigned a weight of 2 for the significant variable in the multivariate analysis and a weight of 1 for the parameter that was significantly different only in the univariate analysis. Sensitivity, specificity, and predictive positive and negative values were reported for each parameter, as well as for the NK-cell clonality score.
Results
Patients
From January 2011 through January 2019, 114 patients were included in the study. The median (interquartile range [IQR]) age was 68 years (57-77), and the male-to-female ratio was 1.47. All blood samples were analyzed with our workflow for suspected CLPD-NK. Molecular assessments and FCM were mainly performed in cases displaying hyperlymphocytosis with LGLs on a blood smear and neutropenia or autoimmune disease. Twenty-five patients (22%) had a past or concomitant malignant disease, 20 (18%) had an autoimmune disease, and 5 (4%) displayed skin symptoms or an elevated blood eosinophil count. Twenty-eight patients (26%) displayed neutropenia (ie, an absolute neutrophil count <1.5 × 109/L). Overall, the median (IQR) proportion of NK cells (as a percentage of lymphocytes) was 47.3% (37.5-61.1).
According to the standard criteria for LGL leukemia that were available at diagnosis, 46 patients were considered to have true CLPD-NK, and 68 were classified as having reactive NK-cell proliferation. Patients’ characteristics are summarized in Table 1. The 2 groups of patients did not differ significantly in age, hemoglobin level, and neutrophil counts. Platelet and NK-cell counts were, respectively, significantly lower and higher in patients with CLPD-NK cells than in those with reactive NK cells.
Flow cytometry characteristics of patients with CLPD-NK
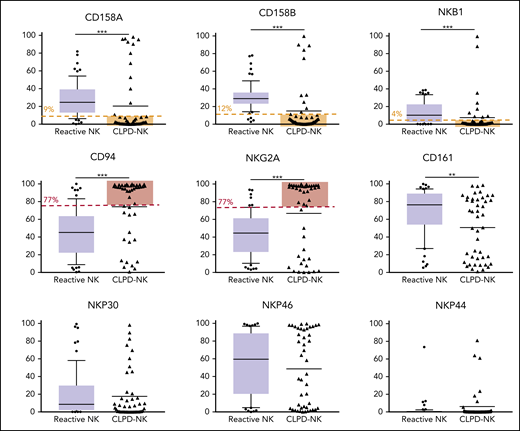
We classified the phenotype of NK cells in patients with CLPD-NK according to their cytolytic potential, by using a published classification.19 Among the patients with CLPD-NK, 24% had a true cytotoxic CD16high/CD57− profile, 53% an intermediate CD16high/CD57+ phenotype, and 22% a CD16low phenotype with low cytolytic potential. We evaluated the expression of 3 KIRs (ie, CD158A [KIR2DL1], CD158B [KIR2DL2/DL3], and NKB1 [KIR3DL1]) and CD94, NKG2A, and CD161 and 3 natural cytotoxicity receptors (NCRs; ie, NKP30 [NCR3], NKP46 [NCR1], and NKP44 [NCR2]) in the 114 patients (Figure 1). The expression of the 3 KIRs was significantly lower in the CLPD-NK group than in the reactive NK group. The expression of CD94 and NKG2A on NK cells was significantly higher in patients with CLPD-NK than in those with reactive NK cells: 94.6% vs 45.2% (P < .001) and 93.4% vs 44.5% (P < .001), respectively. The median percentage of CD161+ NK cells was significantly lower in the CLPD-NK group (63.3%) than in the reactive NK group (76.4%; P = .006). Conversely, the 2 groups did not differ significantly in the expression of NCRs.
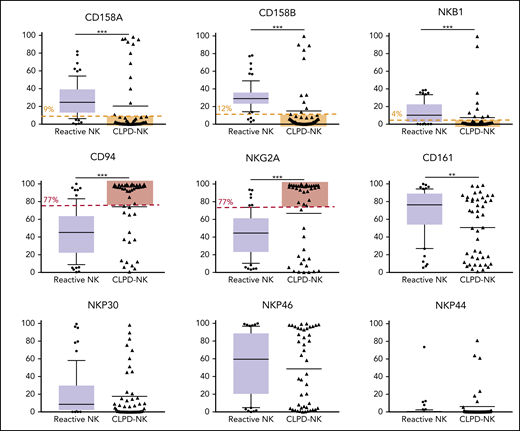
Flow cytometry analysis of NK receptors in the CLPD-NK and reactive NK groups. In the reactive NK group, the expression distribution of each FCM marker is represented by a box plot. The whiskers correspond to the 10th to 90th percentiles. Individual CLPD-NK samples are plotted as triangles. The optimal cutoff defined with the analysis of ROC curves for the NK-cell clonality score are plotted in red for overexpressed antigens or in yellow for underexpressed antigens. Comparisons were performed with the Mann-Whitney U test.
Flow cytometry analysis of NK receptors in the CLPD-NK and reactive NK groups. In the reactive NK group, the expression distribution of each FCM marker is represented by a box plot. The whiskers correspond to the 10th to 90th percentiles. Individual CLPD-NK samples are plotted as triangles. The optimal cutoff defined with the analysis of ROC curves for the NK-cell clonality score are plotted in red for overexpressed antigens or in yellow for underexpressed antigens. Comparisons were performed with the Mann-Whitney U test.
HTS profile
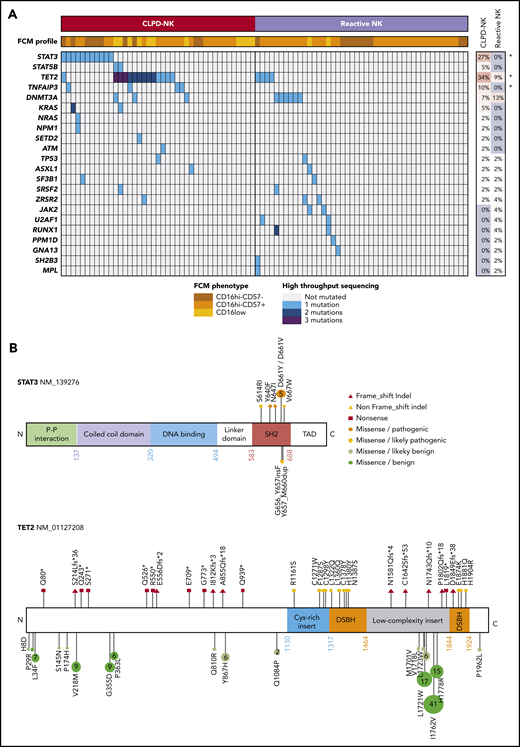
Eighty-six patients (41 with CLPD-NK and 45 with reactive NK-cell proliferation) were characterized by targeted HTS (Figure 2). Overall, we identified 85 clinically pertinent variants in 22 different genes (supplemental Table 4). In the CLPD-NK group, STAT3 was mutated in 11 patients (27%), STAT5 in 2 patients (5%), and TET2 in 14 patients (34%), with 26 different mutations (Figure 2A). Only 1 patient harbored a dual-mutation profile, TET2 and STAT3. Multiple mutations for TET2 were detected only in patients with CLPD-NK. TET2-truncating mutations were equally distributed across the whole gene; however, only missense mutations affecting the functional domains of the TET2 protein were considered to be clinically relevant (Figure 2B). Mutations in STAT3 and STAT5B were restricted to the SH2 domain. Both STAT3 and TET2 mutations with variant allele frequencies >10% were confirmed by Sanger sequencing (supplemental Figure 2). In the CLPD-NK group, we also detected mutations in TNFAIP3 in 4 patients (10%), DNMT3A in 3 patients (7%), and NRAS or KRAS in or 3 patients (7%). Of the patients with reactive NK-cell proliferation, 6 (13%) presented a DNMT3A mutation and 3 (7%) a TET2 mutation. Furthermore, we detected 19 mutations (5 in the CLPD-NK group and 14 in the reactive NK group) in genes described as clinically relevant in myeloid malignancies: JAK2, MPL, PPM1D, ASXL1, SF3B1, SRSF2, ZRSR2, RUNX1, U2AF1, and NPM1. No copy number variations were detected for any of the genes explored. Overall, mutations in the STAT3, TET2, and TNFAIP3 genes were significantly overrepresented in the CLPD-NK group, suggesting their clinical importance in the CLPD-NK diagnosis (Figure 2A).
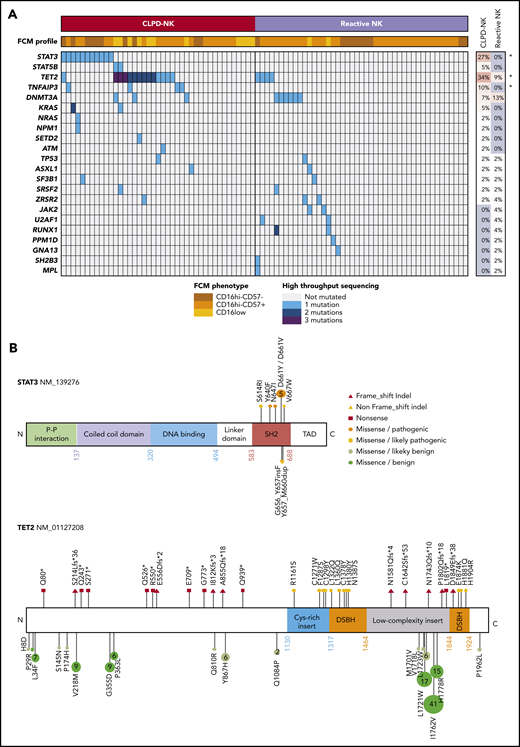
Mutational status of patients with CLPD-NK. (A) A heat map of the FCM profile and mutations detected by targeted HTS for the 41 patients with CLPD-NK and 45 with reactive NK cells. The percentage of mutations for each gene was compared by using Fisher’s exact test. *P < .05. (B) Lolliplots showing the distribution of STAT3 and TET2 mutations detected in this study, with their functional interpretation.
Mutational status of patients with CLPD-NK. (A) A heat map of the FCM profile and mutations detected by targeted HTS for the 41 patients with CLPD-NK and 45 with reactive NK cells. The percentage of mutations for each gene was compared by using Fisher’s exact test. *P < .05. (B) Lolliplots showing the distribution of STAT3 and TET2 mutations detected in this study, with their functional interpretation.
Clinical and phenotypical features of patients with CLPD-NK with TET2 mutations
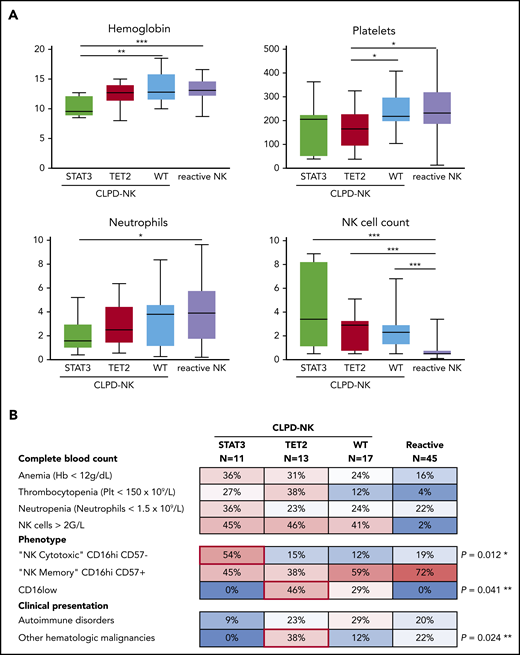
The CLPD-NK group was divided in 3 subgroups based on their molecular profile for STAT3 and TET2: (G1) STAT3-mutated (n = 11), (G2) TET2-mutated (n = 13), and (G3) wild-type (n = 17) (Figure 3). Patients with STAT3 mutations showed a significant decrease in hemoglobin concentration compared with the 2 other subgroups, with a median (IQR) level of 9.6 g/dL (8.8-12.2) vs 12.8 g/dL (11.5-14.5; P = .012). Moreover, the STAT3 mutation subgroup showed a lower neutrophil count than the reactive NK group: 1.6 × 109/L (range, 1.0-3.0) vs 3.9 × 109/L (range, 1.7-5.8), respectively (P = .049). Patients with TET2 mutations had significantly lower platelet counts (range) than did patients with wild-type or reactive NK-cell proliferation: 165 × 109/L (93-229) vs 218 × 109/L (195-300) and 232 × 109/L (184-322) (P = .047 and P = .014), respectively (Figure 3A). On the phenotypical level, 6 (54%) patients had STAT3 mutations with a cytotoxic CD16hi/CD57− profile, and only 4 (13%) patients in the 2 other subgroups (P = .012) had the same phenotype. Patients with TET2 mutations preferentially exhibited a CD16low phenotype (46% vs 17%; P = .041). Finally, patients with TET2 mutations presented another malignant hematologic disease more often than the other subgroups (38% vs 7%, P = .024; Figure 3B). Indeed, 5 patients with TET2 mutations developed either acute myeloid leukemia, myelodysplastic syndrome, pure red cell aplasia, angioimmunoblastic T-cell lymphoma (AITL), or Hodgkin lymphoma. In contrast, only 2 of 28 patients without TET2 mutations (7%) exhibited a second hematopoietic neoplasm (supplemental Table 5).
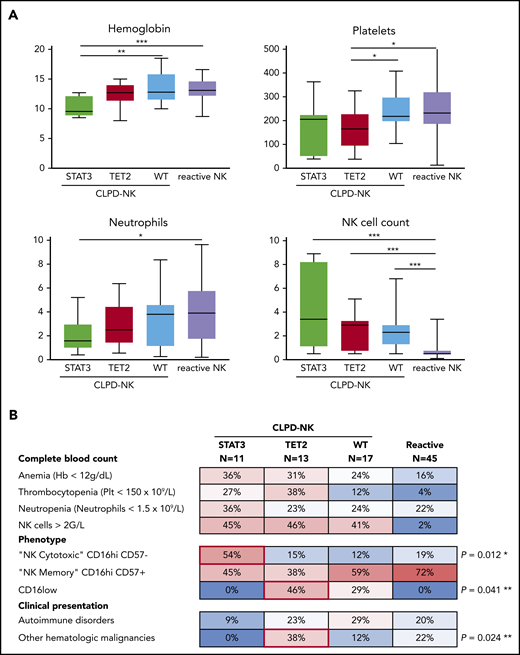
Clinical and phenotypical characteristics of patients according to mutational status. (A) Comparison of complete blood count parameters according to the mutational profile (Mann-Whitney U test). (B) A heat map of the prevalence (percentage) of cytopenia, phenotypical profile, and associated events in patients with CLPD-NK or reactive NK cell proliferation. *P for STAT3-mutated vs the other 2 CLPD-NK groups; **P for TET2-mutated vs the other 2 CLPD-NK groups comparisons (Fisher’s exact test).
Clinical and phenotypical characteristics of patients according to mutational status. (A) Comparison of complete blood count parameters according to the mutational profile (Mann-Whitney U test). (B) A heat map of the prevalence (percentage) of cytopenia, phenotypical profile, and associated events in patients with CLPD-NK or reactive NK cell proliferation. *P for STAT3-mutated vs the other 2 CLPD-NK groups; **P for TET2-mutated vs the other 2 CLPD-NK groups comparisons (Fisher’s exact test).
Establishment of an NK-cell clonality score
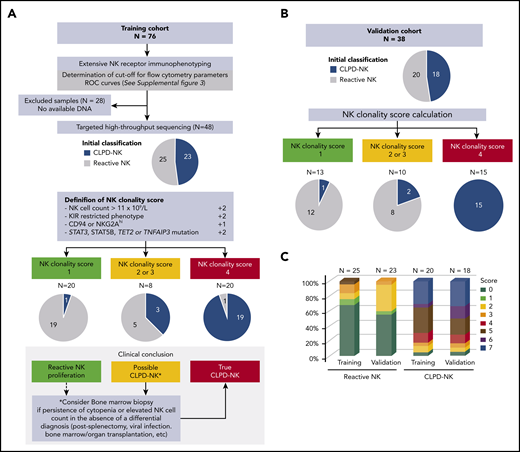
We established a novel NK-cell clonality score to diagnose CLPD-NK in the clinical practice (Figure 4). According to the data of the training cohort (n = 76), NK cell count and CD158A, CD158B, NKB1, CD94, and NKG2A expression levels discriminated CLPD-NK from reactive NK-cell proliferation with optimal cutoffs defined with the analysis of ROC curves. Instead, CD161 and NCR expression failed to separate these 2 entities (supplemental Figure 3). Among the 48 patients assessed with both FCM and HTS analyses, 4 criteria were significantly associated with the CLPD-NK diagnosis: (1) NK cell count >1 × 109/L; (2) a KIR-restricted pattern defined by a loss of expression of at least 2 KIRs (CD158A <9% of NK cells, CD158B <12%, and/or NKB1 <4%); (3) high CD94 or NKG2A expression (>77%), and (4) mutations in STAT3, STAT5B, TET2, and TNFAIP3 genes (Table 2). We further included these 4 criteria in a logistic regression model and attributed a weight of 2 points to parameters deemed significant in this multivariate analysis: NK cell count, the KIR-restricted phenotype, and somatic mutations (Table 2). We assigned a weight of 1 point to the CD94/NKG2Ahigh phenotype. This score ranged from 0 (lowest possible clonality) to 7 (highest possible clonality). We integrated the score into a dedicated diagnostic workflow for patients with clinical or laboratory data suggestive of CLPD-NK (Figure 4A). Twenty (42%) of 48 patients (19 CLPD-NKs and 1 reactive NK cells) had a score of ≥4. Eight (17%) patients (3 CLPD-NKs and 5 reactive NK cells) had a score of 2 or 3. Finally, 20 (42%) patients (1 CLPD-NKs and 19 reactive NK cells) had a score <2. Thus, a ≥4 had a sensitivity of 83% and a specificity of 96% for the diagnosis of CLPD-NK (Table 2). A score <2 could rule out the diagnosis of CLPD-NK with a negative predictive value of 95%. The diagnostic value of the NK-cell clonality score was confirmed in the independent validation cohort (n = 38), showing a positive predictive value of 100% (Figure 4B; Table 2). Between the reactive NK and CLPD-NK groups, the score value was equally distributed in the training and the validation sets (Figure 4C). Overall, bone marrow assessment was routinely performed for 14 patients and evidence an LGL infiltrate in 10 cases (supplemental Table 5). On the complete data set, we retrospectively reanalyzed the medical records of the 2 patients with an NK-cell clonality score less than 2 but who presented with confirmed CLPD-NK disease according to the standard criteria. The first case had inclusion-body myositis without circulating LGL; the second patient was diagnosed with diffuse large B-cell lymphoma and presented with an NK cell count of 0.7 × 109/L with a low level of CD161 as a unique FCM marker, which argued for a final classification in the reactive NK group. Only 1 patient had a score of 5, but he was initially classified in the reactive NK group because of an NK cell count at 0.4 × 109/L. However, he showed neutropenia with an autoimmune disorder, met both FCM criteria and carried a TET2 mutation.
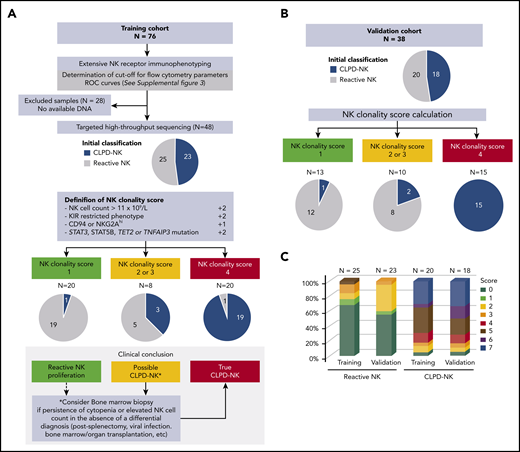
NK-cell clonality score. (A) Flowchart for NK-cell clonality score development based on the training set data (n = 76). The pie charts show the distributions of patients in CLPD-NK (blue) vs reactive NK group (gray) according to standard criteria. Comparison with final assessment according NK-cell clonality score. (B) Validation of NK-cell clonality score on an independent set of patients. (C) Distribution of the NK-cell clonality score between the 2 groups.
NK-cell clonality score. (A) Flowchart for NK-cell clonality score development based on the training set data (n = 76). The pie charts show the distributions of patients in CLPD-NK (blue) vs reactive NK group (gray) according to standard criteria. Comparison with final assessment according NK-cell clonality score. (B) Validation of NK-cell clonality score on an independent set of patients. (C) Distribution of the NK-cell clonality score between the 2 groups.
Clonal hierarchy in CLPD-NK
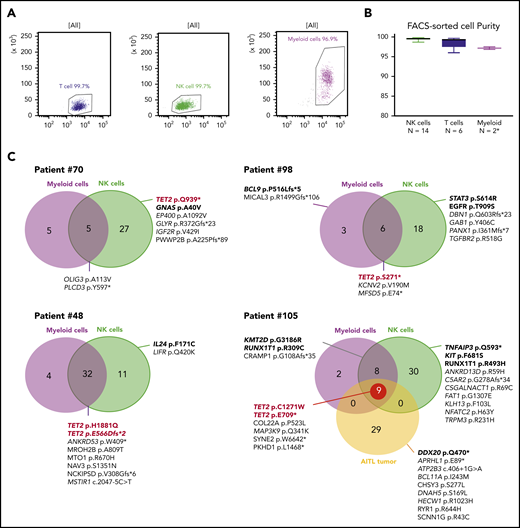
We next studied the clonal hierarchy in 6 patients with TET2 mutations by sequencing fluorescence-activated cell sorting–delineated myeloid and T-, and NK-cell compartments. The purity was higher than 96% in all tested subpopulations (Figure 5A-B). All TET2 mutations in NK cell samples were at a high allelic frequency. In 4 cases (67%), at least 1 TET2 mutation was also detected in myeloid cells, but not in T cells. We performed comparative WES for 4 patients who had sufficient DNA for all 3 compartments; patient 105 had a fourth sample issued from AITL diagnosed 2 years before the CLPD-NK (Figure 5C). Overall, we identified 189 different variants: 86 were restricted to NK cells, 60 were shared between myeloid and NK cells, 9 were restricted to myeloid compartments, and 29 were detected only in the AITL sample (supplemental Table 6). In patient 70, the TET2 mutation was present only in the NK cells but was associated with a GNAS mutation previously described as driving STAT3 pathway activation.20 In patients 98, 48, and 105, the 5 TET2 variants were identified in NK and myeloid cells. Their NK compartments had at least 1 activating mutation in genes involved in signaling pathways described in NK cell diseases: STAT3, TNFAIP3, and IL24. Patient 105 had 2 TET2 mutations in the NK and myeloid compartments that were also detectable 2 years earlier in the AITL tumor (Figure 5C).
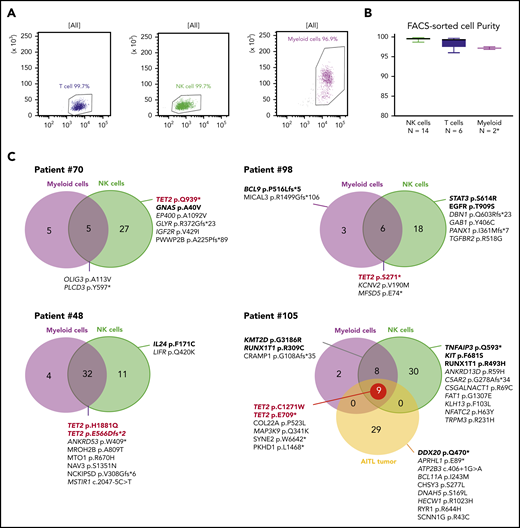
WES of sorted myeloid, T, and NK cells. (A) Evaluation by flow cytometry of the purity of sorted NK, T, and myeloid cells from 1 representative case (patient 105). (B) Purity evaluation of fluorescence activated cell sorting–delineated cells for all samples. *For 2 patients, the number of sorted myeloid cells was too low (<100 000) to perform purity control analysis. (C) WES results for 4 TET2-mutated patients. Venn diagrams represents the number of mutations in NK cells and myeloid cells as compared with the reference sample (T cell). Patient 105 analysis also includes a tumoral sample (AITL) collected 2 years before the occurrence of the CLPD-NK. Mentioned mutations correspond to pathogenic variants and/or variants related in OncoKB database.
WES of sorted myeloid, T, and NK cells. (A) Evaluation by flow cytometry of the purity of sorted NK, T, and myeloid cells from 1 representative case (patient 105). (B) Purity evaluation of fluorescence activated cell sorting–delineated cells for all samples. *For 2 patients, the number of sorted myeloid cells was too low (<100 000) to perform purity control analysis. (C) WES results for 4 TET2-mutated patients. Venn diagrams represents the number of mutations in NK cells and myeloid cells as compared with the reference sample (T cell). Patient 105 analysis also includes a tumoral sample (AITL) collected 2 years before the occurrence of the CLPD-NK. Mentioned mutations correspond to pathogenic variants and/or variants related in OncoKB database.
Methylation profile of CLPD-NK proliferations
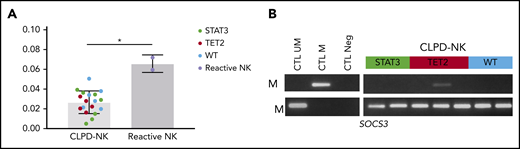
We evaluated the functional impact of TET2 enzyme loss of function. Thus, we measured the overall levels of 5hmC from sorted NK cells issued from 18 samples of CLPD-NK (7, STAT3; 5, TET2; and 6, wild-type) and 2 reactive NK samples. Overall, 5hmC levels were significantly decreased in CLPD-NK compared with reactive NK samples with a median of 0.034% (IQR .030-.039) and 0.066% (.060-.072), respectively (P = .036; Figure 6A). However, no detectable differences were found between the 3 subgroups of patients with CLPD-NK. CpG methylation of selected gene promoters involved in NK cells proliferations were explored for 7 patients with CLPD-NK (2, STAT3; 3, TET2; and 2, wild-type). For KIR2DL1, KIR2DL3, and KIR3DX1 genes, we detected strong methylation for all tested patients with CLPD-NK without specific modulations according to the patient molecular profile (data not shown). We found methylation in the SOCS3 gene in patient 70 (14%) who carried a TET2 mutation that was investigated by WES and RNA-seq (Figure 6B).
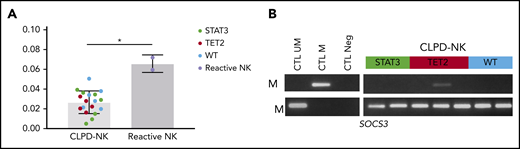
Methylation analysis of CLPD-NK according to molecular profile. (A) Comparison of 5hmC level in the CLPD-NK group vs the reactive NK one (Mann-Whitney U test). (B) Evaluation of SOCS3 methylation by MSP of 7 patients with CLPD-NK.
Methylation analysis of CLPD-NK according to molecular profile. (A) Comparison of 5hmC level in the CLPD-NK group vs the reactive NK one (Mann-Whitney U test). (B) Evaluation of SOCS3 methylation by MSP of 7 patients with CLPD-NK.
Transcriptomic profile of CLPD-NK proliferation
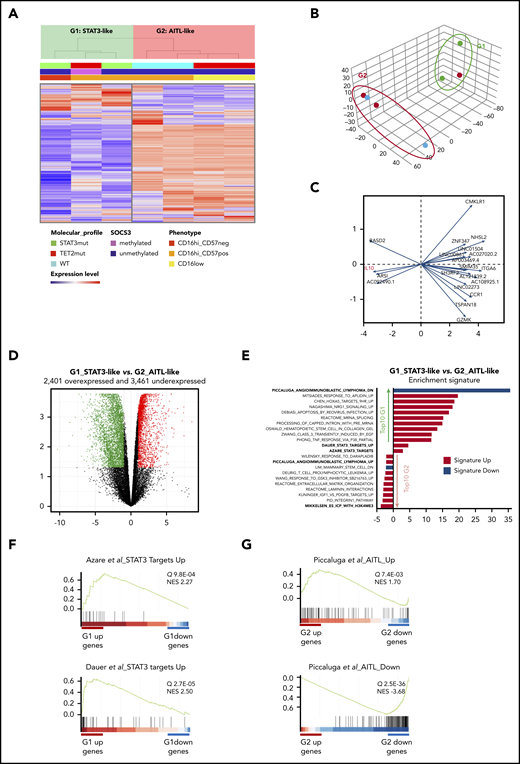
Finally, we studied the transcriptome by RNA-seq of blood-purified NK cells in the 7 patients with CLPD-NK. Both unsupervised clustering and principal component analysis (PCA) of the 1000 most variable genes of the data set identified 2 groups of patients (G1 and G2; Figure 7A-B). Expression of IL10 was highly discriminant between them (Figure 7C). We then examined differentially expressed genes between these 2 groups (G1 vs G2) and identified a set of 2410 overexpressed and 3604 underexpressed genes (fold change >2; q > 0.05; Figure 7D; supplemental Table 7). Gene Set Enrichment Analysis (GSEA) identified 899 and 68 curated signatures in G1 overexpressed and underexpressed genes, respectively (Figure 7E). Integrative analysis of the data showed that the 3 samples that constituted G1 involved the 2 cases with STAT3 mutation, whereas the third case (patient 70) was SOCS3 methylated and mutated for GNAS, both abnormalities are functionally related to a STAT3 pathway activation.20,21 Accordingly, 2 signatures related to STAT3 pathway activation were significantly enriched in this group (Dauer et al,22 P = 8.1E-07; Azare et al,23 P = 5.1E-05; Figure 7E-F). In contrast, among the G2-related signatures, 1 was the most striking and was connected to AITL (Figure 7G).24 Indeed, G2 underexpressed 70 genes that belong to a 131-gene signature called AITL-down (P = 5.8E-40) and overexpressed 35 genes involved in an AITL-up signature of 79 genes (P = 6.3E-04). The G2 group included 2 patients with TET2 mutations and showed lower expression of TET2 (FC 2.4; P = 1.4E-04; supplemental Table 7).
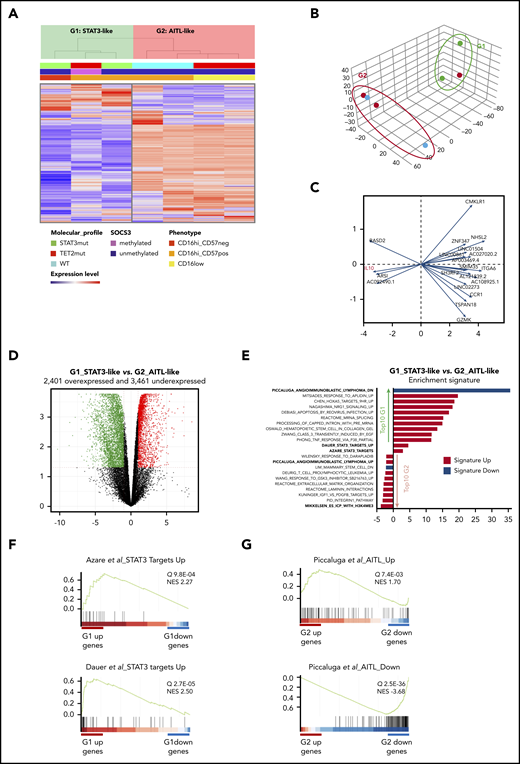
Transcriptional analysis by RNA-sequencing of 7 patients with CLPD-NK. (A) Hierarchical clustering (A) and PCA (B) performed on the 1000 most variable genes of the data set. Samples are clustered into 2 groups (G1 vs G2). (C) PCA gene contribution. (D) Volcano plot representing the significantly overexpressed (red) and underexpressed (green) gene in G1 vs G2 group (q > 0.05; fold change >2). (E) GSEA: STAT3-targets related signatures and top 10 overrepresented signatures among the differentially expressed genes in G1 vs G2, ranked by q value. (F) GSEA plot for STAT3 target signatures in G1. (G) GSEA plot for AITL signature in G2. NES, normalized enrichment score.
Transcriptional analysis by RNA-sequencing of 7 patients with CLPD-NK. (A) Hierarchical clustering (A) and PCA (B) performed on the 1000 most variable genes of the data set. Samples are clustered into 2 groups (G1 vs G2). (C) PCA gene contribution. (D) Volcano plot representing the significantly overexpressed (red) and underexpressed (green) gene in G1 vs G2 group (q > 0.05; fold change >2). (E) GSEA: STAT3-targets related signatures and top 10 overrepresented signatures among the differentially expressed genes in G1 vs G2, ranked by q value. (F) GSEA plot for STAT3 target signatures in G1. (G) GSEA plot for AITL signature in G2. NES, normalized enrichment score.
Discussion
The objective of the present study was to optimize the classification of NK-cell proliferation by leveraging data from NK-receptor phenotyping and HTS. Proven clonality is a mandatory diagnostic criterion for CLPD-NK.15 The human androgen receptor assay (evaluating the methylation pattern of the human androgen receptor gene on chromosome X) is the standard test for NK-cell clonality.25 However, the assay is not widely available in hospital laboratories and is only relevant in female patients. Thus, the FCM profile of NK receptors has been broadly accepted as a surrogate marker for NK-cell clonality.8 In our study, a restrictive KIR pattern, CD161 loss, and elevated expression of the lectin-type heterodimer CD94/NKG2A were the most specific markers for CLPD-NK. These data from a large real-life experience confirmed the published data on the diagnostic value of FCM for determining NK cell proliferation.26,,-29 Conversely, few studies have been reported on the diagnostic value of the natural cytotoxicity receptor in LGL leukemia.27 In our hands, this parameter did not allow for differentiation of CLPD-NK from reactive conditions, as it was similar in both groups and therefore did not constitute strong evidence of NK-cell clonality.
STAT3 mutation, found in up to 30% of the cases, is now considered to be a robust marker to distinguish CLPD-NK from NK reactive conditions.9,10,12 Recently, the mutational landscape of CLPD-NK has been characterized by WES and revealed in 2 STAT3-nonmutated cases the presence of TET2 mutations in a series of 13 patients.12 In the context of T-cell LGL leukemia, 2 reports described TET2 mutations related to clonal hematopoiesis of indeterminate potential.13,30 Our study of CLPD-NK confirmed the prevalence of STAT3 and STAT5B mutations (27% and 5%, respectively).10,11 We identified TET2 mutations in 34% of the cases, constituting a new hallmark of CLPD-NK. We detected additional mutations in genes involved in other signaling pathways, such as TNFAIP3, as previously reported in small series of cases of NK- and T-cell LGL leukemia.31,32 Overall, we found a strong molecular marker of CLPD-NK for 63% of the patients by using a targeting HTS strategy dedicated to hematologic malignancies. reserved
We propose that a combination of FCM and targeting HTS should be considered for the diagnosis of NK cell proliferation. We developed an NK-cell clonality score, integrating NK cell count, the 2 most discriminant phenotypic criteria (KIR restriction and elevated CD94/NKG2A expression), and the detection of at least 1 mutation in STAT3, STAT5B, TET2, and/or TNFAIP3. Assessing clonality with only FCM represents a complex, multiparametric procedure, available only at specialized centers. This score reduces the complexity of FCM interpretation by selecting the most relevant markers and providing optimal thresholds for the final decision. In addition, it includes a simple quantitative parameter (NK-cell count) and detection of mutations that provide objective evidence of clonality. Although the WES assay is readily available at present, targeted HTS is more widely used in routine practice and offers a higher sensitivity for detecting variants in subclonal populations in unsorted samples. In our cohort, a score of 4 or more allows for diagnosis of CLPD-NK with an excellent positive predictive value (>95%). Instead, patients with a score of 0 to 1 were almost exclusively diagnosed as having reactive NK-cell proliferation. We suggest that patients with a score of 2 or 3 fall within a gray area that justifies the performance of a bone marrow biopsy to screen for an interstitial infiltrate of granzyme B+, TIA1+ LGL.4,26,33 The value of this score was confirmed in an independent set of 38 patients and is currently used to validate patient inclusions in an ongoing French national prospective clinical trial (registered on www.clinicaltrials.gov as #NCT01976182).
Patients with CLPD-NK with STAT3 and TET2 mutations have distinct clinical and phenotypical patterns. Traditionally, mature NK cells are divided into 2 entities: (1) CD56hiCD16low regulatory NK cells with greater cytokine-producing ability and (2) CD56dim/CD16hi NK cells with greater cytotoxic function. In the latter, the acquisition of CD57 defines a subset of terminally differentiated memory NK cells.34,35 Consistent with previous studies, the most cytotoxic CD16hiCD57− profile was preferentially observed in STAT3-mutated CLPD-NK, characterized by deeper cytopenia compared with nonmutated cases.9,19,31 Conversely, we showed that patients with CLPD-ND, bearing the TET2 mutation, exhibited a regulatory CD16low phenotype with thrombocytopenia and more often had other hematologic malignancies in their clinical history.
These findings led us to question the clonal hierarchy of mutations in CLPD-NK. Currently, the CLPD-NK pathogenesis model postulates that a first event consisting of an expansion of polyclonal NK cells, induced by an exogenous stimulus, leads to the production of cytokines and then to the selection of a population of clonal NK cells. These expanded NK cells ultimately accumulate genetic alterations and cause clinical symptoms such as cytopenia and autoimmune disorders.36 We proposed an alternative scenario for cases with TET2 mutations. Indeed, our WES data showed TET2 mutations in NK and myeloid cell compartments whereas additional mutations, implicated in NK-cell biology (ie, STAT3, TNFAIP3, IL24, and GNAS), were restricted to NK cells. Our results suggest a multiple-hit model of leukemogenesis in which TET2 mutations can occur in early hematopoietic progenitors and give rise to different hematologic neoplasms. This scenario is somewhat illustrated by the patient who presented with AITL 2 years before CLPD-NK, a disease in which preexisting TET2-clonal hematopoiesis has been postulated.37,38 TET2 is one of the main genes involved in clonal hematopoiesis of intermediate potential.39 About 5% of the patients with LGL leukemia have concurrent myelodysplastic syndrome.7,13 These patients display a high prevalence of TET2, ASXL1, and DNMT3A mutations, suggesting that myelodysplasia and LGL leukemia arise from common clonal hematopoiesis of intermediate potential.38,40 Although NK-cell ontogeny has been considered to be exclusively lymphoid, several studies have shown that NK cells may originate from myeloid progenitors.41,-43 Specifically, common myeloid progenitors and granulocytic-monocytic precursors can give rise to mature NK cells in a cytokine environment supporting NK (IL-3, IL-15, IL-7, SCF, and FLT3-L).42 In accordance with this observation, we hypothesized that the occurrence of TET2 mutations in these progenitors could ultimately lead to an expansion of myeloid and NK cells, highlighting the epidemiological link between CLPD-NK and myeloid malignancies.
The high prevalence of TET2 mutations associated with the detection of multiple truncating or functionally damaging variants in the same sample (presumably associated with biallelic inactivation) suggests that TET2 loss of function could lead to the emergence of CLPD-NK. TET2 induces an oxidation of 5-methylcytosine (5mC) into 5hmC in the first step of an active DNA demethylation process.44 Accordingly, we found a significant decrease in 5hmC levels in CLPD-NK, in accordance with previous studies in other hematologic malignancies.45 Because of the paucity of samples, we could not observe any correlations between 5hmC residues and mutation profile of CLPD-NK as was done in acute myeloid leukemia.45 Furthermore, we did not detect methylation modifications of CpG islands in KIR gene promoters according to the mutation profile, whereas such an element was considered to be a crucial event in the clonal selection of NK cells.46,47 We could speculate that our investigations, limited to some regions because of the MSP approach, do not allow for a complete identification of methylation variations of KIR genes. However, we found a methylation of SOCS3 promoter in 1 patient with a TET2 mutation. SOCS3 is an important regulator of the STAT3 pathway, involved in a negative feedback loop.48 Thus, the epigenetic SOCS3 downregulation may enhance the STAT3 pathway activation demonstrated by our RNA-seq analysis, in agreement with a previous report in T-LGL leukemia.21
The transcriptomic analysis of clonal NK cells issued from CLPD-NKs sheds new light on the pathogenesis of CLPD-NK. Two different groups of patients were identified. The first one, called STAT3-like, was linked to a strong imprint of the STAT3 pathway, whereas the second group, linked to TET2 loss of function, displayed an AITL-like signature.24 These data open up therapeutic perspectives for CLPD-NK. Currently, immunosuppressive therapy is the standard treatment of LGL leukemia,15 but complete response is rare and long-term remission almost nonexistent, regardless of the patient’s mutation status. Thus, CLPD-NK represents an unmet clinical need, and new therapeutic options are necessary for relapsing or treatment-refractory cases. A better understanding of the molecular features of T- or NK- LGL leukemia is likely to accelerate the development of targeted therapies. Specific inhibitors of the JAK-STAT pathway should be preferred for patients with STAT3 mutation or STAT3-like patients. Although the specific JAK3 inhibitor tofacitinib has shown good clinical efficacy, this drug is currently authorized only for patients who have rheumatoid arthritis.49 Chromosomal instability and DNA hypermethylation have been observed in NK cells from the IL-15 transgenic mouse model, suggesting that proteasome inhibitors and demethylating agents may be efficacious for CLPD-NK.50 Hence, 5-azacytidine (already evaluated in the context of AITL) should be tested in cases of TET2 mutation or in AITL-like patients.51
In summary, we confirm that TET2 mutation is a new molecular marker and a potential therapeutic target for CLPD-NK. Our results also provide new insights into the pathogenesis and classification of NK-cell proliferative disorders.
The RNA-seq data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE163093).
For original data, please contact Thierry Lamy at thierry.lamy@univ-rennes1.fr.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgments
The authors thank the Biological Resource Center (Centre de Ressource Biologique-Santé; Rennes, France) for providing biological samples (BB-0033-00056), Cécile Thomas de la Pintière for technical support, and David Fraser (Biotech Communication SARL, Ploudalmézeau, France) for copy-editing support.
This project was funded in part by the Hematology Laboratory at Rennes University Medical Center and the Association pour le Développement de l’Hématologie Oncologie (ADHO).
Authorship
Contribution: C. Pastoret, M.R., T.F., and T.L. designed the study; G.D. and T.L. reviewed the clinical files for initial classification; M.R. and C. Pastoret analyzed the flow cytometry data; S.L.G. performed cell sorting; C. Pastoret and M.-L.B. generated and analyzed the high-throughput sequencing and RNA-seq data; F.D. designed and analyzed methylation experiments; C. Pangault managed the sample collection; G.D., A.T., A.-V.D., V.S., G.L.D., R.V.-M., O.T., A.M., and T.L. provided clinical information; C. Pastoret and T.F. wrote the manuscript; and all authors critically revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thierry Fest, Department of Hematology, CHU Pontchaillou, 2 Rue Henri Le Guilloux, F-35033 Rennes Cedex 9, France; e-mail: thierry.fest@univ-rennes1.fr; and Thierry Lamy, Department of Hematology, CHU Pontchaillou, 2 Rue Henri Le Guilloux, F-35033 Rennes Cedex 9, France; e-mail: thierry.lamy@univ-rennes1.fr.