


Abstract
Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs), polycythemia vera, essential thrombocythemia, and primary myelofibrosis, are hematopoietic stem cell disorders that are defined by activating mutations in signal transduction pathways and are characterized clinically by the overproduction of platelets, red blood cells, and neutrophils, significant burden of disease-specific symptoms, and high rates of vascular events. The focus of this review is to critically reevaluate the clinical burden of thrombosis in MPNs, to review the clinical associations among clonal hematopoiesis, JAK2V617F burden, inflammation, and thrombosis, and to provide insights into novel primary and secondary thrombosis-prevention strategies.
Introduction
The Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs), polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are hematopoietic stem cell disorders that are driven by acquired activating mutations that control the production of blood cells.1 Recent advances in our understanding of the roles of clonal hematopoiesis (CH), JAK2V617F, and inflammation in thrombosis risk provide new insights that will lead to novel primary and secondary thrombosis-prevention strategies.
Clinical scope of thrombotic disease in MPNs
Thrombosis takes the form of macrovascular or microvascular arterial (AT) or venous (VT) events that have unique and often paradoxical features: the majority of thrombotic events occur at or during the years preceding MPN diagnosis; AT events outnumber VT events in the older population, whereas VT events outnumber AT events in the younger population; and female sex is associated with unusual sites, including splanchnic veins, cerebral sinuses, and placental thromboses.2-10 A recent meta-analysis of >13 000 individuals revealed a pooled prevalence of thrombosis at diagnosis of 20%, with a higher prevalence in PV compared with ET or PMF.4 Hultcrantz and colleagues examined thrombotic events in 9429 MPN patients and 35 820 matched population control subjects and confirmed that, although thrombotic events occurred most frequently around the time of MPN diagnosis, the risk of developing additional events persisted throughout the patients’ lifetimes.7 Advanced age and male sex added to the cumulative risk of developing AT and VT events, but the thrombotic risk was higher in the MPN group, regardless of age or sex, compared with a control population. Paradoxically, the highest hazard ratio was for VT events in the youngest age group.7 To put these findings into clinical perspective, during long-term follow-up, having an MPN increased an individual’s risk for developing an AT event to a similar degree as cigarette smoking, and it increased an individual’s risk for VT events to a similar degree as factor V Leiden heterozygosity. Furthermore, associated cardiovascular risk factors that are important in the general population, including tobacco use, dyslipidemia, hypertension, diabetes mellitus, atrial arrhythmias, and hormonal therapy, have also been implicated as contributing to the thrombotic risk in patients with PV and ET.6,11-15
Blood counts and thrombotic risk
In the general population, elevated blood counts are associated with vascular risk. In a prospective study of 108 521 individuals, platelet counts in the top 5th percentile (>398 × 109/L) were associated with a 1.8‐fold higher risk for stroke compared with those in the 25th to 75th percentiles, whereas hematocrit values in the top 5th percentile (ie, >45% in women and >48% in men) were associated with a 1.5‐fold risk for myocardial infarction compared with those in the 25th to 75th percentiles.16 Elevation of the neutrophil count is an indicator of adverse outcomes in acute myocardial infarction and coronary revascularization.17 Because blood counts are influenced by sex, race, age, obesity, smoking, inflammation, iron metabolism, and a myriad of other factors, dissecting the independent effect of blood cell counts on thromboembolic risk is complex.18-21
Within the MPNs, the same external factors modify blood counts, in addition to disease-specific factors, such as mutation type.2,22 The MPNs are defined by elevations in specific cell lineages, such that, although thrombocytosis is common among MPNs, erythrocytosis is specific to PV, and anemia is common in MF. This has compromised attempts at thrombosis risk estimation based on absolute platelet counts or white cell counts alone, particularly when disease classes are analyzed together. Landolfi et al showed an independent association between leukocytosis and AT thrombosis (primarily coronary thrombosis) in a large well-defined group of patients with PV.15 Recently, a meta-analysis in ET and PV patients found a 1.59-fold (95% confidence interval [CI], 1.40-1.80) relative risk for AT thrombosis with leukocytosis; however, this was only significant in ET.23 A recent retrospective study of >500 patients with PV did not find any association between persistently elevated white blood cell (WBC) counts and thrombosis, but it did find an association between disease evolution and myelofibrosis.24 The inconsistent nature of these reports concerning the impact of leukocytosis on the risk for MPN thrombosis likely reflects the limitations of using retrospective studies and combining disease entities to establish such clinical correlative relationships.
Although elevated platelet counts are assumed to be associated with increased thrombosis risk in PV, when adjusted for other clinical and laboratory factors, platelet count does not have an independent association with thrombosis.15 The association between thrombocytosis and thrombosis risk is stronger in ET, but a number of studies have suggested that higher platelet counts may be protective against thrombosis, perhaps as a result of extreme thrombocytosis leading to an acquired form of von Willebrand disease.25 These observations have failed to identify a goal or target platelet count to assure a reduction in thromboembolic risk in patients with PV and ET.
The lack of concordance between thrombosis risk and blood count elevation in MPNs is highlighted best in splanchnic vein thrombosis (SVT). Plasma volume expansion or portal hypertension contributes to lowered blood counts and hematocrit and, therefore, confounds the association between blood counts and thrombosis risk.26-28
JAK2V617F variant allele frequency and thrombosis risk
In the 15 years since the discovery of JAK2V617F as a driver of mutation in MPNs, investigators examined JAK2V617F as a categorical and quantitative risk factor for thrombosis in both MPN cohorts, as well as in CH cohorts from the general population. Many studies have observed a higher risk for thrombotic events in patients with JAK2V617F+ ET or MF compared with patients with JAK2V617F− ET or MF, and JAK2V617F status is now incorporated in the International Prognostic Score for Thrombosis in ET.29-32 In 2020, the specific effect of JAK2V617F on thrombosis risk was refined by the analysis of thrombosis in 1537 patients in China with JAK2V617F+ MPNs: the risk of thrombosis was significantly higher in PV than in ET or PMF, and the incidence of thrombosis in patients with PV with JAK2V617F variant allele frequencies (VAFs) ≥50% was 4.6 times higher than that in patients with a VAF <50%.5 Subsequently, investigations into general population cohorts tested the association between blood count elevations and thrombosis risk and qualitative and quantitative measures of JAK2V617F. Three large studies found that low JAK2V617F VAFs were present in 0.1% to 0.2% of the general population.33-36 Surprisingly, despite the absence of an overt MPN, individuals with JAK2V617F+ CH had significantly higher platelet counts, WBC counts, and hemoglobin concentrations, as well as higher rates of AT and VT events, compared with those with wild-type JAK2. In 2019, Cordua et al provided further insight into the association between JAK2V617F+ or calreticulin-mutation positive (mCALR+) CH and clonal burden in 19 958 participants in the Danish General Suburban Population Study (Figure 1).37 JAK2V617F+ CH was detected in 3.1% of this population with a median VAF of 2.1%, whereas mCALR+ CH was observed in 0.16% with a median VAF of 7.5%. Individuals with mCALR+ CH had higher platelet counts compared with control individuals or those with JAK2V617F+ CH, but not higher WBC counts or increased AT or VT rates. Those with JAK2V617F+ CH and higher VAFs had higher blood cell counts and a greater risk for developing a VT, pulmonary embolism, or a cerebrovascular event, again demonstrating the specific increase in thrombotic events associated with the JAK2V617F+ state and with increasing JAK2V617F+ clonal burden (Figure 1). These findings are reminiscent of individuals with normal blood cell counts who develop thromboses in splanchnic veins or cerebral sinuses; most of these patients harbor JAK2V617F mutations, frequently with very low VAF.38,39 Many such individuals ultimately fulfill MPN clinical diagnostic criteria, but a substantial number remain in the range meeting CH clinical criteria.8,38-40

Prevalence, relative thrombosis risk, and average blood counts stratified by JAK2V617F VAFs from the Danish Suburban Population Study.37 Mean blood counts of 19 313 patients with JAK2V617F− (nonmutated), 508 patients with JAK2V617F+ (<1% VAF) non-MPN, 105 patients with JAK2V617F+ (≥1% VAF) non-MPN, and 16 patients with MPN (14 of whom were JAK2V617F+ with VAF 0.3% to 96%) (upper panel). The numbers in each group (left vertical axis) and the corresponding odds ratios for venous thromboembolism (VTE) and ischemic cerebrovascular disease (ICVD) compared with the nonmutated group (right vertical axis) (lower panels). Hg, hemoglobin. *Significantly different compared with nonmutated; #significantly different compared with JAK2V617F+ <1% VAF.
Prevalence, relative thrombosis risk, and average blood counts stratified by JAK2V617F VAFs from the Danish Suburban Population Study.37 Mean blood counts of 19 313 patients with JAK2V617F− (nonmutated), 508 patients with JAK2V617F+ (<1% VAF) non-MPN, 105 patients with JAK2V617F+ (≥1% VAF) non-MPN, and 16 patients with MPN (14 of whom were JAK2V617F+ with VAF 0.3% to 96%) (upper panel). The numbers in each group (left vertical axis) and the corresponding odds ratios for venous thromboembolism (VTE) and ischemic cerebrovascular disease (ICVD) compared with the nonmutated group (right vertical axis) (lower panels). Hg, hemoglobin. *Significantly different compared with nonmutated; #significantly different compared with JAK2V617F+ <1% VAF.
Inflammation and thrombosis risk
Epidemiological data from the mid-1990s first revealed that inflammation, as assessed by elevated levels of high sensitivity C-reactive protein (hs-CRP) or interleukin-6 (IL-6), played an important role in the development of atherosclerosis, independently of the previously known cardiovascular risk factors.41,42 Furthermore, Lussana et al reported that patients with ET and PV with high levels of hs-CRP had an increased risk for developing thrombotic events that was independent of the driver mutational status.43 Busque et al provided additional insight into the relationship between CH and inflammation in an elderly patient population with coronary artery disease (CAD).44 Their findings indicated that hs-CRP levels were elevated to a greater degree in individuals with CH, supporting the link between inflammation and CH in aging cardiac patients. The acquisition of CH, particularly JAK2V617F+ CH, the expansion of JAK2V617F+ CH into MPN, and AT and VT events share smoking, older age, and a chronic inflammatory state as risk factors.19,33,45 Inflammation drives thrombotic risk related to increased WBC and platelet counts, activation of clotting factors, endothelial cells, platelets, WBCs, and hypoxia sensing and its associated intracellular signaling pathways; inflammation is important in cancer, aging, sickle cell anemia, infection, autoimmunity, and atherosclerotic disease.46-49 JAK2V617F is uniquely connected to thromboinflammation because inflammatory states may predispose to acquiring JAK2V617F; conversely, JAK2V617F signaling also drives inflammatory pathways.50-52 Pedersen et al analyzed 107 969 individuals from the Copenhagen Population Study using a Mendelian randomization approach to test the role of a common loss-of-function IL-6 receptor variant that impairs IL-6 signaling.50 The age- and sex-adjusted risk of acquiring any MPN was 40% lower among carriers of the loss-of-function IL-6 receptor variant. This relative risk reduction was even more prominent in the JAK2V617F+ CH subcohort, further substantiating the concept that acquisition of JAK2V617F+ CH and JAK2V617F+ MPN share the same risk factors (Figure 2).35,50,53
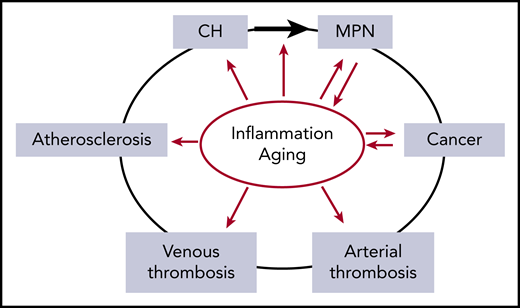
The relationship between inflammation and aging as risk factors (red arrows) for the development of CH, MPN, cancer, thrombosis, and atherosclerosis. Inflammation is implicated in increasing the risk of evolution from CH to MPN (black arrow). In addition, MPN and cancer drive inflammatory states, compounding the effects of inflammation. Inflammation and aging confound the relationship between MPN and thrombosis due to their roles as independent risk factors for both arterial and venous thrombosis.
The relationship between inflammation and aging as risk factors (red arrows) for the development of CH, MPN, cancer, thrombosis, and atherosclerosis. Inflammation is implicated in increasing the risk of evolution from CH to MPN (black arrow). In addition, MPN and cancer drive inflammatory states, compounding the effects of inflammation. Inflammation and aging confound the relationship between MPN and thrombosis due to their roles as independent risk factors for both arterial and venous thrombosis.
CH and thrombosis risk
With the increasing availability of next-generation sequencing to identify mutations in genes that are associated with myeloid malignancies, it became apparent that these mutations were not unique to individuals with blood cancers but rather were common in the normal population, especially in older individuals or those exposed to various forms of cytotoxic therapies.36,54-58 Although the overwhelming majority of individuals with CH never develop a hematological malignancy, these individuals do have an overall increase in mortality primarily because of their increased risk for developing cardiovascular events. Individuals with CH have nearly a doubling of cardiovascular disease prevalence and a fourfold greater risk for developing a myocardial infarction.36,59,60 A number of myeloid gene mutations can be responsible for CH, but CAD is most closely associated with mutations in DNMT3A, TET2, ASXL1, and JAK2V617F. Individuals with mutations in DNMT3A, TET2, or ASXL1 had a 1.7-fold to twofold increase in CAD, whereas individuals with JAK2V617F had a 12.1-fold increase in CAD.59 The degree of elevation in VAF of the mutated gene was also related to the risk of CAD. These same myeloid gene mutations are the most frequent after JAK2 and CALR driver mutations and are observed in up to 50% of patients with PV or ET.61-64 TET2 is most commonly mutated in ET and PV and is associated with thrombosis in some studies. Cerquozzi et al reported an association between TET2 mutations and thrombosis that was independent of age and driver mutational status in a cohort of patients with ET, but not patients with PV, seen at the Mayo Clinic.65 Surprisingly, this association between TET2 mutations and thrombosis was not observed in another patient cohort from the University of Florence that was evaluated in the same study. The mutation order of TET2 and JAK2 may have been an uncontrolled variable in the former study, because Ortmann et al found that patients who acquired the JAK2V617F mutation first had higher thrombosis rates compared with patients who acquired the TET2 mutation first.66
In addition to CH being a risk factor for the development of CAD, Dorsheimer et al showed that patients with established congestive heart failure (CHF) with TET2 or DNMT3A CH had an increase in mortality due to progressive CHF rather than additional acute ischemic events.67 Furthermore, there was a dose-response association between VAF and an inferior clinical outcome, suggesting that the presence of DNMT3A or TET2 mutations likely contributes to disease progression in patients with CHF. These clinical data are consistent with studies in mouse models that indicated that TET2 and DNMT3A loss-of-function mutations in myeloid cells promote cardiac dysfunction. A growing number of studies have indicated that loss-of-function mutations in these 2 genes lead to the development of a generalized inflammatory state.68 Low-density lipoprotein receptor–deficient mice, which are prone to develop atherosclerosis, were genetically engineered to lack TET2 in marrow cells. They developed atherosclerotic plaques in their aortic walls that had both increased size and numbers of macrophages.59 The macrophages isolated from these animals generated increased levels of cytokines and chemokines ex vivo relative to macrophages from TET2 wild-type mice, indicating that TET2 acts as a negative transcriptional regulator of the proinflammatory response. In particular, IL-1β levels were increased in atherosclerotic plaques and macrophages within these plaques in the TET2-deficient context. Fuster et al showed that TET2 deficiency contributed to IL-1β production by enhancing activation of the NLRP3 inflammasome components, in addition to promoting IL-1β transcription.69 They also showed that IL-1β played a pivotal role in endothelial cell activation and correlated the upregulation of IL-1β with upregulation of P-selectin expression in the aorta. Individuals with TET2 CH display elevated plasma levels of IL-8, an inflammatory chemokine that mediates monocyte adhesion to activated endothelium and is a downstream target of NF-κB. The reported upregulation of P-selectin by IL-1β is of particular importance to MPNs because P-selectin and von Willebrand factor expression are upregulated in JAK2V617F+-engineered endothelial cells, and because JAK2V617F+ endothelial cells have been reported in patients with MPNs.70-72 Such findings provide an experimental foundation for the clinical observations that indicate that vascular disease is mediated directly by MPN-specific mutational events that increase blood cell counts, alter cell function, and mediate inflammatory signaling. These studies also serve as a rationale for targeting IL-1β, IL-6, IL-8, NF-κB, and P-selectin to reduce thrombotic risk in patients with MPN.
Myelosuppression and thrombosis prevention
Hematocrit control is essential to lower blood viscosity and thrombosis risk, and the thrombosis reduction benefit of strict hematocrit control in PV was shown in a prospective randomized trial in which phlebotomy alone or in concert with cytoreductive therapy (which included hydroxyurea [HU] in the majority of patients) was used to achieve hematocrit targets.73,74 Thrombosis risk reduction via control of platelet or WBC counts in PV is more difficult to demonstrate and to address in a therapeutic trial because of the promiscuous effects of myelosuppression in controlling of hematocrit, as well as reducing WBC and platelet counts.75 The efficacy of cytoreduction in PV for preventing thrombosis has been extrapolated from studies involving patients with ET, despite the known differences in JAK2V617F and CALR mutation prevalence, JAK2V617F VAFs, and differences in WBC count and hematocrit levels between patients with ET and PV.76,77
In 114 patients with high-risk ET who were randomized to HU vs observation, HU was effective in preventing thrombosis.76 In another randomized controlled trial of patients with high-risk ET who were given low-dose aspirin plus anagrelide or HU, despite a similar degree of control in platelet counts between the 2 groups, those receiving low-dose aspirin plus HU had a reduced risk for AT compared with the aspirin plus anagrelide group, which ultimately was ascribed to the JAK2V617F+ ET subgroups (PT1 trial).29,77 By contrast, although the overall number of VT events was low, the rate of VT in the patients treated with anagrelide was fourfold lower relative to those treated with HU (P = .006). Patients with JAK2V6127F+ required lower HU doses to normalize platelet counts than did patients who lacked the mutation, indicating that the patients with JAK2V6127F+ were particularly sensitive to HU. However, the rates of death from any cause and death from thrombotic or hemorrhagic causes were not significantly different between the 2 groups. Furthermore, in the ANAHYDRET Study, in which previously untreated patients with high-risk ET were randomized to anagrelide or HU, a significant difference between the incidence of thromboses or bleeding events could not be discerned; this study indicated that a myelosuppressive agent that reduces WBC counts (HU) does not contribute to a further reduction in the number of thrombotic events, in ET at least.78 Comparison of the PT1 trial (809 patients) and the ANAHYDRET Study (259 patients) is difficult because of the differences in size of the patient populations and the design of the 2 trials: the PT1 Trial was designed to detect the superior agent, whereas the ANAHYDRET Study was designed to show noninferiority. Most recently, a randomized study that compared HU plus aspirin therapy with aspirin alone in 382 patients with moderate-risk ET did not detect a difference in the composite end point, which included AT or VT events.79 It is important to note that event rates were low, WBC counts were mostly normal, and JAK2V617F VAF was not available for a subgroup analysis. It is also important to note that studies of myelosuppressive therapies in PV and ET are usually done with the addition of aspirin, thereby confounding the interpretation of specific benefit of platelet or WBC control on additional thrombosis risk reduction.
The increased risk of leukemia or second cancers as a result of exposure to 32P or alkylating agents in the MPN is well established, and this concern extends to HU.80-82 The use of alkylating agents after HU therapy is associated with an unacceptable incidence of MPN blast phase,81 and long-term follow-up of busulfan and HU–treated ET patients found higher second cancer rates and no survival benefit.83 A recent large prospective study (EXCELS) examined 3649 patients with high-risk ET who were treated at physician discretion to receive HU or anagrelide; a higher risk for developing acute myelogenous leukemia was found in the HU-treated cohort.84 Thus, although these studies’ conclusions may be confounded by their retrospective nature or by other uncontrolled factors, enthusiasm for HU as a thrombosis risk-reduction agent is tempered by the neoplastic risk incurred by decades of exposure.
Large numbers of patients with PV have now been enrolled in prospective clinical trials of recombinant interferon and ruxolitinib with reasonable follow-up. The RESPONSE trial, which compared ruxolitinib, a JAK1/2 inhibitor, with best available therapy in 222 patients with PV who were resistant or intolerant to HU with 5-year follow-up, showed that the number of thromboembolic events in each arm was few, making it difficult to have statistical certainty that ruxolitinib therapy effectively reduced the numbers of thrombotic events.85 A recent meta-analysis of ruxolitinib treatment in 663 patients with PV found that the annual incidence of thrombosis was 3.09 per year (95% CI, 1.22-4.96) for those receiving ruxolitinib, 5.51 (95% CI, 3.72-7.30) for best available therapy, with a 0.56 relative risk reduction for ruxolitinib, although this risk reduction was not statistically significant.86
Long-acting interferons have been heralded as an approach that might alter disease at the hematopoietic stem cell level, normalize blood cell counts, and dampen inflammatory cytokine levels, all of which might reduce thrombotic events.87 Interferon therapy has been shown to prevent thrombosis in contexts in which HU has not been effective. Mascarenhas et al reported an absence of recurrent thrombotic events in a single-arm trial of 20 patients with SVTs treated with pegylated interferon.88 In contrast, failure of HU to prevent recurrent SVTs was shown by De Stefano et al89 in a 1500 MPN and thrombosis patient cohort, as well as by Sant’Antonio et al90 in a 518 MPN and SVT patient cohort. Recently, a randomized controlled trial of ropeginterferon vs HU in recently diagnosed patients with PV showed superior rates of hematological and molecular responses in the ropeginterferon treatment group.91 As expected for this cohort of patients with recently diagnosed PV, very few major vascular events were recorded, and a relationship between clinical and molecular response and benefits in terms of a reduction in vascular events could not be observed, despite 36 months of follow-up.
Novel approaches to thromboprophylaxis in patients with MPN
The backbone of therapy for reducing the incidence of thrombotic events in patients with PV continues to be maintenance of hematocrit levels <45% in males and, as suggested by some investigators, <42% in females.73,80 Strict control of hematocrit involves periodic therapeutic phlebotomy and/or the use of myelosuppressive therapies and it can be associated with intervals when the hematocrit levels exceed 45%, thereby increasing thrombosis risk. One promising type of therapy to control hematocrit involves the administration of a recombinant form of a pivotal mediator of iron trafficking, hepcidin. Administration of minihepcidins to PV mouse models normalized hematocrit and reduced the degree of splenomegaly.92 We and other investigators have hypothesized that a hepcidin mimetic agent would block the intestinal absorption of iron and promote the sequestration of iron in tissue macrophages, thereby reducing therapeutic phlebotomy requirements, maintaining hematocrit <45% to lessen thrombosis risk, and reversing iron deficiency–associated symptoms.93 PTG-300 is a hepcidin mimetic agent that resulted in a 70% reduction in transferrin saturation from baseline, without significant adverse events, in 62 healthy individuals. The efficacy and safety of PTG-300 in phlebotomy-requiring patients with PV are being evaluated in a 3-part phase 2 trial clinical trial (PTG-300-04), with early experience indicating minimal adverse events, prolonged therapeutic phlebotomy-free periods, and relief of systemic symptoms.
P-selectin is a cell adhesion molecule that is expressed by activated endothelial cells and platelets and plays an essential role in the initial recruitment of WBCs to the site of vessel injury as a thrombotic event develops. Data from several groups, including ours, report JAK2V617F+ endothelial cells in peripheral and splenic vessels in a subset of patients with MPN.71,72 Guy et al94 and Guadall et al70 showed that JAK2V617F+ endothelial cells promote thrombosis by upregulating endothelial cell P-selectin expression. HU therapy limited the expression of P-selectin by JAK2V617F+ endothelial cells, suggesting that the effects of HU on reducing thrombotic risk might be beyond that of mere normalization of blood cell counts. Furthermore, in JAK2V617F-knockin mice, administration of a P-selectin blocking antibody prevented the pathologically increased leukocyte adhesion associated with JAK2V617F+ endothelial cells and completely eliminated thrombus formation.94 The relevance of these data and its translation to clinical practice have been highlighted by a study of crizanlizumab, a humanized monoclonal antibody that binds to P-selectin and blocks its interaction with its ligand P-selectin glycoprotein ligand 1. Ataga et al reported that crizanlizumab therapy led to a 45.3% reduction in the annual rate of vaso-occlusive episodes in a randomized trial of patients with sickle cell disease.95 Evaluation of this agent in patients with MPN is in the planning stages.
Although the use of low-dose aspirin to prevent recurrent thrombotic events in patients with PV and ET is widespread, the data for such practices are limited. Landolfi et al reported that, in a randomized trial of 518 patients with PV followed for 3 years, treatment with a single daily dose of low-dose aspirin vs placebo reduced the incidence of thrombotic events without increasing the incidence of bleeding events.96 Recently, the widespread use of aspirin therapy for patients with low-risk ET has been questioned and abandoned by many skilled investigators. Alvarez-Larrán et al reported a retrospective study of patients with low-risk ET that indicated that low-dose aspirin therapy in patients with mutated CALR did not affect the risk of developing thrombotic episodes; in fact, it increased the incidence of bleeding episodes.97 However, these same investigators suggested that, in patients with JAK2V617+ with cardiovascular risk factors and leukocytosis, low-dose aspirin therapy might represent inadequate antiplatelet therapy. Rocca et al attributed such limitations of aspirin therapy to accelerated platelet production in patients with ET, which they speculated might reduce the duration of platelet cyclooxygenase-1 inhibition.98 In a clever biomarker-based trial, these investigators tried to overcome these limitations by increasing the frequency of aspirin administration to 2 or 3 times a day. They provide provocative data indicating that twice-daily low-dose aspirin therapy can lead to more adequate suppression of platelet activation in patients with ET without increasing gastrointestinal toxicity. Whether this alternative dosing schedule will result in further reductions in AT and VT events is the subject of continued investigations.
Statins are known to lower the incidence of thrombosis in individuals as a result of their powerful lipid-lowering effects, as well as their anti-inflammatory actions.99 Statins are known to have pleiotropic effects, including blunting monocyte and endothelial cell production of chemokines (eg, MCP-1, IL-6, IL-8, and RANTES), as well as affecting monocyte infiltration of atherosclerotic plaques. Statins reduce monocyte CD11β expression and directly bind to LFA1, thus inhibiting the monocyte-endothelial interactions that lead to atherosclerotic plaque formation.
Targeting the production or action of inflammatory cytokines is a logical and intriguing approach that is in concert with efforts of the vascular medicine community.41 Kleppe et al demonstrated in an elegant series of single-cell analyses that mutated and nonmutated MPN cells contributed to this increased cytokine production (IL-6, IL-8, and tumor necrosis factor-α).100 The CANTOS trial randomized 10 000 patients with a prior myocardial infarction and high hs-CRP levels to 3 doses of canakinumab, an IL-1β inhibitor, or placebo. In the absence of lipid-lowering effects and after 3.7 years, canakinumab treatment was associated with a significant reduction in the incidence of recurrent cardiovascular events.101 Unfortunately, neutropenia, thrombocytopenia, and a significantly greater number of deaths due to infection or sepsis were observed in patients treated with canakinumab. The toxicity profile of this agent might be less daunting in treating patients with PV and ET in whom leukocytosis and thrombocytosis are prevalent, and a reduction of white cells and platelets is frequently a therapeutic target. Additional agents that might be used include IL-6– or IL-8–neutralizing antibodies, or agents such as ladraxin, an oral agent that blocks the receptors for IL-8, CXCR1, and CXCR2.
Kleppe et al102 and Fisher et al103 presented convincing data that NF-κB, which serves as the transcriptional regulator of proinflammatory cytokine production, is active in MPN cells. An alternative approach to downregulating atherosclerosis-associated inflammation was first raised by Brown et al, who implicated the epigenetic regulator BRD4 in NF-κB–induced inflammation.104 BRD4/BET proteins play an important role in NF-κB–driven macrophage and endothelial cell–associated inflammation; their inhibition suppresses leukocyte rolling, adhesion, and transmigration in the endothelium, as well as atherosclerosis. Kleppe et al applied these findings to MPN mouse models and showed that BET inhibitors alone, as well as in combination with a JAK2 inhibitor, could dramatically reduce the burden of proinflammatory cytokines.102 A BET inhibitor, Constellation 0610, is being evaluated for the treatment of patients with MF, with promising effects on anemia, degree of splenomegaly, constitutional symptoms, and bone marrow fibrosis.
Alternative approaches to clinical trial design for prevention of thrombosis in MPNs
MPNs are similar to other chronic diseases and conditions with high thrombosis rates, such as sickle cell anemia, cancer, and aging, in that the thrombotic risk is confounded by patient-specific, disease-specific, genomic-specific, and environmental and therapeutic exposures.64,105-107 MPNs are clonal hematopoietic stem cell disorders in which morbidity and mortality are attributed to thrombosis, as well as to disease evolution to myelofibrosis, myelodysplasia, bone marrow failure, MPN blast phase, infection, development of secondary cancers, and aging. Each of these complications also raises the risk of developing thromboses. This association between contributory factors and the development of thrombotic events in MPN is highlighted in the recent study by De Stefano et al, who showed that patients with MPN who developed AT events, but not VT events, were at higher risk for developing a second malignancy (Figure 2).108 As a result of low thrombosis rates after diagnosis, and the fact that trials are also directed to treating these diseases and addressing their natural history, a single study end point to reduce thrombosis is frequently not possible when pursuing a clinical evaluation of a drug for patients with MPN. More recently, both large prospective trials that established the benefit of ruxolitinib or ropeginterferon over HU in hematocrit control or blood count and molecular response were not powered to examine thrombosis end points; powering them to do so would delay their execution and the reporting of important benefit to clonal suppression and disease modification.85,91 The validity of achievement of complete hematological remission (normalization of blood cell counts) alone should be questioned as a valid surrogate end point for reducing thrombotic risk in patients with MPN, and even more so now because we understand that JAK2V617F VAF on its own exerts thrombotic risk (Figure 1), even when blood counts are within the normal range.
Designing a therapy or trial that is specific to treat thrombosis risk, which is a very distal and, at times, disparate end point that does not address the most high-risk aspect of the disease, is not in the best interest of patients with MPN. Because PV and ET are diseases that are associated with survival that is measured in decades, studies of individual or combinations of therapeutic agents would require careful examination of large numbers of patients for prolonged periods of time. This dilemma has led some investigators to raise the possibility that such studies can best be achieved by using composite surrogate end points, such as normalization of blood cell counts, deep molecular remissions with elimination of MPN driver mutations and other CH-associated mutations, and a dampening of the levels of proinflammatory cytokines.
The last 10 years have unveiled the role of inflammation as causal and consequential to the development of CH, MPNs, and thrombosis (Figure 2). We can now frame thrombosis risk as being driven by elevated blood cell counts, as well as by specific inflammatory cytokines that are elaborated by MPN-specific driver mutations and host-specific inflammatory responses. Therapies that have direct or indirect effects on depleting or eradicating the MPN malignant clone are assumed to be able to reduce the MPN-associated thrombotic risk as a result of the elimination of the cascade of events that are prothrombotic and, ultimately, a consequence of the MPN clone. MPN therapeutics should focus on MPN clonal suppression and targeted anti-inflammatory therapeutics.
Acknowledgments
The authors thank the faculty who participated in the International Thrombosis in MPNs Workshop in Charleston, SC; in particular, Jyoti Nangalia, for contributions that formed the foundation of this review.
Authorship
Contribution: A.R.M. and R.H. designed the research and wrote the manuscript; and Y.Z.G. designed the research and edited the manuscript.
Conflict-of-interest disclosure: A.R.M. has acted as a consultant for PharmaEssentia. R.H. has acted as a consultant for Protagonist Therapeutics. Y.Z.G. declares no competing financial interests.
Correspondence: Alison R. Moliterno, Hematology Division, Department of Medicine, Johns Hopkins University School of Medicine; Ross Research 1025, 720 Rutland Ave, Baltimore, MD 21205; e-mail: amoliter@jhmi.edu.


