Key Points
VWF fragments containing the D3 domain enabled FVIII passage from the subcutaneous space into vascular circulation.
Slow absorption from the subcutaneous depot prolonged the apparent FVIII half-life and prolonged protection from traumatic bleeds.

Abstract
Conventional treatment of hemophilia A (HA) requires repetitive IV injection of coagulation factor VIII (FVIII). Subcutaneous administration of FVIII is inefficient because of binding to the extravascular matrix, in particular to phospholipids (PLs), and subsequent proteolysis. To overcome this, recombinant dimeric fragments of von Willebrand factor (VWF) containing the FVIII-stabilizing D3 domain were engineered. Two fragments, called VWF-12 and VWF-13, demonstrated high binding affinity to recombinant human FVIII (rhFVIII) and suppressed PL binding in a dose-dependent manner. High concentrations of VWF fragments did not interfere with the functional properties of full-length VWF in vitro. The HA mouse model was used to study the effects of VWF-12 or VWF-13 on the in vivo pharmacokinetics of rhFVIII, demonstrating (1) no significant impact on rhFVIII recovery or half-life after a single IV administration; (2) enhanced bioavailability (up to 18.5%) of rhFVIII after subcutaneous administration; and (3) slow absorption (peak concentration, 6 hours) and prolonged half-life (up to 2.5-fold) of rhFVIII after subcutaneous administration. Formation of anti-FVIII antibodies was not increased after administration of rhFVIII/VWF-12 subcutaneously compared with rhFVIII IV. A single subcutaneous dose of rhFVIII/VWF-12 provided protection in the HA tail-bleeding model for up to 24 hours. In summary, recombinant VWF fragments support FVIII delivery through the subcutaneous space into vascular circulation without interfering with VWF or FVIII function. Slow resorption and excretion of FVIII after subcutaneous administration highlight the potential application of VWF fragments for subcutaneous FVIII prophylaxis in HA.
Introduction
Patients with congenital hemophilia A (HA) are usually treated with IV injections of coagulation factor VIII (FVIII) to prevent spontaneous bleeding and to restore hemostasis in case of bleeding. Conventional prophylaxis with standard plasma-derived or recombinant FVIII concentrates involves administration 2 to 3 times per week, due to the short FVIII half-life of ∼12 hours.1 These frequent IV injections represent an enormous burden for patients, in particular for infants, small children, and people with poor venous access. FVIII treatment by other administration routes could potentially improve HA management and quality of life for affected patients.
Therapeutic proteins enter the systemic circulation after subcutaneous administration either directly, by diffusion into blood capillaries (small molecular weight [MW] proteins ≤16 kDa), or indirectly through lymphatic vessels (high MW proteins >16 kDa).2,3 Low bioavailability can be due to restricted movement through the extracellular space, depending on molecular size and charge, enhanced clearance by proteolytic enzymes, or properties of excipients.4
Subcutaneous administration of FVIII, a large phospholipid (PL)-binding protein of 170 to 250 kDa, results in low absorption rates of <2% in F8−/− mice.5-7 Fatouros and colleagues reported that binding to PL membranes and the subsequent proteolytic cleavage contributed to the low FVIII bioavailability.8 von Willebrand factor (VWF) is known to protect FVIII from PL binding and proteolysis in the circulation.8,9 This protective capacity is linked to specific binding of the VWF D3 domain to the C1 domain of FVIII.10,11 However, coadministration of full-length VWF (fl-VWF) with FVIII is unlikely to increase FVIII bioavailability after subcutaneous administration due to binding of fl-VWF to extracellular matrix (ECM). Subcutaneous administration of plasma-derived VWF does not lead to significant absorption into the circulation in a mouse model.7
As an approach to overcome low FVIII bioavailability after subcutaneous administration, small recombinant VWF fragments were engineered to protect FVIII from PL binding and improve FVIII absorption into vascular circulation. Here, we characterize 2 VWF fragments in vitro and examine their effects on the in vivo pharmacokinetic (PK) and pharmacodynamic (PD) properties of recombinant human FVIII (rhFVII) in the HA mouse model.
Methods
Cloning, expression, and purification of recombinant VWF fragments and rhFVIII
Genes encoding VWF sequences were synthesized by GeneArt (Thermo Fisher Scientific, Darmstadt, Germany) and cloned into the pDSG expression vector (IBA Lifesciences, Göttingen, Germany), containing a twin Strep-tag. The VWF-12 construct contained amino acids 1 to 1268 of the VWF sequence fused to a 31-aa sequence (aa 1238-1268) repeated twice at the C terminus. The VWF-13 construct contained amino acids 1 to 1905 of the VWF sequence. Both constructs included the VWF propeptide to ensure dimerization. VWF fragments were transiently expressed in HEK293 cells and purified by Strep-tactin affinity chromatography (IBA GmbH, Göttingen, Germany). Typical yields of transient expression for VWF-12 and VWF-13 were 3.6 and 18.5 mg/L cell culture supernatant, respectively. The molecular weights of VWF-12 and VWF-13 were determined by SDS-PAGE. The rhFVIII used in this study was simoctocog alfa (Octapharma AG, Switzerland).
Surface plasmon resonance analysis
Binding of VWF-12 and VWF-13 to rhFVIII was measured using BIACORE 3000 (GE Healthcare, Uppsala, Sweden) according to Sandberg et al.12 In brief, the VWF fragments or fl-VWF (Sekisui Diagnostics, Maidstone, United Kingdom) were coated on a CM5 chip, and rhFVIII was injected over the surface. The running and binding buffer was 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 150 mM NaCl, 5 mM CaCl2 and the regeneration was performed using 20 mM HEPES, 150 mM NaCl, 350 mM CaCl2.
Interaction between rhFVIII and PL monolayer was measured on a hydrophobic association (HPA) chip. Preparation of PL vesicles and coating on a HPA sensor chip were performed according to Cooper et al.13 In brief, 1,2-dioleoyl-sn-glycerol-3-phospho-l-serine (DOPS) (Sigma-Aldrich, Darmstadt, Germany), sodium salt and 1,2-dioleoyl-sn-glycerol-3-phosphocholine (DOPC) (Sigma-Aldrich, Darmstadt, Germany) dissolved in CH2Cl2 were mixed in a 1:3 (w/w) ratio, dried with N2 and dissolved in HEPES buffered saline (HBS; Applichem, Darmstadt, Germany). After 10 minutes of sonication, the mixture underwent 10 freeze/thaw cycles in liquid nitrogen. Small unilammelar vesicles were prepared by extrusion, ie, the liquid was passed 20 times through a 100 nm polycarbonate membrane. The chip was coated by subsequent injections of H2O, octylglucoside and PL vesicles or bovine serum albumin (BSA; Sigma-Aldrich) for the reference flow cell. After rhFVIII binding, the surface was regenerated by NaOH injection.
Partial de-O-glycosylation of VWF-12
Five hundred micrograms of VWF-12 were digested with 200 U of α2-3,6,8,9 neuraminidase A and 400 000 U of O-glycosidase (New England Biolabs, Frankfurt, Germany) in 50 mM sodium phosphate buffer overnight at 37°C (VWF-12deOglyco). For control, VWF-12 was incubated in buffer without enzymes (VWF-12 control). Samples were purified using affinity chromatography with immobilized anti-VWF nanobody (Thermo Fischer Scientific) and size exclusion chromatography (NAP-5 columns; GE Healthcare) followed by concentration with centrifugal filter device (Amicon Ultra-4, PLGC Ultracel-PL Membran, 10 kDa; Merck, Darmstadt, Germany). The concentration of the sample was determined by micro-BCA protein assay kit (Thermo Fisher Scientific).
ECM binding and proteolytic degradation
Cell culture treated 96-well plates (TPP; Trasadingen, Switzerland) were coated with 64 µL per well Cultrex Basement Membrane Extracts (R&D Systems, Wiesbaden, Germany) consisting of laminin I, type IV collagen, entactin, and heparan sulfate proteoglycan at 37°C for 1 hour. After 1 hour of blocking with 0.4% bovine serum albumin (BSA) in 5 mM HEPES, pH 7.4, 140 mM NaCl, 5 mM CaCl2, 2 mM MgCl2, samples of 50 µL containing rhFVIII alone or with VWF fragments were incubated at 37°C for 1 hour. FVIII concentration was measured in supernatant using FVIII antigen (FVIII:Ag) ELISA (see "FVIII assays").
Human neutrophil elastase (Sigma-Aldrich) was used at concentrations of 0.3 or 22 nM to digest 100 nM rhFVIII alone or in the presence of VWF fragments or BSA in a 1:3 molar ratio for 1 hour at room temperature (RT). rhFVIII was detected after sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining or western blotting onto nitrocellulose membrane and staining with sheep anti-FVIII antibody (SAF8C-HRP; Affinity Biologicals, Ancaster, ON, Canada).
Animals
Fully inbred, genetically modified F8−/− mice (B6; 129S4-F8tm2Kaz) were used in this study.14,15 Genotype was confirmed by polymerase chain reaction (PCR) using DNA isolated from stool pellets.16 All mice were aged 10 ± 2 weeks at the beginning of the experiments. Mice were bred and housed in the animal housing facility at Hannover Medical School. Studies were carried out in accordance to the German animal welfare act and were approved by the regulatory authority.
Pharmacokinetics
PK profiles of rhFVIII with or without VWF fragments were investigated after a single dose of 200 IU/kg (IV) or 1000 IU/kg (subcutaneous). rhFVIII (0.59 nmol/1000 IU) was mixed in a 1:3.1 ratio with VWF-12 (1.83 nmol or 291 µg per 1000 IU rhFVIII), VWF-13 (1.83 nmol or 549 µg/1000 IU rhFVIII), or control buffer. The 1:3.1 ratio corresponds to 6.2 binding sites in the dimeric fragments per rhFVIII molecule. Samples were obtained before and at 1, 3, 6, 9, 24, 32, or 48 hours after injection by retro-orbital puncture of 5 mice for each of the 8 time points (ie, 40 mice per group, because each mouse was sampled only once to exclude an effect of sampling on rhFVIII PK). Blood was collected using a mini capillary (Minivette POCT 100 µL; Sarstedt AG & Co KG, Nümbrecht, Germany) containing sodium citrate 3.2% (1:10) (Sarstedt). Plasma was separated by centrifugation for 15 minutes at 2000g and stored at −75°C.
FVIII assays
FVIII chromogenic assay (FVIII:C) was performed using the Coamatic Factor VIII Kit (Haemochrom Diagnostica, Essen, Germany) according to the manufacturer’s instructions with the exception of using a 1:7 prediluted sample buffer. Standard curves were generated using 1 IU/mL rhFVIII serially diluted in pooled mouse plasma of naive F8−/− mice (N = 7) and stored at −75°C. FVIII:C in the plasma samples was calculated from the standard curve. rhFVIII antigen (FVIII:Ag) was quantified using Asserachrom VIII:Ag enzyme-linked immunosorbent assay (ELISA; Diagnostica Stago, Düsseldorf, Germany) according to the manufacturer’s instructions, with the exception that, due to volume restrictions, the total assay volume was reduced to 100 µL per well. The limits of detection for FVIII:C and FVIII:Ag assays were determined by repeated analysis of FVIII-deficient plasma samples (N = 48 and N = 16 from 6 and 8 individual mice, respectively).
VWF-12 quantification
VWF-12 was quantified by ELISA as follows: Strep-Tactin XT coated microplate (IBA GmbH) was blocked with blocking buffer (1% BSA in Dulbecco phosphate-buffered saline [PBS]; Sigma-Aldrich) at RT for 2 hours. Plasma samples were diluted in blocking buffer and applied to the plate. After 2-hour incubation at 37°C, VWF-12 was detected with horseradish peroxidase (HRP)-conjugated antibody (polyclonal rabbit anti-human VWF HRP; Agilent, Santa Clara, CA, USA; diluted 1/2000). After incubation, the plate was washed 3 times with 0.1% Tween in PBS. The signal was detected using a 3, 3′,5, 5′-tetramethylbenzidine Liquid Substrate (TMB; Sigma-Aldrich), the reaction with HRP was terminated using 1 M HCl. Absorbance was read at 450 nm using a Multiskan FC Microplate Photometer (Thermo Fisher Scientific). The standard curve was prepared in a concentration range 1.65 to 50 ng/mL, and the mice plasma samples were diluted 1/42 or higher.
VWF assays
Standard VWF activity assays were used to study the impact of VWF-12 on the interaction between platelet glycoprotein Ib (gp Ib) and VWF. These included (1) ristocetin cofactor assay (VWF:RCo), (2) gp Ib–binding assay with ristocetin (VWF:GPIbR), and (3) gp Ib–binding assay with gain-of-function mutated gp Ib in the absence of ristocetin (VWF:GPIbM).17 Plasma-derived fl-VWF (37 nM) and increasing amounts of VWF-12 ranging from 0 to 3700 nM (100-fold excess) were used.
To further study the platelet-VWF interaction in the presence of VWF-12, an in vitro flow chamber system was used as described previously.18 In brief, hydrophilic flow-chambers (μ-slide VI 0.1; Ibidi GmbH, Munich, Germany) were coated with 25 μL per flow channel of 0.1 mg/mL human pepsin-solubilized collagen III (Southern Biotechnology). Platelet and red blood cell (RBC) concentrates were obtained from Haema AG (Berlin, Germany). Platelets were labeled with 0.5 μM 3.3′-dihexyloxacarbocyanine iodide (DiOC6), washed and resuspended in platelet buffer (10 mM HEPES, 136 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 10 mM glucose, 0.3% [wt/vol] BSA) and used at 250 × 109/L. RBCs were washed with platelet buffer, and used at 50% hematocrit. Plasma-derived fl-VWF at a physiological concentration of 1 IU/mL (37 nM) and increasing amounts of VWF-12 ranging from 0 to 125 nM were added to the mixture of labeled platelets and RBCs immediately before perfusion. Perfusion studies were performed using a neMESYS pump system (cetoni GmbH, Korbußen, Germany) with 5-minute perfusion at 1500 s−1. Microscopic analysis was performed with a fluorescence microscope equipped with AxioCam and AxioVision 4.8.2 SP2 software. Eleven pictures of each channel were taken every 30 seconds over 5 minutes. The surface covered by platelets was subsequently evaluated using Bioflux Software 2.4 (Fluxion Biosciences, San Francisco, CA).
Immunogenicity
Plasma from F8−/− mice treated with four weekly doses of 200 IU/kg of either rhFVIII IV or rhFVIII/VWF-12 subcutaneously was assessed for anti-FVIII binding immunoglobulin G (IgG) as previously described.19 Splenocytes were retrieved from F8−/− mice after 4 weekly doses of rhFVIII IV 200 IU/kg and were restimulated (0.6 × 106 cells per well on 96-well plates) with rhFVIII (1 or 3 IU/mL) either alone or in the presence of VWF-12 in ratios of 1:1 or 1:3.1 for 6 days. Anti-FVIII IgG-secreting cells were detected by enzyme-linked immunosorbent spot assay as previously described.19,20
Pharmacodynamics
The tail-clip bleeding model was used in F8−/− mice treated with a single dose of either 200 IU/kg rhFVIII IV or 1000 IU/kg rhFVIII/VWF-12 subcutaneously. Mice were anesthetized intraperitoneally with 5 mg/kg xylazine and 100 mg/kg ketamine or with 3 mg/kg xylazine, 100 mg/kg ketamine and 2 mg/kg midazolam, and placed on a warming plate to maintain body temperature. Three millimeters of the tail tip were removed with a scalpel, and the tail was immediately placed into a tube containing 31.5 mL of prewarmed (37°C) saline. After 30 minutes, the tail was removed and EDTA added to the saline at a final concentration of 0.05 M to prevent clotting.
Blood loss was determined by measuring the hemoglobin (Hb) concentration in the solution using a microplate spectrophotometer (Ledetect96 Microplate Reader; Landgraf Laborsysteme HLL GmbH, Langenhagen, Germany). Tubes were centrifuged for 20 minutes at 4000g. Supernatant was removed and erythrocytes were resuspended in 2 mL of 1× lysis buffer (BD Pharm Lyse; BD Biosciences, Heidelberg, Germany). After incubation for 5 minutes at RT, tubes were centrifuged for 5 minutes at 10 000g. Supernatant containing Hb was stored at −75°C. Absorbance of the samples was measured at wavelength 550 nm. Mouse Hb (Cusabio, Houston, TX) was used as standard.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (versions 6.0 and 8.0, GraphPad Software, Inc, La Jolla, CA). Correlation between FVIII:C and FVIII:Ag was described using the Pearson correlation coefficient. Treatment groups were compared using 2-way analysis of variance (ANOVA) with Bonferroni correction for multiple comparison. PK was described by reporting peak concentration (cmax), time to peak (tmax), and area under the concentration curve (AUC) as determined using Prism 6. Apparent half-life (t1/2) was estimated by including time points greater than or equal to tmax and a 1-phase exponential decay model. Bioavailability (F) was calculated based on the AUC and dose (D) after IV and subcutaneous (SC) injection using the equation: F = AUCSC × DIV/(AUCIV × DSC).
Results
Characterization of recombinant VWF fragments
The structures of VWF-12 and VWF-13 constructs are depicted in Figure 1. Both constructs are dimers of VWF fragments with the molecular weights of 153and 263 kDa, respectively, containing the D′D3 domains implicated in FVIII binding.21 For VWF-12, the terminal 31-aa sequence (1238-1268) was repeated twice on the C terminus to enrich the fragment in O-glycosylation sites. VWF-13 contained the A1 to A3 domains of VWF in addition to the FVIII binding site.

Schematic representation of recombinant constructs. (A) Simplified domain structure of mature fl-VWF monomer. The FVIII-binding site in the D′D3 domains is indicated above. O- and N-linked glycosylation is indicated by open and full circles, respectively. (B) VWF-12 is a dimer of the D′D3 domains and 3 repeats of a 31-aa fragment downstream of D3 that is rich in O-linked glycosylation sites. (C) VWF-13 dimer contains the D′D3 and A1-A2-A3 domains of VWF. CTCK, C-terminal cystine knot.
Schematic representation of recombinant constructs. (A) Simplified domain structure of mature fl-VWF monomer. The FVIII-binding site in the D′D3 domains is indicated above. O- and N-linked glycosylation is indicated by open and full circles, respectively. (B) VWF-12 is a dimer of the D′D3 domains and 3 repeats of a 31-aa fragment downstream of D3 that is rich in O-linked glycosylation sites. (C) VWF-13 dimer contains the D′D3 and A1-A2-A3 domains of VWF. CTCK, C-terminal cystine knot.
Consistent with literature, specific binding of rhFVIII to fl-VWF had a mean ± standard deviation (SD) dissociation constant of KD = 0.24 ± 0.02 × 10−9 M.22-24 Affinity of rhFVIII to VWF-12 (KD = 0.93 ± 0.05 × 10−9 M) or VWF-13 (KD = 1.15 ± 0.15 × 10−9 M) was slightly lower than to fl-VWF, but still in the nanomolar range. Representative association/dissociation curves are shown in supplemental Figure 1 (available on the Blood Web site).
The ability of VWF-12 and VWF-13 to inhibit PL binding of rhFVIII was analyzed by surface plasmon resonance (Figure 2A; supplemental Figure 2). For reference, an equimolar ratio of fl-VWF inhibited FVIII binding to PL by ∼50% consistent with previous literature.25 BSA as an irrelevant protein control did not influence rhFVIII binding in excess concentration. VWF-12 inhibited rhFVIII-PL interaction in a dose-dependent manner, with equimolar, 2.5- and 5-fold excess concentrations inhibiting PL binding by 80%, 91%, and 95%, respectively. The larger fragment VWF-13 provided less suppression of PL binding of rhFVIII at equimolar concentrations (52%).

Characterization of VWF fragments. (A) Surface plasmon resonance (SPR) analysis of rhFVIII binding to PL monolayer in the presence of fl-VWF, VWF-12, VWF-13, or bovine serum albumin (BSA). Molar ratios are given in the table below the figure. (B) VWF:GPIbM assay of fl-VWF in the presence of increasing concentrations of VWF-12, human serum albumin (HSA), or VWF-12 alone. Concentrations in nanomoles per liter are shown in the table below the figure. (C) Collagen binding of washed human platelets under flow in the absence or presence of VWF with increasing concentrations of VWF-12. Mean ± standard deviation (SD) shown in all panels.
Characterization of VWF fragments. (A) Surface plasmon resonance (SPR) analysis of rhFVIII binding to PL monolayer in the presence of fl-VWF, VWF-12, VWF-13, or bovine serum albumin (BSA). Molar ratios are given in the table below the figure. (B) VWF:GPIbM assay of fl-VWF in the presence of increasing concentrations of VWF-12, human serum albumin (HSA), or VWF-12 alone. Concentrations in nanomoles per liter are shown in the table below the figure. (C) Collagen binding of washed human platelets under flow in the absence or presence of VWF with increasing concentrations of VWF-12. Mean ± standard deviation (SD) shown in all panels.
Partial de-O-glycosylation of VWF-12 resulted in decreased protection of rhFVIII from binding to PL (supplemental Figure 3A) or ECM proteins (supplemental Figure 3B), and protected less from proteolytic degradation by elastase (supplemental Figure 4) suggesting the dense glycosylation of VWF-12 could be involved in preventing rhFVIII interaction with subcutaneous matrix constituents.
VWF-12 did not interfere with VWF function, as demonstrated in the VWF:GPIbM (Figure 2B), VWF:GPIbR and VWF:RCo (data not shown) activity assays. Of note, VWF-12 did not show intrinsic VWF activity in these assays. Platelet adhesion to collagen under flow was not affected by VWF-12 (Figure 2C). In summary, these results indicated that VWF-12 could protect rhFVIII from binding to PL and ECM and from proteolytic degradation but does not interfere with VWF function.
PK assessment
Single-dose PK profiles were evaluated after IV or subcutaneous administration of rhFVIII in the absence or presence of VWF-12 or VWF-13. After IV administration, PK profiles of FVIII:Ag in the absence or presence or VWF-12 or VWF-13 were very comparable (Figure 3A). cmax was observed at the first sampling time point, 1 hour after administration, and was slightly higher in the presence of VWF-12 and VWF-13 as compared with rhFVIII alone. FVIII:Ag then declined rapidly with a half-life of ∼2.6 to 2.8 hours without significant effect of VWF-12 or VWF-13.
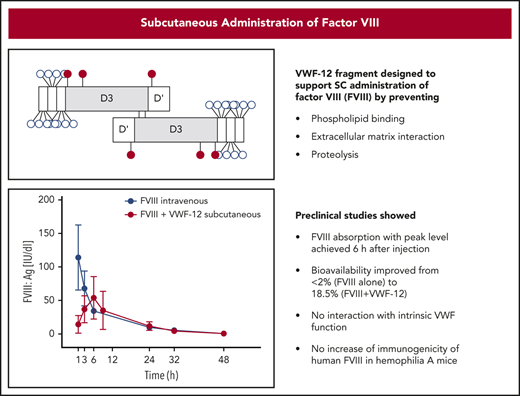
PK profiles of FVIII:Ag in the presence or absence of recombinant VWF fragments in F8−/− mice. (A) Single-dose IV rhFVIII (200 IU/kg) alone or in combination with VWF-12 or VWF-13. (B) Single-dose subcutaneous (SC rhFVIII (1000 IU/kg) with or without VWF-12 or VWF-13. (C) Direct comparison of IV rhFVIII (200 IU/kg), subcutaneous rhFVIII (1000 IU/kg), with VWF-12 (1:3.1) and subcutaneous rhFVIII (1000 IU/kg) with VWF-13. Mean ± SD from 5 mice per group and time point.
PK profiles of FVIII:Ag in the presence or absence of recombinant VWF fragments in F8−/− mice. (A) Single-dose IV rhFVIII (200 IU/kg) alone or in combination with VWF-12 or VWF-13. (B) Single-dose subcutaneous (SC rhFVIII (1000 IU/kg) with or without VWF-12 or VWF-13. (C) Direct comparison of IV rhFVIII (200 IU/kg), subcutaneous rhFVIII (1000 IU/kg), with VWF-12 (1:3.1) and subcutaneous rhFVIII (1000 IU/kg) with VWF-13. Mean ± SD from 5 mice per group and time point.
After subcutaneous administration, rhFVIII alone was poorly absorbed to circulation with a bioavailability of 1.5% (Figure 3B). With the addition of VWF-12, rhFVIII was slowly absorbed with a maximum concentration of 54 IU/dL reached after 6 hours. FVIII:Ag levels were consistently higher than baseline at each time point between 1 hour and 32 hours after administration. As a result of slow absorption and excretion, the apparent plasma half-life of rhFVIII was ∼7.2 hours, a 2.5-fold increase over the half-life observed for rhFVIII alone after IV administration. VWF-13 also improved absorption of rhFVIII but was clearly less effective compared with VWF-12. Higher doses of VWF-12 (rhFVIII to VWF-12 ratio of up to 1:150) did not significantly improve absorption of rhFVIII (supplemental Figure 5A).
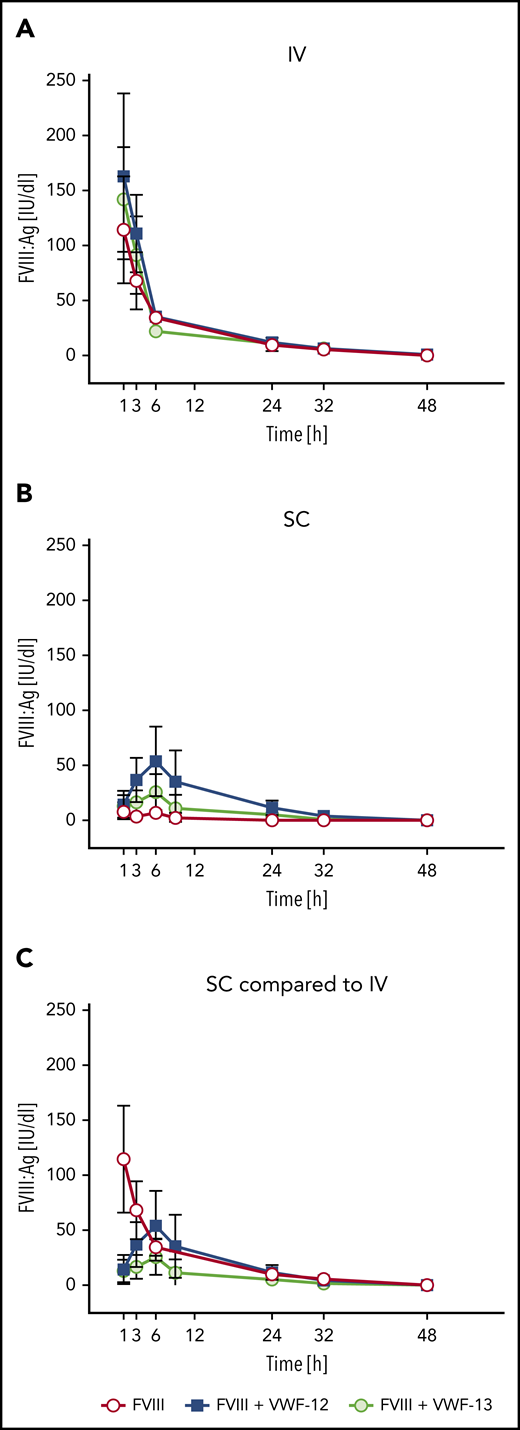
Comparing the subcutaneous profiles with the profile of IV rhFVIII alone (Figure 3C), VWF-12 and VWF-13 increased the bioavailability of subcutaneous rhFVIII to 18.5% and 7.2%, respectively (Table 1). PK profiles of rhFVIII (FVIII:C) were very similar (data not shown). Close correlation was observed for antigen and activity assays over a broad range of activities irrespective of the presence of VWF-12 or VWF-13 and application route (Figure 4). Given the capacity of VWF-13 to suppress PL binding and to support subcutaneous bioavailability of rhFVIII, it was not considered in further studies.
Correlation between FVIII:Ag and FVIII:C. Data are derived from all time points of the PK profiles shown in Figure 3. (A) IV administration. (B) Subcutaneous (SC) administration. Linear regression coefficients and linear regression lines are shown for the individual treatment groups.
Correlation between FVIII:Ag and FVIII:C. Data are derived from all time points of the PK profiles shown in Figure 3. (A) IV administration. (B) Subcutaneous (SC) administration. Linear regression coefficients and linear regression lines are shown for the individual treatment groups.
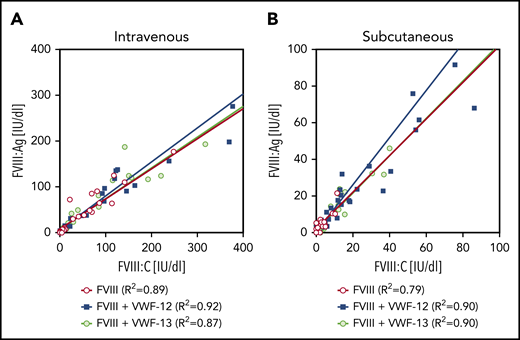
The PK of VWF-12 fragment itself was assessed after a single subcutaneous (291 µg/kg) or IV (58 µg/kg) injection in complex with rhFVIII (Figure 5). Absorption of VWF-12 from the subcutaneous compartment was similar to rhFVIII with a peak concentration of 0.7 ± 0.1 µg/mL reached after 9 hours. The VWF-12 half-life after IV injection was 19 hours; it was increased to an apparent half-life of about 40 hours after subcutaneous administration. Dose-corrected bioavailability of VWF-12 after subcutaneous administration was 53%. PK of VWF-12 was dose-proportional over a broad range of doses (supplemental Figure 5B).
PK profile of VWF fragment VWF-12. Mice received a single dose of IV (200 IU/kg, red) or subcutaneous (SC) (1000 IU/kg, blue) rhFVIII/VWF-12. Mean ± SD from 5 mice per group and time point.
PK profile of VWF fragment VWF-12. Mice received a single dose of IV (200 IU/kg, red) or subcutaneous (SC) (1000 IU/kg, blue) rhFVIII/VWF-12. Mean ± SD from 5 mice per group and time point.
Immunogenicity
The formation of anti-FVIII IgG was compared in naive F8−/− mice receiving either rhFVIII IV or an equal dose of rhFVIII/VWF-12 subcutaneous (Figure 6A). As expected in this model, all mice developed anti-FVIII antibodies. However, antibody formation was significantly slower when mice received rhFVIII/VWF-12 subcutaneous (ANOVA for intergroup comparison P = .023). Anti-FVIII IgG titers tended to be lower until week 4 but reached similar levels in week 5. VWF-12 did not significantly influence the formation of FVIII-specific antibody-secreting cells from memory B cells when these exposed to different concentrations of rhFVIII (Figure 6B). In summary, these data indicated that VWF-12 itself or the subcutaneous administration of rhFVIII/VWF-12 did not increase the immunogenicity of rhFVIII in the F8−/− mouse model.

Anti-FVIII antibody formation and restimulation of FVIII-specific memory B cells. (A) F8−/− mice received 4 doses (weeks 1-4) of rhFVIII IV or rhFVIII+VWF-12 subcutaneous (200 IU/kg, 20 mice per group). Anti-FVIII IgG titers were determined by ELISA. Results are shown for individual mice as scatter plots on a logarithmic scale. Data were analyzed by 2-way ANOVA with Bonferroni correction for multiple comparisons. ****P < .0001. (B) Splenocytes from rhFVIII-immunized mice were restimulated with rhFVIII (1 or 3 IU/ml) in the absence or presence of VWF-12 in different molar ratios. Mean ± SD are shown from 2 independent experiments (group 1 and 2), each with 3 mice per group and triplicate analysis of pooled cells.
Anti-FVIII antibody formation and restimulation of FVIII-specific memory B cells. (A) F8−/− mice received 4 doses (weeks 1-4) of rhFVIII IV or rhFVIII+VWF-12 subcutaneous (200 IU/kg, 20 mice per group). Anti-FVIII IgG titers were determined by ELISA. Results are shown for individual mice as scatter plots on a logarithmic scale. Data were analyzed by 2-way ANOVA with Bonferroni correction for multiple comparisons. ****P < .0001. (B) Splenocytes from rhFVIII-immunized mice were restimulated with rhFVIII (1 or 3 IU/ml) in the absence or presence of VWF-12 in different molar ratios. Mean ± SD are shown from 2 independent experiments (group 1 and 2), each with 3 mice per group and triplicate analysis of pooled cells.
PD assessment
Hemostatic efficacy of subcutaneous rhFVIII in the presence of VWF-12 was analyzed in a murine HA tail-bleeding model (Figure 7). Bleeding was quantified by measuring Hb loss after injury. Few or no bleeding occurred if mice received rhFVIII IV ≤2 hours before injury (median Hb loss, 0.13 g/dL). In contrast, untreated F8−/− mice exsanguinated during observation and a median Hb loss of 4.6 g/dL was determined. subcutaneous rhFVIII/VWF-12 progressively reduced bleeding to a median Hb loss of 2.4 g/dL (after 1 hour), 0.7 g/dL (after 6 hours), and 0.4 g/dL (after 9 hours). Mice treated 24 hours before injury showed an Hb loss of 3.6 g/dL that was still below untreated control mice.
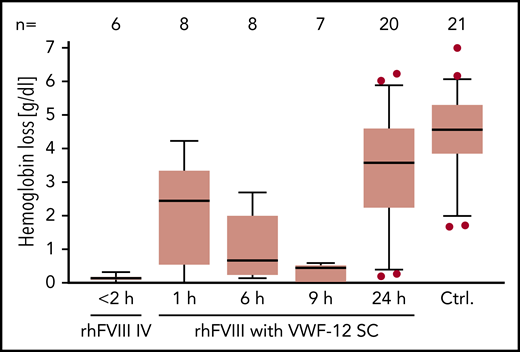
PD profile of rhFVIII with or without VWF-12. F8−/− mice received a single dose of IV rhFVIII (200 IU/kg) alone, subcutaneous rhFVIII/VWF-12 (1000 IU/kg), or no treatment (Ctrl.) Time in hours from treatment to bleeding challenge is indicated. Hemoglobin loss is shown over 30 minutes from tail clipping with higher values indicating more bleeding. Boxes show median and interquartile ranges; whiskers show the 10th and 90th percentiles. Number of mice per group is shown in the top line. Outliers were determined using the ROUT procedure and are represented by dots.
PD profile of rhFVIII with or without VWF-12. F8−/− mice received a single dose of IV rhFVIII (200 IU/kg) alone, subcutaneous rhFVIII/VWF-12 (1000 IU/kg), or no treatment (Ctrl.) Time in hours from treatment to bleeding challenge is indicated. Hemoglobin loss is shown over 30 minutes from tail clipping with higher values indicating more bleeding. Boxes show median and interquartile ranges; whiskers show the 10th and 90th percentiles. Number of mice per group is shown in the top line. Outliers were determined using the ROUT procedure and are represented by dots.
Discussion
In recent years, considerable progress has been made in developing treatments for HA that are less burdensome for the patient. Extended half-life products, including polyethylene glycol (PEG)-conjugated FVIII molecules and FVIII-Fc fusion protein, have enabled dosing frequency to be reduced but still require IV administration.26 Emicizumab, a bispecific antibody mimicking the function of activated FVIII, can be administered subcutaneous at 1- to 4-week intervals.27,28 Emicizumab is effective for bleed protection in the presence of neutralizing antibodies (inhibitors) against FVIII and is considered as a breakthrough in the management of HA patients with inhibitors.29 For the majority of HA patients, who do not have inhibitors, it is unclear whether long-term treatment with nonfactor products such as emicizumab will demonstrate comparable efficacy and safety to FVIII prophylaxis. Long-term studies are needed to address the impact of nonfactor treatments on joint health, safety in emergency situations or surgery, and immunological tolerance toward FVIII.30 At the same time, the development of novel strategies to improve delivery of FVIII protein for prophylaxis and tolerance induction appears justified.
In the present study, we explored the potential of recombinant VWF fragments to support delivery of rhFVIII into the vasculature after subcutaneous administration. Because the low bioavailability of natural FVIII after subcutaneous administration is largely due to PL binding in the extravascular space,8 VWF fragments were designed to inhibit PL binding, without interfering with FVIII function.
The binding affinity of the VWF fragments for rhFVIII was in the nanomolar range, but slightly lower than that of fl-VWF. This may be due in part to an absence of stabilizing effects from interaction of the D3 and CK domains.10 Nevertheless, VWF-12 and VWF-13 administered in moderate excess over rhFVIII were sufficient to inhibit PL binding and support delivery from the subcutaneous compartment to the blood plasma.
The bioavailability of subcutaneous rhFVIII was higher in the presence of VWF-12 than with VWF-13. Possible explanations for this difference are: a lower level of PL binding inhibition by VWF-13; the higher molecular weight of the noncovalent rhFVIII/VWF-13 complex resulting in reduced mobility through the extracellular space31 ; collagen binding to the A3 domain present in VWF-1332 ; a lower rate of fragment O-glycosylation potentially protecting from degradation as it does for natural VWF.33,34 The latter is supported by the observation that a partially de-O-glycosylated form of VWF-12 protected rhFVIII less from PL and ECM binding and provided less resistance to proteolysis. Further studies would be needed to address all differences between VWF-12 and VWF-13. However, given the promising characteristics of VWF-12, analysis of VWF-13 was discontinued.
Single-dose PK analysis demonstrated favorable characteristics of the rhFVIII/VWF-12 complex when administered in a 1:3.1 molar ratio. Higher doses of VWF-12 did not significantly improve speed of absorption, peak levels, half-life or bioavailability. Due to the dimeric nature of VWF-12, the 1:3.1 ratio corresponds to ∼6 binding sites per rhFVIII molecule. Natural VWF in human plasma provides an average of 50 binding sites per FVIII molecule,23 similar to mouse VWF.10 Considering the plasma concentrations and binding constants, we expect FVIII to transfer binding from VWF-12 to endogenous VWF once in the circulation. A different approach of using VWF sequences to improve PK of FVIII has been used in BIVV001, a recombinant FVIII fusion protein containing the VWF D’D3 region in addition to other primary amino acid structure modifications including the immunoglobulin Fc fragment, a F8 R1648A missense mutation and two strings of XTEN, a hydrophilic protein polymer.35 BIVV001 does no longer bind to fl-VWF and the covalently attached D’D3 sequence will only be removed from this molecule after thrombin activation of FVIII. BIVV001 had a threefold to fourfold longer half-life than rhFVIII after IV injection in mice,35 and a bioavailability of 20% after subcutaneous injection.6 The bioavailability of rhFVIII/VWF-12 observed after subcutaneous injection in our studies was very similar suggesting that the additional modifications in BIVV001 are not necessary for subcutaneous delivery of rhFVIII.
Our data indicate that rhFVIII delivered with VWF-12 is fully functional after absorption from the subcutaneous space. FVIII antigen and chromogenic activity values were closely correlated in samples drawn after subcutaneous administration, indicating that rhFVIII retained activity despite passage through the lymphatic system. Administration of subcutaneous rhFVIII/VWF-12 to mice demonstrated a prophylactic effect on bleeding prevention for up to 24 hours, further supporting the functional integrity of rhFVIII after subcutaneous administration.
The slow rate of absorption of rhFVIII/VWF-12 from the subcutaneous compartment into the circulation and the 2.5-fold prolonged half-life suggest the use of rhFVIII/VWF-12 for routine prophylaxis in severe HA. Provided similar PK characteristics were observed in human studies, sufficient trough levels for clinical use might be achievable with daily or less frequent subcutaneous injections. subcutaneous administration has potential to facilitate prophylaxis in newborns, small children and patients with difficult venous access compared with conventional IV prophylaxis. In case of breakthrough bleeds, subcutaneous FVIII could be combined with IV FVIII without the risk of adverse effects due to interference of the 2 drugs.
The immunogenicity of rhFVIII administered subcutaneous is a potential concern. In the hemophilia mouse model used for the current study, there was no indication of increased immunogenicity when rhFVIII was administered together with VWF-12 through the subcutaneous route as compared with an equal dose of rhFVIII given alone IV. Although it was promising to see that anti-FVIII antibody formation was slowed down in mice receiving rhFVIII/VWF-12 subcutaneous, it is acknowledged that the F8−/− mouse model is not ideal to draw conclusions on immunogenicity in humans and definite proof has to come from clinical trials. It may also be interesting to explore the potential of subcutaneous FVIII to establish immune tolerance against FVIII in previously untreated patients, in whom standard prophylaxis is considered unfeasible. Although there is a perception that subcutaneous administration of therapeutic proteins could possibly pose an increased risk of adverse immune reactions, there are very few comparative studies and data to support this hypothesis. Recent reviews from other therapeutic areas highlight the importance of further clinical research delineating the impact of administration route on the risk of antidrug antibody formation.36-38
In summary, coadministration of rhFVIII with the recombinant fragment VWF-12 improved the bioavailability of rhFVIII after subcutaneous administration in mice. VWF-12 did not interfere with the function of VWF or rhFVIII and did not seem to increase immunogenicity of rhFVIII in mice. These data lay the foundation for clinical studies assessing the PK and safety of rhFVIII/VWF-12 in patients with HA.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This work was supported by research funding from Octapharma GmbH.
Authorship
Contribution: N.V.-H., O.O. and B.S.-W. designed and conducted experiments, analyzed and interpreted data, and wrote the manuscript; S.W. designed experiments and analyzed and interpreted data; and C.K. and A.T. supervised the project and revised the manuscript.
Conflict-of-interest disclosure: A.T. obtained funding for this research from Octapharma and received honoraria for lectures and consultancy. B.S.W. and C.K. are employees of Octapharma GmbH. The remaining authors declare no competing financial interests.
Correspondence: Andreas Tiede, Department of Hematology, Hemostasis, Oncology and Stem Cell Transplantation, Carl-Neuberg-St 1, Hannover Medical School, D-30625 Hannover, Germany; e-mail: tiede.andreas@mh-hannover.de.