Key Points
Platelets are required throughout embryogenesis and into neonatal life to prevent intracerebral hemorrhage.
Timing of the onset of thrombocytopenia determines the location of intracerebral bleeds.

Abstract
Intracerebral hemorrhage (ICH) has a devastating impact on the neonatal population. Whether thrombocytopenia is sufficient to cause ICH in neonates is still being debated. In this study, we comprehensively investigated the consequences of severe thrombocytopenia on the integrity of the cerebral vasculature by using 2 orthogonal approaches: by studying embryogenesis in the Nfe2−/− mouse line and by using biologics (anti-GP1Bα antibodies) to induce severe thrombocytopenia at defined times during development. By using a mouse model, we acquired data demonstrating that platelets are required throughout fetal development and into neonatal life for maintaining the integrity of the cerebral vasculature to prevent hemorrhage and that the location of cerebral hemorrhage is dependent on when thrombocytopenia occurs during development. Importantly, this study demonstrates that fetal and neonatal thrombocytopenia-associated ICH occurs within regions of the brain which, in humans, could lead to neurologic damage.
Introduction
Platelets are anucleate blood cells derived from the megakaryocyte lineage. The most basic function of platelets is to elicit a rapid clotting response, most dramatically after trauma, but also as a homeostatic mechanism. Regarding the developmental role of platelets, severe thrombocytopenia (dangerously low numbers of circulating platelets) is most commonly caused by fetal and neonatal alloimmune thrombocytopenia (FNAIT). In FNAIT, transfer of maternal anti-platelet antibodies to the offspring results in rapid platelet destruction with consequences ranging from minor skin bleeds to major bleeding events in the brain, most commonly intracerebral hemorrhage (ICH).1 Treatment of FNAIT includes antenatal intravenous immunoglobulin (Ig) to suppress platelet destruction and postnatal platelet transfusion. In patients who do not have FNAIT, it has been reported that there is a poor correlation between severity of thrombocytopenia and risk of hemorrhage.2 When taken with the observation that platelet transfusions given to neonates with thrombocytopenia did not decrease the incidence of ICH,2,3 the role of platelets in preventing ICH in humans is unclear.
To investigate the developmental consequences of thrombocytopenia, many genetic disruption mouse models have been used in combination with a megakaryocyte-specific (platelet factor 4 [Pf4]) cre transgene to induce megakaryocyte and platelet death or to conditionally inactivate molecular regulators of platelet function. Both approaches have shown that disruption of platelets results in reproducible and sustained bleeding in the body of the embryo, particularly causing bleeding in the skin, blood-filled lymphatic vessels, and edema.4-9 In these models (which include deletion of Mcl1, Clec1b, Nbeal2, Unc13d, and Itga2b), ICH is detectable at embryonic day 12.5 (E12.5), but it seems to resolve as development proceeds. This suggests at least some involvement of platelets in preserving the integrity of the early fetal cerebral vasculature.
Observations from studying the Nfe2−/− mouse line argue against a clear developmental requirement for platelets. NF-E2 is a transcriptional master regulator of platelet biogenesis; in its absence, megakaryocytes are produced throughout development but platelet production is blocked.10-12 On the basis of publicly available gene expression data sets, Nfe2 is broadly expressed in the hematopoietic lineages but is not appreciably expressed within the endothelium (supplemental Figures 1 and 2, available on the Blood Web site); thus, it is expected that the phenotype of Nfe2−/− mice derives from defects in the hematopoietic system. Nfe2−/− neonates are born with <10% of normal platelet numbers at the expected Mendelian frequency but die within a few days as a result of systemic bleeding (including profound ICH).11 The observation that intralymphatic or ICH did not occur in utero11 led to the conclusion that platelets are dispensable during embryogenesis. Interestingly, it was observed that Nfe2−/− pups delivered by caesarean section did not have significant bleeding. This indicates that platelets are not required to preserve vascular integrity but are required to survive the trauma of birth.
Studies using a mouse model of FNAIT suggested that anti-platelet antibodies such as anti-β3 integrin can directly target the cerebral vasculature, and that it is the direct destruction of the cerebral vasculature rather than thrombocytopenia that causes fetal and neonatal ICH.13 Of note, ICH was not observed when anti-GP1Bα immunization was used to induce fetal and neonatal thrombocytopenia.13 Therefore, the question of whether thrombocytopenia is sufficient to drive neonatal ICH remains unresolved, but it is of high clinical relevance.
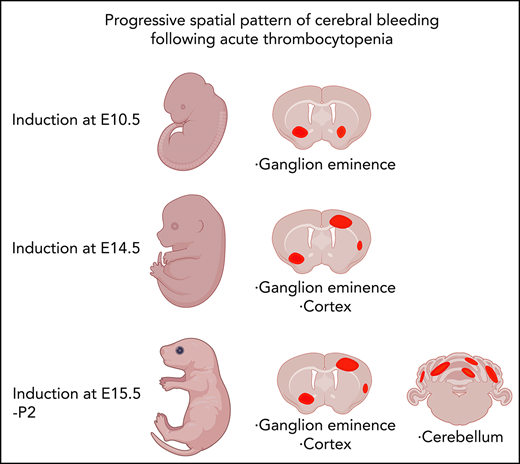
To understand whether thrombocytopenia is sufficient for ICH to occur, we have investigated 2 orthogonal mouse models of developmental thrombocytopenia: (1) the Nfe2−/− mouse line, which provides a genetic model of constitutive severe thrombocytopenia, and (2) anti-GP1Bα antibody (which specifically targets platelets without binding to the endothelium) to induce acute thrombocytopenia at specific developmental stages. By comprehensively investigating development of the cerebral vasculature in these thrombocytopenic contexts we have demonstrated that although platelets are not required to establish the cerebral vasculature, platelets are critical throughout prenatal and neonatal development to preserve its integrity. Of particular interest, we demonstrate that bleeding within regions of the brain that are of clinical importance in humans (the ganglionic eminence, cerebral cortex, and cerebellum) follows a striking pattern that is driven by the developmental timing of thrombocytopenia. These findings establish that thrombocytopenia is sufficient to drive ICH.
Methods
Mice
Nfe2+/− mice were maintained on a C57BL/6 background or a C57BL/6 and FVB/NJ mixed background (backcrossed for 2 generations). Mice treated with antibodies were on either a C57BL/6 or a 129/Sv background. Experimental procedures were approved by the Walter and Eliza Hall Institute Animal Ethics Committee. Developmental stages were determined morphologically by Theiler’s criteria. Timed matings were set up overnight, and the morning of a positive plug was designated E0.5. Embryos were collected between E10.5 and E18.5. When required, tail tips were collected in polymerase chain reaction tail buffer (Sigma) for genotyping. All experiments included comparative littermate wild-type controls or comparative controls from litters generated from an Nfe2+/− intercross collected on the same experimental day.
Analysis of circulating fetal platelets
Peripheral blood was collected as previously described10,14 from individual embryos in EDTA 7% fetal calf serum in calcium and magnesium-free Dulbecco’s phosphate-buffered saline. Samples were stained with antibodies (anti-CD41, anti-CD45, and anti-TER119) at room temperature. Flow cytometry was performed by using a BD LSR Fortessa system. Platelets were defined as CD45–TER119–CD41+ cells with a low forward scatter and side scatter profile. Data were analyzed using FlowJo software (TreeStar). Severe thrombocytopenia was defined as platelet numbers below 10% of the comparative control number (shown by a dashed line in relevant figure panels). This threshold was derived from the maximum percentage of circulating platelets observed in Nfe2−/− fetuses,10,12 which we considered to be the benchmark for severe thrombocytopenia.
Sample preparation and imaging
Whole embryos or dissected tissues were fixed with 4% paraformaldehyde overnight at 4°C. Generally, primary antibody incubations were performed overnight at 4°C, and secondary antibody incubations were performed for 6 hours at room temperature or overnight at 4°C. For analysis of fetal skin, samples were dissected, fixed, stained, and then flat mounted onto slides. For preparation of thick sections, fixed samples were embedded in 3% low-melting-temperature agarose and were oriented for coronal sectioning; 150- to 250-μm sections were generated using a Leica VT1000 S vibratome. Samples were stained with various combinations of anti-CD41-fluorescein isothiocyanate (MWReg30), anti-CD31-phycoerythrin (MEC13.3), anti-Ter119-allophycocyanin, anti-NG2 (purified), and goat anti-rabbit-Alexa-647. For confocal microscopy, tiled z-stack images were captured using a Zeiss LSM780 inverted confocal microscope. For light sheet microscopy, fixed samples were stained with directly conjugated antibodies for 3 days, washed, and then embedded in 3% ultra clean agarose. Mounted samples were cleared using the clear, unobstructed brain/body imaging cocktails and computational analysis (CUBIC) clearing method15 and were imaged on the Zeiss Z Light Sheet microscope. Data were processed and analyzed using Imaris (Bitplane) or Fiji (National Institutes of Health) software.
Hemorrhage scoring criteria
After assessment of gross morphology, hemorrhage in the head was identified as abnormal pooling of blood around the cerebral neural tissue or deep within the neural epithelium; hemorrhage in the body was identified as the abnormal collection of blood in the skin or the presumptive lymphatic system. From confocal imaging data, hemorrhage was identified by the presence of Ter119+ erythroid cells that were not contained within CD31+ cerebral vessels or by the presence of Ter119+ erythroid cells within the LYVE1+ lymphatic vasculature.
Induction of thrombocytopenia in utero
As previously described,16,17 5 μg/g of body weight of anti-GP1Bα antibody (Emfret clone R300) or IgG control antibody (Emfret clone R301) was intraperitoneally injected into pregnant female mice. Embryos were collected at 6 to 48 hours posttreatment. Cerebral bleeds were scored from thick vibratome sections. Postnatal day 0 (P0) to P1 pups were injected with 5 μg/g anti-GP1Bα antibody or IgG control in a 15-μL volume into the superficial temporal facial vein by using a Hamilton fine needle and syringe. Pups were collected at 6 or 48 hours after injection, and cerebral bleeds were scored.
Statistical analysis
Prism 9 (GraphPad) was used for data analysis and graph production. Data are represented as mean ± standard deviation (SD), and analyzed using the Student t test (2-way, unpaired). One-way analysis of variance (ANOVA; using Tukey’s P-value adjustment) was used for multiple comparisons. Contingency table analysis was performed using the 2-tailed χ2 test for the comparison of bleed frequencies. The number of independent experimental mice was designated by “n.” The value P < .05 shown in either the figure or the figure legend indicates statistically significant differences.
Results
Cerebral hemorrhage in Nfe2−/− neonates arises in utero
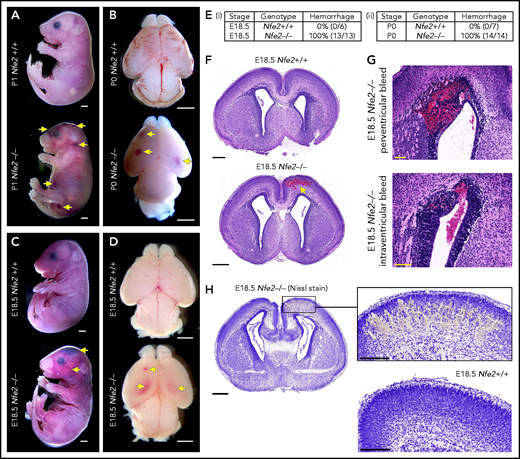
Nfe2−/− mice are severely thrombocytopenic throughout embryogenesis, as such, they are a useful model for understanding whether thrombocytopenia results in in utero ICH. To this end, we performed a comprehensive analysis of prenatal Nfe2−/− mice. Consistent with previous descriptions,11 P0 to P1 Nfe2−/− pups exhibited signs of bleeding in the skin and within internal organs (Figure 1A; supplemental Figure 3). Examination of brains from Nfe2−/− mice confirmed the presence of hemorrhage (Figure 1B). When fetuses were collected at E18.5 (24 hours before birth), we made the surprising finding that all embryos examined had large bleeds in the head and the body (Figure 1C-E). Brains dissected at E18.5 showed obvious cerebral and intraventricular bleeds (Figure 1D). Further histologic studies showed that all ICH-associated types of bleeds were present, including cerebral, periventricular, and intraventricular hemorrhage (Figure 1F-G). Nissl staining of the brain tissue revealed the structural changes caused by the presence of large bleeds (Figure 1H). This phenotype had previously been attributed to the trauma of birth,11 but we found that it occurred in utero. This finding led us to ask, How early in development does ICH occur in a thrombocytopenic context?
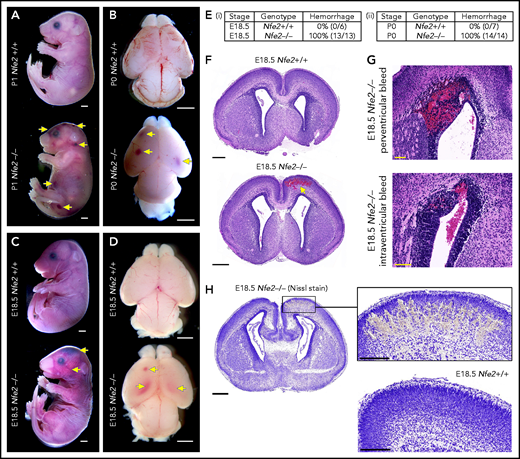
Hemorrhage in Nfe2−/− mice occurs in utero. (A) P1 Nfe2+/+ and Nfe2−/− pups born by natural delivery. Yellow arrows point to representative head and body bleeds seen in the Nfe2−/− mice (Nfe2+/+, n = 8; Nfe2−/−, n = 13). Scale bars, 1 mm. (B) P0 Nfe2+/+ and Nfe2−/− dissected brains. Yellow arrows point to bleeds in both frontal lobes. Scale bars, 1 mm. (C) E18.5 Nfe2+/+ and Nfe2−/− mice delivered by caesarean section. Yellow arrows point to bleeds. Scale bars, 1 mm. (D) E18.5 Nfe2+/+ and Nfe2−/− dissected brains. Yellow arrows point to frontal and ventricle bleeds in the Nfe2−/− pups. Scale bars, 1 mm. (Ei-ii) Tables comparing the percentage of bleeds between E18.5 Nfe2+/+ pups (Nfe2+/+ n = 10, Nfe2−/− n = 10) and P0 Nfe2−/− pups (Nfe2+/+ n = 8, Nfe2−/− n = 13). (F-G) Histologic analysis demonstrates multiple bleed types present in the E18.5 Nfe2−/− dissected brains, but not in the Nfe2+/+ brain. Representative bleeds are shown in the cerebral cortex along with periventricular and intraventricular bleeds. Scale bars, 500 µm (F); 100 µm (G). (H) Nissl staining of E18.5 Nfe2−/− (n = 3) coronal brain section and the corresponding region in the Nfe2+/+ brain (n = 3). Scale bar, 500 µm (inset, 200 µm).
Hemorrhage in Nfe2−/− mice occurs in utero. (A) P1 Nfe2+/+ and Nfe2−/− pups born by natural delivery. Yellow arrows point to representative head and body bleeds seen in the Nfe2−/− mice (Nfe2+/+, n = 8; Nfe2−/−, n = 13). Scale bars, 1 mm. (B) P0 Nfe2+/+ and Nfe2−/− dissected brains. Yellow arrows point to bleeds in both frontal lobes. Scale bars, 1 mm. (C) E18.5 Nfe2+/+ and Nfe2−/− mice delivered by caesarean section. Yellow arrows point to bleeds. Scale bars, 1 mm. (D) E18.5 Nfe2+/+ and Nfe2−/− dissected brains. Yellow arrows point to frontal and ventricle bleeds in the Nfe2−/− pups. Scale bars, 1 mm. (Ei-ii) Tables comparing the percentage of bleeds between E18.5 Nfe2+/+ pups (Nfe2+/+ n = 10, Nfe2−/− n = 10) and P0 Nfe2−/− pups (Nfe2+/+ n = 8, Nfe2−/− n = 13). (F-G) Histologic analysis demonstrates multiple bleed types present in the E18.5 Nfe2−/− dissected brains, but not in the Nfe2+/+ brain. Representative bleeds are shown in the cerebral cortex along with periventricular and intraventricular bleeds. Scale bars, 500 µm (F); 100 µm (G). (H) Nissl staining of E18.5 Nfe2−/− (n = 3) coronal brain section and the corresponding region in the Nfe2+/+ brain (n = 3). Scale bar, 500 µm (inset, 200 µm).
Sensitivity to in utero ICH occurs concurrently with cerebral vascularization
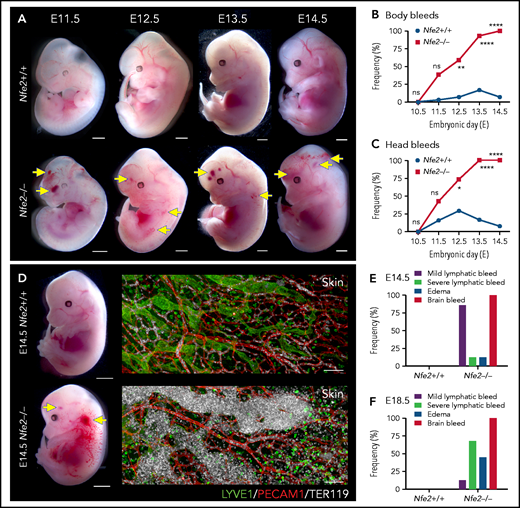
The first platelets appear in the peripheral blood between E9.5 and E10.5.10,14,18 Thus, any developmental process that requires platelets has the potential to be affected from mid-gestation. We collected litters of mice between E10.5 and E14.5 and compared the gross appearance of wild-type and Nfe2−/− littermates. At E10.5, wild-type and Nfe2−/− embryos were indistinguishable (supplemental Figure 4). Focal frontal lobe hemorrhage was observed in Nfe2−/− mice from E11.5, and the penetrance of ICH increased as development proceeded (Figure 2A-C). Although hemorrhage was observed in a portion of wild-type mice between E11.5 and E13.5, by E12.5 the presence of ICH and body bleeds was a distinguishing feature of the Nfe2−/− genotype (Figure 2B-C). We observed degrees of severity in ICH among Nfe2−/− mice, which ranged from focal hemorrhage (Figure 2A) to more severe and widespread hemorrhage (supplemental Figure 5).
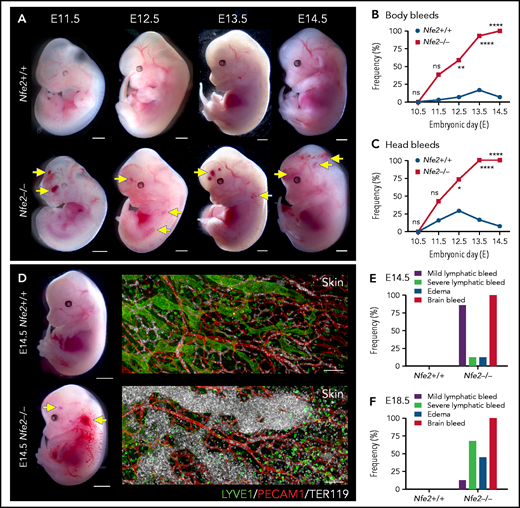
Cerebral hemorrhage in Nfe2−/− mice occurs as early as E11.5 and persists through development. (A) Representative images of Nfe2−/− and age-matched Nfe2+/+ control embryos at E11.5 (Nfe2+/+ n = 16; Nfe2−/− n = 25), E12.5 (Nfe2+/+ n = 13; Nfe2−/− n = 18), E13.5 (Nfe2+/+ n = 16; Nfe2−/− n = 14), and E14.5 (Nfe2+/+ n = 14; Nfe2−/− n = 14). Scale bars, 1 mm. (B-C) Scored bleeding phenotypes in the Nfe2+/+ and Nfe2−/− embryo body (B) or head (C) from E10.5 to E14.5. (D) Bleeding into the lymphatic sacs and lymphatic vessels is seen at E14.5 in the Nfe2−/− but not in the Nfe2+/+ mice. Immunofluorescence staining of E14.5 Nfe2+/+ skin showing that erythrocytes (TER119, white) are restricted to the blood vessels (PECAM1, red), whereas erythrocytes are readily detected within dilated lymphatic vessels (LYVE1, green) in Nfe2−/− mice. Embryo scale bars, 2 mm. Skin confocal scale bars, 100 µm. (E-F). Frequency of bleed types at E14.5 (Nfe2+/+ n = 14; Nfe2−/− n = 12) (E) and E18.5 in Nfe2+/+ (n = 10) and Nfe2−/− embryos (n = 10) (F). Scale bars for whole embryos, 1 mm; scale bars for skin, 150 μm. Contingency table analysis was performed using the 2-tailed χ2 test for the comparison of bleed frequencies (B-C). *P < .05; ****P < .0001. ns, not significant.
Cerebral hemorrhage in Nfe2−/− mice occurs as early as E11.5 and persists through development. (A) Representative images of Nfe2−/− and age-matched Nfe2+/+ control embryos at E11.5 (Nfe2+/+ n = 16; Nfe2−/− n = 25), E12.5 (Nfe2+/+ n = 13; Nfe2−/− n = 18), E13.5 (Nfe2+/+ n = 16; Nfe2−/− n = 14), and E14.5 (Nfe2+/+ n = 14; Nfe2−/− n = 14). Scale bars, 1 mm. (B-C) Scored bleeding phenotypes in the Nfe2+/+ and Nfe2−/− embryo body (B) or head (C) from E10.5 to E14.5. (D) Bleeding into the lymphatic sacs and lymphatic vessels is seen at E14.5 in the Nfe2−/− but not in the Nfe2+/+ mice. Immunofluorescence staining of E14.5 Nfe2+/+ skin showing that erythrocytes (TER119, white) are restricted to the blood vessels (PECAM1, red), whereas erythrocytes are readily detected within dilated lymphatic vessels (LYVE1, green) in Nfe2−/− mice. Embryo scale bars, 2 mm. Skin confocal scale bars, 100 µm. (E-F). Frequency of bleed types at E14.5 (Nfe2+/+ n = 14; Nfe2−/− n = 12) (E) and E18.5 in Nfe2+/+ (n = 10) and Nfe2−/− embryos (n = 10) (F). Scale bars for whole embryos, 1 mm; scale bars for skin, 150 μm. Contingency table analysis was performed using the 2-tailed χ2 test for the comparison of bleed frequencies (B-C). *P < .05; ****P < .0001. ns, not significant.
Blood-filled lymphatics is a component of the Nfe2−/− phenotype
As discussed above, a concern often raised with the proposed role of platelets in maintaining the separation of blood and lymphatic vascular systems during embryogenesis is the absence of an in utero Nfe2−/− bleeding phenotype. We noted, as has been observed in other models of fetal megakaryocyte and platelet perturbation, that blood was first observed in the vessels of jugular lymphatic sacs of Nfe2−/− fetuses from E12.5. By E14.5, widespread blood-filled lymphatic vessels were observed (Figure 2D). In addition to ICH and blood-filled lymphatic vessels, some evidence of edema was observed at E14.5 (Figure 2E). By E18.5, the incidence of severe lymphatic bleeding and edema had increased (Figure 2F). These findings corroborate the importance of platelets for the prevention of blood-filled lymphatic vessels and the onset of severe edema.
ICH is initiated within the ganglionic eminence and then develops to involve the cortex and ventricles
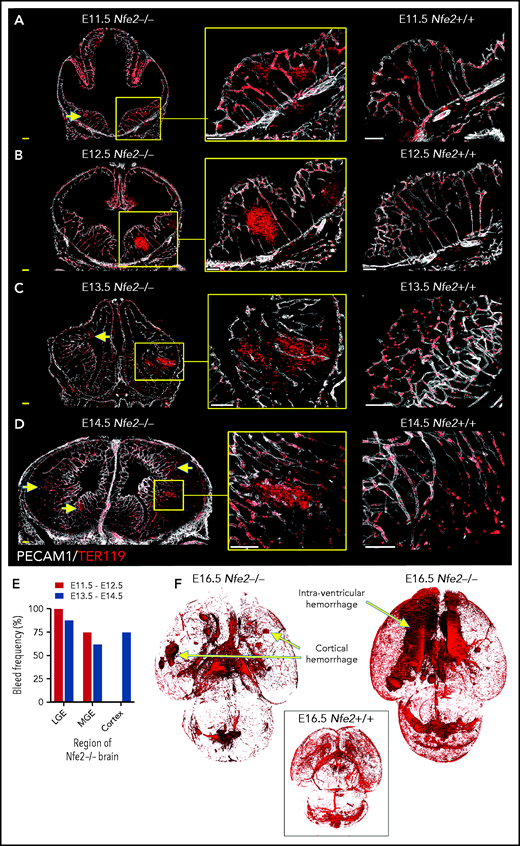
To understand the spatial axis of in utero ICH, we performed large-scale 3D confocal imaging by using thick coronal sections of brains collected from E11.5 to E18.5 wild-type and Nfe2−/− fetuses and made the intriguing discovery that the location of ICH evolved as embryogenesis proceeded. Between E11.5 and E12.5, hemorrhage was restricted to the ganglionic eminence. This was detected at E11.5 as micro bleeds that developed into large, fully penetrant bleeds by E12.5 (Figure 3A-B). From E13.5 onward, the pattern of hemorrhage included the medial and lateral ganglionic eminence and began to include the cortex (Figure 3C-E). Focal hemorrhage within the ganglionic eminence or cortex was not detected within E11.5 to E14.5 wild-type mice. Light sheet microscopy was used to generate full-volume images of wild-type and Nfe2−/− E16.5 brains. From these data, we observed that the spatial pattern of hemorrhage continued to evolve over developmental time, with the inclusion of intraventricular hemorrhage at E16.5 (Figure 3F).
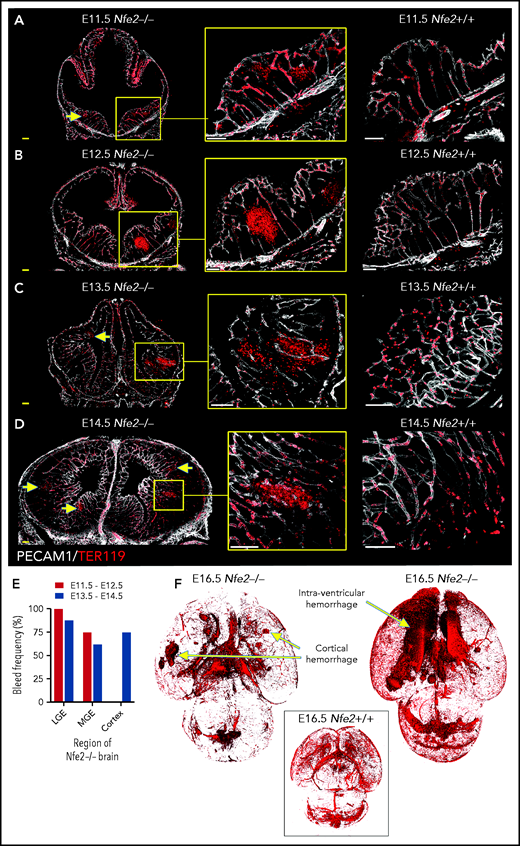
ICH first occurs in the ganglionic eminence and progresses to ventricle and cortical involvement. (A-D) Representative confocal images from thick sections of Nfe2+/+ (n = 3 per stage) and Nfe2−/− (n = 3 per stage) brains at (A) E11.5, (B) E12.5, (C) E13.5, and (D) E14.5. Sites of ICH are indicated by yellow arrows. TER119 (red), erythrocytes; PECAM1 (white), cerebral vasculature. Scale bars for E11.5 to E13.5, 150 μm; inset, 50 μm; scale bars for E14.5, 200 μm; inset, 50 μm . (E) Summary of hemorrhage types between E11.5 and E12.5 (n = 5) and between E13.5 and E14.5 (n = 8) scored from vibratome cut coronal brain sections. LGE, lateral ganglionic eminence, MGE, medial ganglionic eminence. (F) Light sheet microscopy showing spatial location of hemorrhage in the E16.5 Nfe2−/− embryos, including multiple cerebral bleeds and large ventricle hemorrhage (n = 2). No bleeds can be seen in the E16.5 Nfe2+/+ brain control (n = 2).
ICH first occurs in the ganglionic eminence and progresses to ventricle and cortical involvement. (A-D) Representative confocal images from thick sections of Nfe2+/+ (n = 3 per stage) and Nfe2−/− (n = 3 per stage) brains at (A) E11.5, (B) E12.5, (C) E13.5, and (D) E14.5. Sites of ICH are indicated by yellow arrows. TER119 (red), erythrocytes; PECAM1 (white), cerebral vasculature. Scale bars for E11.5 to E13.5, 150 μm; inset, 50 μm; scale bars for E14.5, 200 μm; inset, 50 μm . (E) Summary of hemorrhage types between E11.5 and E12.5 (n = 5) and between E13.5 and E14.5 (n = 8) scored from vibratome cut coronal brain sections. LGE, lateral ganglionic eminence, MGE, medial ganglionic eminence. (F) Light sheet microscopy showing spatial location of hemorrhage in the E16.5 Nfe2−/− embryos, including multiple cerebral bleeds and large ventricle hemorrhage (n = 2). No bleeds can be seen in the E16.5 Nfe2+/+ brain control (n = 2).
Thrombocytopenia does not impair the establishment of the cerebral vasculature
The ICH observed in Clec1b−/− embryos is caused by defective vasculature patterning (tortuous cerebral vascular appearance) associated with diminished pericyte coverage of the cerebral vasculature and possible impaired pericyte-endothelial attachment.5 We next asked whether the cerebral hemorrhage in Nfe2–/– mice involved similar broad changes to vascular structure.
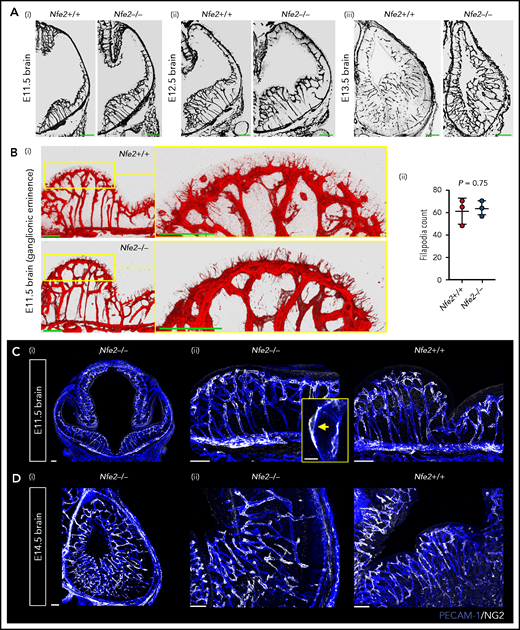
Between E10.5 and E13.5, cerebral vessel ingression occurs from the perineural vascular plexus into the neural epithelium in a ventral-to-dorsal direction.19,20 We observed that Nfe2−/− vessels seemed to sprout in a wild-type-like pattern between E11.5 and E13.5 (Figure 4A). Tip cells guide the growth of blood vessel sprouts from preexisting vasculature, and in the developing brain, periventricular vessels extend filipodia toward the ventricle in the ganglionic eminence region.21 Thus, another indicator of normal vessel development is the number of filipodia that extend from tip cells and the direction of growth. We found that similar to wild-type controls, the filipodia of Nfe2−/− tip cells in the lateral ganglionic eminence were directed toward the lateral ventricular surface (Figure 4Bi). Quantification of filipodia numbers further confirmed that Nfe2−/− embryos were undergoing normal cerebral vascular development (Figure 4Bii).
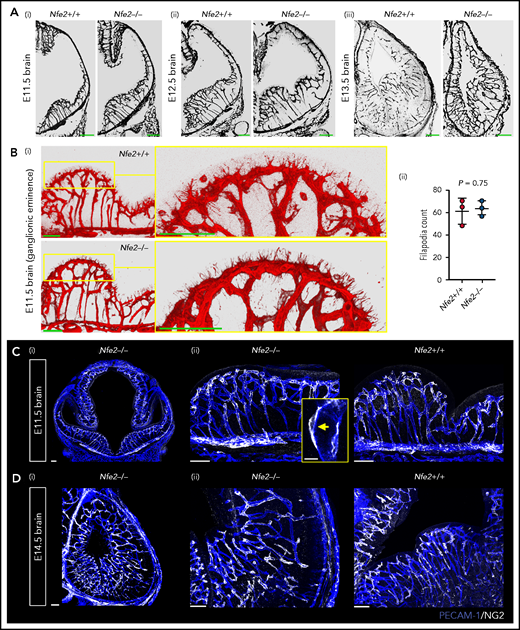
Cerebral vasculature development and pericyte recruitment is not disrupted in Nfe2−/− mice. (A) Coronal brain sections stained with PECAM-1 from E11.5 to E13.5 embryos reveal the stages of blood vessel development in Nfe2+/+ and Nfe2−/− mice (n = 3 embryos per genotype per stage; scale bars, 100 μm). (B) Identification (i) and quantification (ii) and of tip cell filipodia in the ganglion eminence of Nfe2+/+ and Nfe2−/− mice (n = 3 per genotype). Horizontal bar = mean; error = SD (ii). Scale bars, 50 μm). Data are represented as mean ± SD, and analyzed using Student t test (2-way, unpaired). (C) Pericyte (NG2, white) coverage of the E11.5 cerebral vasculature (PECAM-1, blue) in the head (scale bars, 100 μm) (i) and the ganglionic eminence (scale bars, 50 μm) (n = 3 per genotype) (ii). Inset, optical section of a pericyte in close association with a vessel. (D) Pericyte (NG2, white) coverage of the E14.5 cerebral vasculature (PECAM-1, blue) in the head (scale bars, 100 μm) (i) and ganglionic eminence (scale bars, 50 μm) (ii) (n = 3 per genotype).
Cerebral vasculature development and pericyte recruitment is not disrupted in Nfe2−/− mice. (A) Coronal brain sections stained with PECAM-1 from E11.5 to E13.5 embryos reveal the stages of blood vessel development in Nfe2+/+ and Nfe2−/− mice (n = 3 embryos per genotype per stage; scale bars, 100 μm). (B) Identification (i) and quantification (ii) and of tip cell filipodia in the ganglion eminence of Nfe2+/+ and Nfe2−/− mice (n = 3 per genotype). Horizontal bar = mean; error = SD (ii). Scale bars, 50 μm). Data are represented as mean ± SD, and analyzed using Student t test (2-way, unpaired). (C) Pericyte (NG2, white) coverage of the E11.5 cerebral vasculature (PECAM-1, blue) in the head (scale bars, 100 μm) (i) and the ganglionic eminence (scale bars, 50 μm) (n = 3 per genotype) (ii). Inset, optical section of a pericyte in close association with a vessel. (D) Pericyte (NG2, white) coverage of the E14.5 cerebral vasculature (PECAM-1, blue) in the head (scale bars, 100 μm) (i) and ganglionic eminence (scale bars, 50 μm) (ii) (n = 3 per genotype).
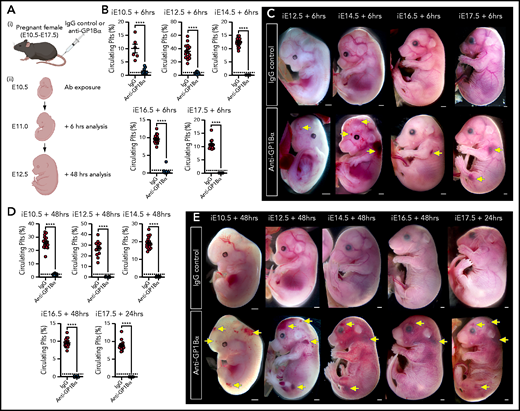
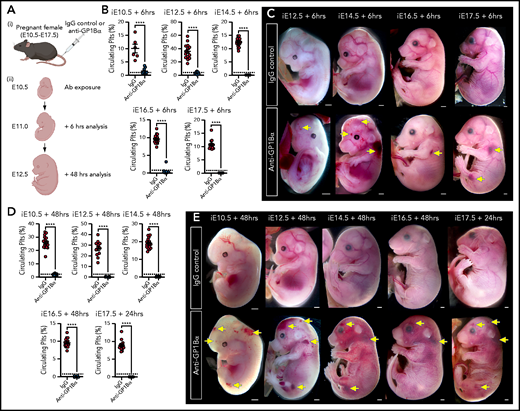
Timed induction of in utero thrombocytopenia. (A) Experimental scheme: mice were injected with anti-GP1Bα or IgG control antibodies between E10.5 and E17.5 and analyzed at 6 hours and 48 hours after injection for platelet numbers and hemorrhage phenotypes. (B) Quantification of circulating platelets as a percentage of peripheral blood at 6 hours after injection at E10.5 to E17.5. (C) Images of embryos 6 hours after treatment (E12.5: IgG n = 12, GP1Bα n = 15; E14.5: IgG n = 18, GP1Bα n = 17; E16.5: IgG n = 14, GP1Bα n = 12; E17.5: IgG n = 16, GP1Bα n = 8). Scale bars, 1 mm. (D) Circulating platelets 48 hours posttreatment. (E) E10.5 to E16.5 embryos 48 hours and E17.5 embryos 24 hours posttreatment (E10.5: IgG n = 16, GP1Bα n = 8; E12.5: IgG n = 12, GP1Bα n = 17; E14.5: IgG n = 17, GP1Bα n = 16; E16.5: IgG n = 18, GP1Bα n = 14; E17.5: IgG n = 17, GP1Bα n = 14). Scale bars, 1 mm. (B,D) Data are represented as mean 6 SD and analyzed using the Student t test (2-way, unpaired). **** P < .0001. Ab, antibody. Yellow arrows indicate sites of hemorrhage.
Timed induction of in utero thrombocytopenia. (A) Experimental scheme: mice were injected with anti-GP1Bα or IgG control antibodies between E10.5 and E17.5 and analyzed at 6 hours and 48 hours after injection for platelet numbers and hemorrhage phenotypes. (B) Quantification of circulating platelets as a percentage of peripheral blood at 6 hours after injection at E10.5 to E17.5. (C) Images of embryos 6 hours after treatment (E12.5: IgG n = 12, GP1Bα n = 15; E14.5: IgG n = 18, GP1Bα n = 17; E16.5: IgG n = 14, GP1Bα n = 12; E17.5: IgG n = 16, GP1Bα n = 8). Scale bars, 1 mm. (D) Circulating platelets 48 hours posttreatment. (E) E10.5 to E16.5 embryos 48 hours and E17.5 embryos 24 hours posttreatment (E10.5: IgG n = 16, GP1Bα n = 8; E12.5: IgG n = 12, GP1Bα n = 17; E14.5: IgG n = 17, GP1Bα n = 16; E16.5: IgG n = 18, GP1Bα n = 14; E17.5: IgG n = 17, GP1Bα n = 14). Scale bars, 1 mm. (B,D) Data are represented as mean 6 SD and analyzed using the Student t test (2-way, unpaired). **** P < .0001. Ab, antibody. Yellow arrows indicate sites of hemorrhage.
We next investigated the pericyte coverage of the vasculature in the Nfe2−/− ganglionic eminence. Pericytes are an essential component for the maintenance of cerebral vascular integrity, and severe ICH occurs if pericyte-endothelial interaction is disrupted.5,22 In humans, it has been suggested that paucity in pericyte coverage in the ganglionic eminence could sensitize the region to hemorrhage.23,24 We found that pericyte coverage of the vasculature in the E11.5 ganglionic eminence (Figure 4C) and the E14.5 cerebral cortex was comparable between wild-type and Nfe2−/− fetuses (Figure 4D). Furthermore, pericytes were readily observed to be tightly associated with the Nfe2−/− cerebral vasculature (Figure 4C). These data indicate that thrombocytopenia-associated hemorrhage does not result from broad changes to cerebral vasculature growth, patterning, or pericyte coverage.
In utero induction of thrombocytopenia recapitulates the Nfe2−/− bleeding phenotype
Anti-GP1Bα antibodies specifically target platelets16 and have been shown to induce severe fetal thrombocytopenia when injected into pregnant adult mice.17 We adopted this in utero induction method to independently test the model that thrombocytopenia is sufficient to drive ICH and to gain insight into the temporal requirement for platelets.
To this end, we injected 5 μg/g body weight of anti-GP1Bα or IgG control antibody into pregnant mice between E10.5 and E17.5 (Figure 6A). Six hours after administration of anti-GP1Bα was enough time to induce severe fetal thrombocytopenia (between 0.2% and 16% of control platelet numbers; Figure 6B; supplemental Table 1). Assessment of the gross appearance of treated litters revealed the presence of Nfe2−/−-like bleeding features at each of the induced stages, including signs of hemorrhage in the skin and signs of ICH (Figure 6C). At 48 hours posttreatment, thrombocytopenia persisted at 0.2% to 8% of control platelet numbers (Figure 6D; supplemental Table 1), with hemorrhage becoming markedly more severe at all of the developmental stages tested as characterized by multiple sites of hemorrhage in the skin (Figure 6E).
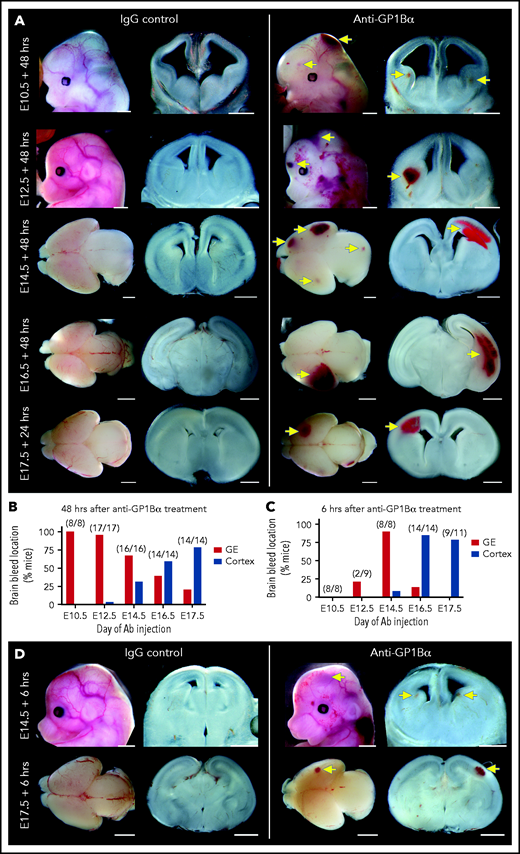
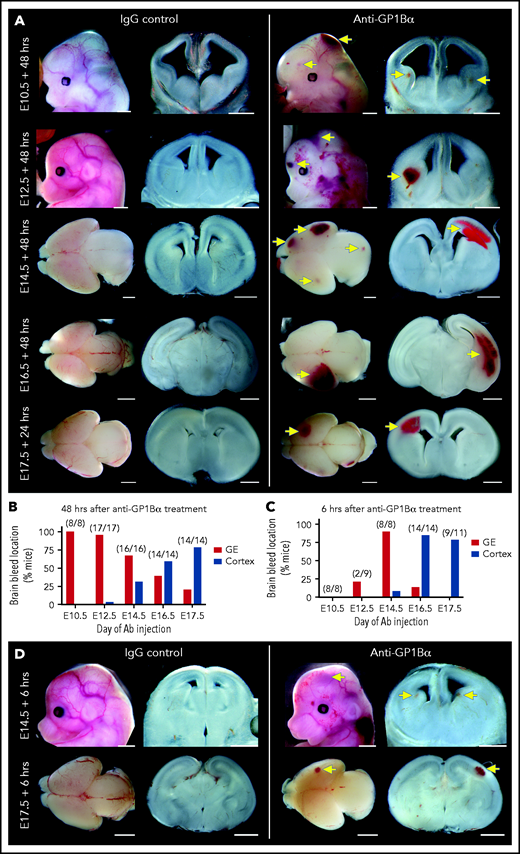
Identification of a spatial pattern of ICH. (A) Representative images of E10.5 to E17.5 heads and brains 48 hours after treatment with anti-GP1Bα or IgG (scale bars, 1 mm). (B) Frequency of hemorrhage in the ganglionic eminence (GE) and the cortex of treated E10.5 to E17.5 mice after 48 hours (E10.5: IgG n = 16, GP1Bα n = 8; E12.5: IgG n = 12, GP1Bα n = 17; E14.5: IgG n = 17, GP1Bα n = 16; E16.5: IgG n = 18, GP1Bα n = 14; E17.5: IgG n = 17, GP1Bα n = 14). Total number of mice with ICH is provided in parentheses. (C) Frequency of hemorrhage in the GE and the cortex of treated E10.5 to E17.5 mice after 6 hours (E10.5: IgG n = 8, GP1Bα n = 8; E12.5: IgG n = 9, GP1Bα n = 8; E14.5: IgG n = 7, GP1Bα n = 8; E16.5: IgG n = 7, GP1Bα n = 14; E17.5: IgG n = 6, GP1Bα n = 11). Total number of mice with ICH is provided in parentheses. (D) Representative images of hemorrhage in E14.5 (IgG n = 6, GP1Bα n = 7) and E17.5 (IgG n = 4, GP1Bα n = 6) embryos 6 hours posttreatment (scale bars, 1 mm). Yellow arrows indicate sites of hemorrhage.
Identification of a spatial pattern of ICH. (A) Representative images of E10.5 to E17.5 heads and brains 48 hours after treatment with anti-GP1Bα or IgG (scale bars, 1 mm). (B) Frequency of hemorrhage in the ganglionic eminence (GE) and the cortex of treated E10.5 to E17.5 mice after 48 hours (E10.5: IgG n = 16, GP1Bα n = 8; E12.5: IgG n = 12, GP1Bα n = 17; E14.5: IgG n = 17, GP1Bα n = 16; E16.5: IgG n = 18, GP1Bα n = 14; E17.5: IgG n = 17, GP1Bα n = 14). Total number of mice with ICH is provided in parentheses. (C) Frequency of hemorrhage in the GE and the cortex of treated E10.5 to E17.5 mice after 6 hours (E10.5: IgG n = 8, GP1Bα n = 8; E12.5: IgG n = 9, GP1Bα n = 8; E14.5: IgG n = 7, GP1Bα n = 8; E16.5: IgG n = 7, GP1Bα n = 14; E17.5: IgG n = 6, GP1Bα n = 11). Total number of mice with ICH is provided in parentheses. (D) Representative images of hemorrhage in E14.5 (IgG n = 6, GP1Bα n = 7) and E17.5 (IgG n = 4, GP1Bα n = 6) embryos 6 hours posttreatment (scale bars, 1 mm). Yellow arrows indicate sites of hemorrhage.
Induction of thrombocytopenia at discrete development stages drives a spatial pattern of ICH
We performed a detailed assessment of the spatial distribution of ICH using thick coronal sections of treated brains. We discovered that when thrombocytopenia was induced between E10.5 and E12.5, the most frequent site of hemorrhage was within the ganglionic eminence, but when it was induced between E14.5 and E17.5, the location of ICH shifted toward a cortical bias (Figure 7A-B). Analysis of mice 6 hours after treatment revealed that the hemorrhagic lesion occurred rapidly after the induction of thrombocytopenia (Figure 7C-D).
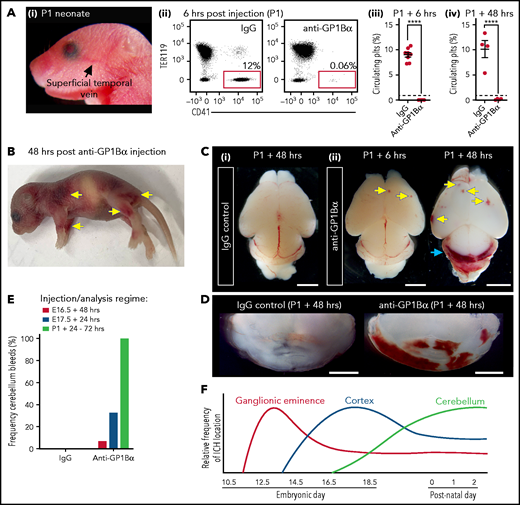
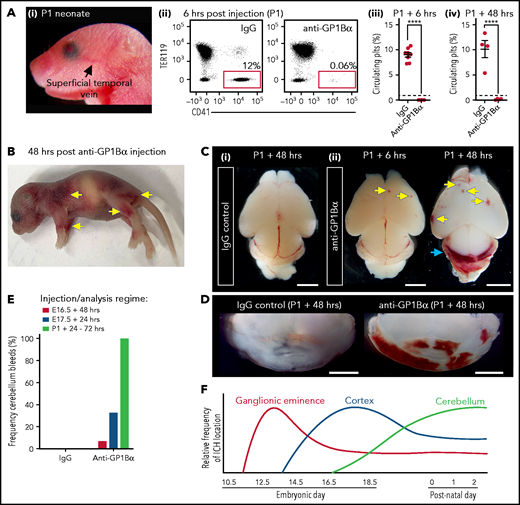
Induction of thrombocytopenia in neonates results in cerebellar hemorrhage. (A) (i) Image highlighting the facial vein of a P1 mouse used to deliver anti-GP1bα and IgG control antibodies. (ii) Representative flow cytometry plots of P1 peripheral blood stained with markers of erythrocytes (TER119) and platelets (CD41). (iii) Quantification of circulating platelets at 6 hours (IgG n = 8, GP1Bα n = 8) and 48 hours (IgG n = 4, GP1Bα n = 4) posttreatment. (B) Representative image of a P1 neonate 48 hours after treatment with anti-GP1bα (n = 7 embryos). Yellow arrows indicate sites of hemorrhage. (C) (i-iii) Representative images of brains 6 hours (IgG n = 8, GP1Bα n = 8) and 48 hours (IgG n = 4, GP1Bα n = 4) posttreatment. Yellow arrows indicate sites of cortical hemorrhage; the blue arrow highlights cerebellar hemorrhage (scale bars, 1 mm). (D) Coronal sections of cerebellum 48 hours posttreatment (bars, 1 mm). (E) Frequency of cerebellar hemorrhage in mice treated at E16.5 (IgG n = 10, GP1Bα n = 16), E17.5 (IgG n = 10, GP1Bα n = 12), or P1 (IgG n = 4, GP1Bα n = 7). (F) Summary of changes in ICH location during embryogenesis. ICHs in the ganglionic eminence, cortex, and cerebellum occur sequentially during development in a thrombocytopenic context.
Induction of thrombocytopenia in neonates results in cerebellar hemorrhage. (A) (i) Image highlighting the facial vein of a P1 mouse used to deliver anti-GP1bα and IgG control antibodies. (ii) Representative flow cytometry plots of P1 peripheral blood stained with markers of erythrocytes (TER119) and platelets (CD41). (iii) Quantification of circulating platelets at 6 hours (IgG n = 8, GP1Bα n = 8) and 48 hours (IgG n = 4, GP1Bα n = 4) posttreatment. (B) Representative image of a P1 neonate 48 hours after treatment with anti-GP1bα (n = 7 embryos). Yellow arrows indicate sites of hemorrhage. (C) (i-iii) Representative images of brains 6 hours (IgG n = 8, GP1Bα n = 8) and 48 hours (IgG n = 4, GP1Bα n = 4) posttreatment. Yellow arrows indicate sites of cortical hemorrhage; the blue arrow highlights cerebellar hemorrhage (scale bars, 1 mm). (D) Coronal sections of cerebellum 48 hours posttreatment (bars, 1 mm). (E) Frequency of cerebellar hemorrhage in mice treated at E16.5 (IgG n = 10, GP1Bα n = 16), E17.5 (IgG n = 10, GP1Bα n = 12), or P1 (IgG n = 4, GP1Bα n = 7). (F) Summary of changes in ICH location during embryogenesis. ICHs in the ganglionic eminence, cortex, and cerebellum occur sequentially during development in a thrombocytopenic context.
To understand whether sensitivity to thrombocytopenia persists beyond gestation, we treated P1 neonates with anti-GP1Bα or IgG control antibody via the superficial temporal vein (Figure 8Ai). As observed with in utero treatment, delivery of anti-GP1Bα resulted in the rapid induction of severe thrombocytopenia (Figure 8Aii-iv). Consistent with the postnatal Nfe2−/− phenotype, multiple sites of hemorrhage in the skin were observed (Figure 8B), and all mice exhibited ICH by 48 hours posttreatment (Figure 8C-E). These data indicate that the trauma of natural delivery is not necessary to explain postnatal hemorrhage but that thrombocytopenia alone is sufficient for this to occur. Furthermore, scoring the location of ICH in the thrombocytopenic E16.5 to P1 brain revealed that hemorrhage within the cerebellum is a late developmental sensitivity (Figure 8C-E). These data confirmed that thrombocytopenia was sufficient to drive ICH and revealed that the spectrum of ICH observed was driven by a temporal axis (Figure 8F) (ie, the location of ICH is dependent on both the absence of platelets and when the absence occurs).
Discussion
Understanding whether platelets have a direct role in preventing ICH is critical because diagnosing and treating thrombocytopenia in prenatal and neonatal individuals is possible, thus preventing thrombocytopenia-associated ICH and ICH-associated morbidities such as cerebral palsy. Uncertainty regarding the role of platelets in ICH derives from the following observations: (1) previous investigations of the Nfe2−/− mouse line suggesting that absence of platelets during embryogenesis did not lead to in utero damage,11 and (2) the observation that ICH caused by anti-β3 integrin antibodies is not driven by thrombocytopenia but by directly targeting the cerebral vasculature.13
From our detailed investigation of prenatal and neonatal Nfe2−/− mice, we discovered that the neonatal bleeding phenotype arose during development rather than after natural delivery. In addition to the prenatal onset of ICH, we observed a high frequency of blood-filled lymphatic vessels in Nfe2−/− fetuses. These data establish the requirement of platelets for preventing ICH and also reconcile the disparate findings of the original description of the Nfe2−/− mouse line11 with the phenotype of other platelet-specific mutant mouse lines.4-9,25,26
To test the temporal requirement for platelets during embryogenesis and into early neonatal life, we induced thrombocytopenia at precise developmental stages. We found that the conceptus becomes sensitive to thrombocytopenia between E10.5 and E12.5, concomitant with the emergence of the fetal platelet mass.10,14,18 Furthermore, all of the developmental stages exhibited sensitivity to thrombocytopenia. This indicates that platelets are required during the fetal and neonatal periods to prevent bleeding; loss of platelets drives ICH at this time. Although it is unknown how early in human development platelets emerge, the first megakaryocytes appear at week 5 of gestation.27 Because we have shown that, within 2 days of platelet emergence,14,18 prevention of ICH is dependent on platelets in mice, it is possible that severe thrombocytopenia during early human life could cause significant ICH. This suggests that the timing of treatment of potential FNAIT-affected pregnancies should be further evaluated.
Of note, because the severity of thrombocytopenia achieved in both of the mouse models we investigated was likely to be more severe than that suffered by patients enrolled in previous clinical trials,2,3 it is possible that platelet transfusions in these trials seemed to be ineffective because the recipient platelet counts were above the threshold required to prevent spontaneous bleeding.
That thrombocytopenia-associated ICH in Nfe2−/− mice did not present with obvious signs of abnormal vascular patterning or pericyte coverage, and that the induction of thrombocytopenia with anti-GP1Bα resulted in ICH without the opportunity to impede general vascular development suggest that the role of platelets is to preserve the integrity of the cerebral vasculature at ICH hotspots. Although it is self-evident that once ICH is initiated, the inability to produce blood clots would result in continued hemorrhage, it is unclear why the absence of platelets would cause ICH. One possible explanation is that small sites of hemorrhage appear as blood-filled vessels remodel. This occurs during the remodeling of the mesenteric vasculature.28 Because we observed hemorrhage in the brains of wild-type embryos (Figure 2C), this might also be a feature of normal cerebral vascular development. Accordingly, the role of platelets might be to prevent innocuous microbleeds from developing into instances of pathological ICH. We speculate that the onset of FNAIT and the timing of cerebral vessel development may influence the location and severity of ICH.
CLEC-2-podoplanin signaling is crucial for maintaining the separation of lymphatic and blood vascular systems.5,7,8 This axis operates through the reciprocal expression of CLEC-2 (encoded by Clec1b) on platelets and the podoplanin (encoded by Pdpn) expressed by the lymphatic (but not blood) vascular endothelium.5,7 Because cerebral vascular endothelium does not express podoplanin, it has been proposed that CLEC-2-podoplanin signaling operates in the developing brain via interaction between circulating platelets and the podoplanin-expressing neuroepithelium.5 Although physical interaction between circulating megakaryocytes and the neuroepithelium has been recently described in the E10.5 embryo,29 we did not observe platelets interacting within the neuroepithelium; thus, it is unclear whether opportunity for such an interaction exists during normal fetal development. Furthermore, the key features of the Clec1b−/−phenotype (tortuous vascular patterning and disrupted pericyte coverage and interaction of the cerebral vasculature) were not observed in either the Nfe2−/− or anti-GP1Bα models of thrombocytopenia, and ICH in Pdpn−/− mice is reported to be restricted to the E10.5 to E12.5 developmental stages.29 This indicates that the underlying mechanism(s) of Clec1b−/−-associated ICH and thrombocytopenia-associated ICH are distinct, in which case the molecular mechanism(s) by which platelets preserve cerebral vascular integrity remain unknown. Of note, although Gp1bα−/− mice are viable, they exhibit a bleeding phenotype in the adult mouse30 ; therefore, the ICH observed after treatment with anti-GP1Bα antibody could be a result of both perturbed GP1Bα signaling and platelet clearance. Accordingly, it is possible that ICH would be less severe with other anti-platelet antibodies.
Regarding the differences between our study and previous reports, it is possible that the ICH we observed after anti-GP1Bα treatment differs from a previous report13 because of the degree of thrombocytopenia achieved. Although fetal platelet counts were not established in the study by Yougbare et al,13 assessment of the reported neonatal platelet counts suggests that their immunization approach might not have achieved the severity of thrombocytopenia required to compromise hemostasis.31 We found that transfer of the Nfe2−/− allele to FVB/NJ mice (supplemental Figure 6) and the induction of thrombocytopenia on a 129/Sv background (supplemental Figure 7) with anti-GP1Bα treatment results in severe thrombocytopenia and ICH. It is unclear why our results differ from those in previous reports of the Nfe2−/− prenatal phenotype,11,32 but the differences are not caused by a hypersensitivity of C57BL/6 embryos to thrombocytopenia. We found that dissection of brains from the late fetal stages (E16.5 onward) was essential for identifying ICH in Nfe2−/− mice, so unless this was performed in previous studies, the prenatal ICH phenotype in Nfe2−/− mice could previously have been overlooked.
It is unclear what structural abnormalities in the mouse brain could result from fetal and neonatal ICH. With the caveat of the known differences in platelet biology between mice and humans (reviewed in Rasmussen and Ahlen33 ), given the observation that ICH within the human ganglionic eminence significantly reduced the proportion of cycling cells,34 it is possible that thrombocytopenia-driven ICH with the ganglionic eminence could affect normal brain development. Of interest, cerebral palsy is associated with structural lesions in the brain (white matter damage of immaturity and cortical and subcortical damage),35 and cerebellar hemorrhage is associated with multiple neuropathies including cerebral palsy.36-40 Because severe thrombocytopenia induced at any stage of mouse fetal and neonatal development was sufficient to drive ICH within regions of the brain that in humans give rise to significant neuropathies, further studies to understand how in utero thrombocytopenia and ICH correlate with neurologic outcome in the human population are critical.
Acknowledgments
The authors thank Marnie Blewitt and Antoine Terreaux for critical comments on the manuscript, and Mirella Dottori and Samantha Chandler (consumer advocate) for insightful advice. This work was supported by the Australian Research Council Stem Cells Australia Program, and by grants from the National Health and Medical Research Council (NHMRC) (1128993 and 1129012), Cerebral Palsy Alliance (PG12418), Independent Research Institutes Infrastructure Support Scheme, NHMRC (361646), the Australian Cancer Research Fund, and Victorian State Government Operational Infrastructure Support. S.T. was supported by a fellowship from the Lorenzo and Pamela Galli Charitable Trust.
Authorship
Contribution: A.F. and S.T. conceived the study, designed and performed experiments, and analyzed data; S.L. designed and performed experiments and analyzed data; M.D. performed experiments; C.B. and O.S. provided essential input into experimental design and data analysis; and all authors contributed to manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Samir Taoudi, Walter and Eliza Hall Institute , 1G Royal Parade, Parkville, VIC 3052, Australia; e-mail: taoudi@wehi.edu.au.
For original data, please contact Samir Taoudi via e-mail at taoudi@wehi.edu.au.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.