Key Points
Asciminib demonstrated superior efficacy vs bosutinib and an improved safety profile in patients with CML-CP after at least 2 prior TKIs.
Asciminib has the potential to transform standard of care in this population through its novel mechanism of action as a STAMP inhibitor.

Abstract
Patients with chronic myeloid leukemia in chronic phase (CML-CP) resistant/intolerant to ≥2 tyrosine kinase inhibitors (TKIs) are at high risk of experiencing poor outcomes because of disease biology and inadequate efficacy and/or safety of current therapies. Asciminib, a first-in-class BCR-ABL1 inhibitor Specifically Targeting the ABL Myristoyl Pocket (STAMP), has the potential to overcome resistance/intolerance to approved TKIs. In this phase 3, open-label study, patients with CML-CP previously treated with ≥2 TKIs were randomized (2:1) to receive asciminib 40 mg twice daily vs bosutinib 500 mg once daily. Randomization was stratified by major cytogenetic response (MCyR) status at baseline. The primary objective was to compare the major molecular response (MMR) rate at week 24 for asciminib vs bosutinib. A total of 233 patients were randomized to asciminib (n = 157) or bosutinib (n = 76). Median follow-up was 14.9 months. The MMR rate at week 24 was 25.5% with asciminib and 13.2% with bosutinib. The difference in MMR rate between treatment arms, after adjusting for MCyR at baseline, was 12.2% (95% confidence interval, 2.19-22.30; 2-sided P = .029). Fewer grade ≥3 adverse events (50.6% vs 60.5%) and adverse events leading to treatment discontinuation (5.8% vs 21.1%) occurred with asciminib than with bosutinib. The study showed a superior efficacy of asciminib compared with that of bosutinib, together with a favorable safety profile. These results support the use of asciminib as a new therapy in patients with CML-CP who are resistant/intolerant to ≥2 prior TKIs. This trial was registered at www.clinicaltrials.gov as #NCT03106779.
Introduction
Adenosine triphosphate (ATP)–competitive tyrosine kinase inhibitors (TKIs) have transformed chronic myeloid leukemia (CML) from a fatal disease to one associated with near-normal life expectancy.1-5 However, some patients do not respond to TKI therapy (primary resistance), lose response (secondary resistance),6,7 or experience intolerance.4,8,9 Available therapies for patients with resistance to or intolerance (R/I) of ≥2 TKIs are often limited by modest efficacy, safety concerns, or both.4,8 Following the failure of ≥1 second-generation TKI (2G-TKI), the use of alternative 2G-TKIs rarely results in optimal or durable responses.10-13 Ponatinib, a third-generation TKI, is effective in patients who received prior therapies, but it is associated with high iatrogenic cardiovascular risk.14
All approved TKIs for CML bind to the ATP site of the BCR-ABL1 oncoprotein to inhibit aberrant kinase activity.15-17 Asciminib is a novel, first-in-class Specifically Targeting the ABL Myristoyl Pocket (STAMP) inhibitor that potently inhibits the kinase activity of BCR-ABL1 via allosteric binding.15-17 It has potential to maintain activity against most ABL1 kinase domain mutations (eg, T315I) that confer resistance to approved TKIs.15-18 Asciminib previously demonstrated relatively high and durable response rates (48% of patients achieved or maintained major molecular response [MMR] by 12 months and 91% of them maintained MMR at the time of analysis) and a favorable safety and tolerability profile in a phase 1 study in heavily pretreated patients with CML.18 Here, we report the results of the primary analysis from ASCEMBL (NCT03106779)—an open-label, randomized, phase 3 trial comparing asciminib with bosutinib in patients with CML in chronic phase (CML-CP) previously treated with ≥2 TKIs.
Bosutinib, an ATP-competitive 2G-TKI,19 demonstrated clinical efficacy in heavily pretreated patients with CML in prospective studies11,12 and is approved for use in patients with Philadelphia chromosome–positive CML-CP with R/I to prior TKIs.20,21 A prospective study of bosutinib in patients who received ≥2 TKIs yielded cumulative major cytogenetic response (MCyR) and complete cytogenetic response (CCyR) rates of 36% and 28%, respectively, after a median treatment duration of 8.6 months12 and a cumulative MMR rate of 15% after a median treatment duration of 8.3 months.11 When the ASCEMBL study design was being developed, the approved bosutinib dose for third-line (3L) therapy and beyond was 500 mg once daily.11,12,20,21 Ponatinib also demonstrated clinical efficacy in patients receiving up to 4 prior TKIs.14 However, the optimal dose of ponatinib was being reassessed in the ongoing OPTIC trial,22 and its practical use warranted caution and potential avoidance in patients with cardiovascular comorbidities.23 Therefore, based on best available evidence and feasibility at the time ASCEMBL was initiated, bosutinib was chosen for the comparator treatment over ponatinib. We hypothesized that asciminib could provide a superior response rate to bosutinib based on the efficacy of asciminib in the phase 1 study.18
Methods
Study overview
The study was designed collaboratively by the sponsor (Novartis Pharmaceuticals) and lead study investigators. The protocol was approved by the sites’ institutional review boards and conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent. An independent Data Monitoring Committee reviewed safety data approximately every 6 months. The sponsor collected data and analyzed them in conjunction with authors. All authors contributed to the development and writing of the manuscript. All authors and representatives of the sponsor reviewed and amended the manuscript and vouch for the accuracy and completeness of the data and fidelity of the study to the protocol.
Patients
Eligible patients were ≥18 years of age, with CML-CP previously treated with ≥2 TKIs. Patients must have experienced treatment failure (lack of efficacy) as defined in the 2013 European LeukemiaNet (ELN) recommendations for patients receiving a second-line (2L) TKI24 or intolerance of the most recent TKI therapy at the time of screening (see supplemental Appendix on the Blood Web site for details). At screening,BCR-ABL1 transcript levels on the international scale (BCR-ABL1IS) must have been ≥1%. After a protocol amendment, for patients with intolerance, BCR-ABL1IS >0.1% was required at screening. Patients with known bosutinib-resistant BCR-ABL1 mutations of T315I or V299L detected at any time before study entry were ineligible (supplemental Figure 1).
Trial design, randomization, and treatments
In this randomized, open-label, active-controlled, multicenter, phase 3 trial, eligible patients were randomized in a 2:1 ratio (based on a computer-generated randomization list via a Web-based system) to receive either asciminib 40 mg twice daily or bosutinib 500 mg once daily. Randomization was stratified by MCyR status at baseline.
Per protocol, patients meeting lack of efficacy criteria must permanently discontinue study treatment. Lack of efficacy criteria are based on 2013 ELN recommendations for 2L TKI therapy24 (supplemental Appendix). Following a protocol amendment (14 December 2018), patients failing bosutinib treatment from a lack of efficacy were offered the possibility of switching to asciminib. Patients who discontinued treatment with bosutinib because of intolerance (or any reason other than lack of efficacy) were not allowed to switch to asciminib. Efficacy data collected for patients on asciminib after the switch from bosutinib are not part of the primary analysis presented in this manuscript.
Statistical analysis
The primary objective of the study was to assess whether asciminib is superior to bosutinib in patients with CML-CP previously treated with ≥2 TKIs; the primary endpoint was the rate of MMR (BCR-ABL1IS ≤0.1%) at week 24; to be counted as being in MMR at week 24 for the primary end point, patients must have been on study treatment with BCR-ABL1IS ≤0.1% at week 24 and must not have met any treatment failure criteria before week 24. Treatment failure was defined as lack of efficacy or treatment discontinuation for any reason.
A total sample size of 222 was planned to have a 90% power to detect a 20% difference in the MMR rates at week 24 at a significance level of 0.05 (2-sided test), assuming an MMR rate of 15% for bosutinib at week 24 based on previous studies.11,25,26
The MMR rate at week 24 was calculated based on the full analysis set (supplemental Table 1). The Cochrane-Mantel-Haenszel χ2 test, stratified by the cytogenetic response status (MCyR vs no MCyR) at baseline, was used to compare MMR rates between the treatment groups, at the 5% level of significance (2-sided test). The Mantel-Haenszel estimates of the common risk difference and the corresponding 95% confidence intervals (CIs) are presented, as well as the MMR rates and 95% CIs based on the Pearson-Clopper method, for each treatment arm. The cumulative incidence of MMR was calculated considering discontinuation from study treatment of any reason and without prior achievement of MMR as a competing risk.
Subgroup analyses were performed to assess homogeneity of the treatment effect. A multivariate analysis using a logistic regression was performed to assess the treatment effect after adjusting for important demographic and disease characteristics between treatment groups. Secondary efficacy endpoints are described in the supplemental Appendix. These end points are reported descriptively for both treatment arms and no P values were calculated.
Results
Patients
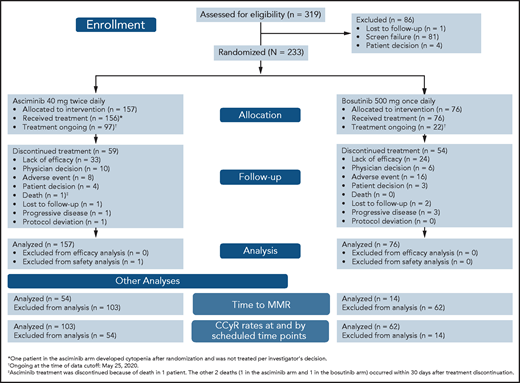
From 15 November 2017 to 4 December 2019, 233 patients with CML-CP previously treated with ≥2 TKIs were randomized 2:1 to receive asciminib (n = 157) or bosutinib (n = 76) (Figure 1). One patient assigned to the asciminib arm developed cytopenia after randomization and did not initiate study treatment per investigator’s decision. At baseline, 68 (29.2%) patients were in MCyR: 46 (29.3%) in the asciminib arm and 22 (28.9%) in the bosutinib arm. The cutoff date for this analysis was 25 May 2020. Median follow-up for all randomized patients was 14.9 months from randomization to cutoff. At cutoff, all randomized patients had completed their 24-week visit or discontinued earlier. Treatment was ongoing in 97 (61.8%) and 22 (28.9%) patients receiving asciminib and bosutinib, respectively: 59 (37.6%) and 54 (71.1%) patients, respectively, discontinued therapy. Before completing 24 weeks of therapy, 16.6% and 32.9% of patients had discontinued asciminib and bosutinib, respectively, most frequently because of adverse events (AEs) (asciminib, 4.5%; bosutinib, 14.5%) and lack of efficacy as per protocol (asciminib, 4.5%; bosutinib, 6.6%). After 24 weeks, the most frequent reason for discontinuation in both treatment arms was lack of efficacy. Other reasons for discontinuation are listed in supplemental Table 2.
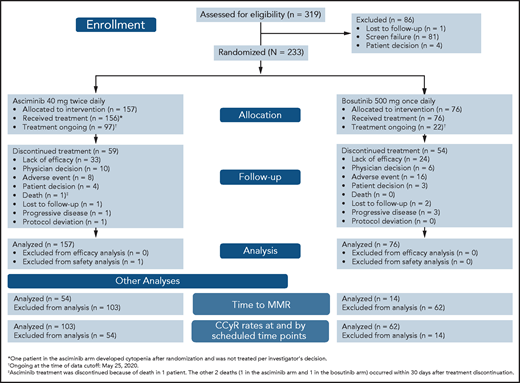
CONSORT diagram for ASCEMBL (data cutoff 25 May 2020). Efficacy analyses were based on all randomized patients. Safety analyses were based on patients who received ≥1 dose of study treatment.
CONSORT diagram for ASCEMBL (data cutoff 25 May 2020). Efficacy analyses were based on all randomized patients. Safety analyses were based on patients who received ≥1 dose of study treatment.
Although baseline demographics and disease characteristics were mostly balanced between treatment arms, differences in sex distribution and certain disease characteristics were observed (Table 1) despite randomization. More male patients were on asciminib than on bosutinib (52.2% vs 40.8%). More patients experienced intolerance of their prior TKI on asciminib (37.6%) than on bosutinib (28.9%), and fewer patients experienced lack of efficacy on their last TKI on asciminib (60.5%) than on bosutinib (71.1%) before study entry. More patients received asciminib than bosutinib as 3L therapy (52.2% vs 39.5%); hence, fewer received asciminib than bosutinib as fourth- and subsequent-line therapy (47.8% vs 60.5%).
At baseline, 20 (12.7%) patients on asciminib and 10 (13.2%) on bosutinib had ≥1 BCR-ABL1 mutation, including 4 patients (3 on asciminib and 1 on bosutinib) with the T315I mutation and 1 patient on bosutinib with the V299L mutation. T315I and V299L mutations were unknown at study entry. Patients with these mutations were discontinued from study treatment.
At data cutoff, the median duration of exposure to asciminib was 43.4 (range, 0.1-129.9) weeks and to bosutinib was 29.2 (range, 1.0-117.0) weeks. The median relative dose intensity was 99.7% (range, 41% to 100%) and 95.4% (range, 36% to 100%) for asciminib and bosutinib, respectively; and the median dose intensity was 79.8 (range, 33-80) and 478.6 (range, 181-566) mg/d, respectively.
Efficacy
The MMR rate at week 24 was 25.5% with asciminib vs 13.2% with bosutinib, meeting the primary objective of the study. The difference in MMR rates at week 24 between asciminib and bosutinib, after adjusting for MCyR status at baseline, was 12.2% (95% CI, 2.19-22.30; 2-sided P = .029).
The response rate with asciminib was superior to that with bosutinib across major demographic and prognostic subgroups. In particular, the MMR rate at week 24 was greater with asciminib than with bosutinib, regardless of line of therapy, and in patients who discontinued their last TKI because of lack of efficacy (Figure 2; supplemental Figure 2). The results from the multivariate analysis assessing the treatment effect showed that the odds of achieving MMR, after adjusting for MCyR at baseline, sex, lines of prior therapy, and reason for discontinuation of last TKI (2.38; 95% CI, 1.06-5.35), was almost identical to that observed after adjusting for only MCyR at baseline (the randomization stratification factor) (2.35; 95% CI, 1.08-5.12) (supplemental Table 3). These results indicate a consistent treatment effect, independent of the demographic and prognostic variables tested, including those that were imbalanced between the treatment arms at baseline.
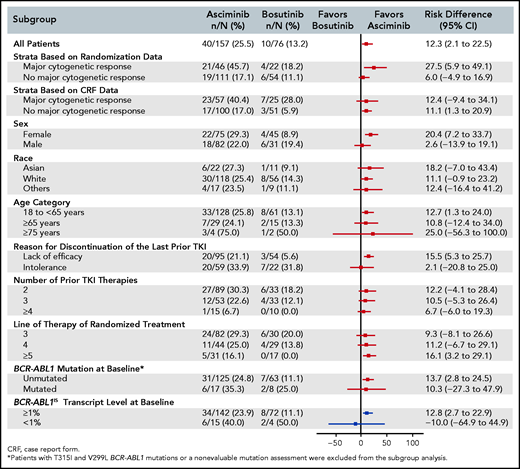
Risk difference (95% CI) for MMR at week 24 from subgroup analyses. A forest plot shows the risk difference with 95% confidence intervals for MMR rate at week 24 from subgroup analyses.
Risk difference (95% CI) for MMR at week 24 from subgroup analyses. A forest plot shows the risk difference with 95% confidence intervals for MMR rate at week 24 from subgroup analyses.
Regardless of BCR-ABL1 levels at baseline, more patients on asciminib (63.1%) than on bosutinib (43.4%) had BCR-ABL1IS ≤10% at week 12, and more patients on asciminib (49.0%) than on bosutinib (23.7%) had BCR-ABL1IS ≤1% at week 24. MR4 (BCR-ABL1IS ≤0.01%) and MR4.5 (BCR-ABL1IS ≤0.0032%) rates at week 24 were 10.8% and 8.9% in the asciminib arm and 5.3% and 1.3% in the bosutinib arm, respectively.
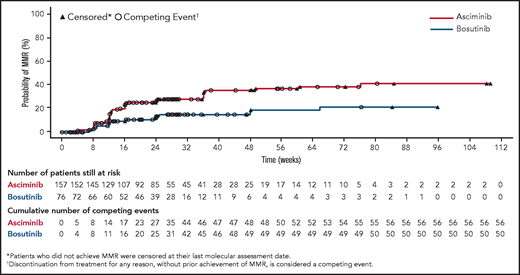
The cumulative incidence of MMR by week 24 was 25.0% with asciminib vs 12.0% with bosutinib (Figure 3). The CCyR rate at week 24 in patients without CCyR at baseline was 40.8% with asciminib vs 24.2% with bosutinib. The difference in CCyR rates at week 24 between the 2 arms, after adjusting for MCyR status at baseline, was 17.3% (95% CI, 3.62-30.99).

Cumulative incidence of MMR. The cumulative incidence curve shows the probability of achieving MMR over time in each treatment arm calculated using a competing risk analysis.
Cumulative incidence of MMR. The cumulative incidence curve shows the probability of achieving MMR over time in each treatment arm calculated using a competing risk analysis.
Among patients with mutations identified at baseline (excluding those with T315I or V299L mutations), 6 of 17 patients on asciminib and 2 of 8 patients on bosutinib were in MMR at week 24. Of the patients not in MMR at week 24, 10 in the asciminib arm and 4 in the bosutinib arm discontinued treatment (supplemental Table 4).
Safety
Of the 156 patients receiving asciminib and 76 patients receiving bosutinib evaluable for safety, 140 (89.7%) and 73 (96.1%) patients, respectively, experienced all-grade AEs regardless of study drug relationship. Grade ≥3 AEs occurred in 79 (50.6%) and 46 (60.5%) patients, respectively (Table 2). Treatment-related AEs (per investigator assessment) were reported in 99 (63.5%) and 67 (88.2%) patients receiving asciminib and bosutinib, respectively (supplemental Table 5).
The proportion of patients who experienced AEs leading to treatment discontinuation was lower with asciminib (5.8%) than with bosutinib (21.1%). The most common AEs leading to treatment discontinuation included thrombocytopenia (all-grade, 3.2%; grade ≥3, 3.2%) with asciminib and increased alanine aminotransferase (ALT) (all-grade, 5.3%; grade ≥3, 3.9%) with bosutinib (supplemental Table 6).
Thirty-three (21.2%) and 32 (42.1%) patients in the asciminib and bosutinib arms, respectively, had ≥1 dose reduction due to AEs; 60 (38.5%) and 43 (56.6%) patients, respectively, had ≥1 dose interruption because of AEs. Five (6.6%) patients on bosutinib had dose escalation to 600 mg by week 12 because of a suboptimal response as per protocol (supplemental Appendix; supplemental Table 7).
Grade ≥3 laboratory hematologic abnormalities that were more common with asciminib than with bosutinib included decreased leukocytes (9.6% vs 5.3%), neutrophils (19.2% vs 15.8%), and platelets (22.4% vs 11.8%). Grade ≥3 laboratory biochemical abnormalities that were more common with asciminib than with bosutinib included increased amylase (1.3% vs 0%), triglycerides (7.1% vs 2.6%), and urate (6.4% vs 2.6%). Grade ≥3 laboratory biochemical abnormalities that were less frequent with asciminib than with bosutinib included increased ALT (0.6% vs 15.8%), aspartate aminotransferase (AST) (0.6% vs 6.6%), creatine kinase (1.9% vs 5.3%), and pancreatic lipase (3.8% vs 5.3%) (supplemental Table 8). Pancreatitis, a dose-limiting toxicity in the phase 1 trial of asciminib,18 was not reported on treatment in either arm.
Four patients (2.6%) in the asciminib arm and 1 patient (1.3%) in the bosutinib arm died during the study. In the asciminib arm, 2 deaths occurred on treatment (defined as death occurring during treatment or within 30 days after the end of treatment) from arterial embolism and ischemic stroke (1 each). Two deaths occurred after asciminib discontinuation during survival follow-up (both from CML). In the bosutinib arm, 1 patient died on treatment from septic shock. A 62-year-old White woman who died of arterial embolism had received asciminib for 8 months, then discontinued because of lack of efficacy and initiated ponatinib treatment; 15 days after the last dose of asciminib and on day 7 of ponatinib treatment, an ischemic event of the distal bowel was observed. She had been treated with imatinib, dasatinib, and nilotinib before study entry, and her relevant medical history included arterial hypertension and gastric bypass. A 65-year-old Black man who died of ischemic stroke had been receiving asciminib for 2 months. He had been treated with imatinib, nilotinib, and dasatinib before study entry, and his relevant medical history included arterial hypertension and left ventricular hypertrophy as per electrocardiogram, which was identified at screening. A 42-year-old White man who died of septic shock had been receiving bosutinib for 2.8 months. He had been treated with imatinib, nilotinib, and dasatinib before study entry; he discontinued bosutinib because of disease progression to accelerated phase, and the onset of septic shock occurred 25 days after the last bosutinib dose.
The frequency of arterial-occlusive events (AOEs) was 3.2% (n = 5) with asciminib and 1.3% (n = 1) with bosutinib. In addition to the 2 fatal AOEs described previously, 3 other AOEs were reported on asciminib: myocardial ischemia (n = 2) and coronary artery disease (n = 1). Both myocardial ischemia events were based on ST and T changes observed after dosing in electrocardiograms performed as per protocol, at day 1 and day 8, respectively, after randomization and were without clinical manifestations; both patients discontinued the study for lack of efficacy, at 3.6 and 5.6 months after experiencing the AE. Coronary artery disease was reported after a routine coronary arteriography was performed because of the medical history of the patient, and was without clinical manifestations. Medical history included hypertension, chronic heart failure, and hyperlipidemia. The patient was on study treatment at cutoff. On bosutinib, acute coronary syndrome was reported in a patient with a history of myocardial infarction; the patient was on study treatment at cutoff.
Discussion
In this first randomized controlled study conducted in patients with CML-CP previously treated with ≥2 TKIs, asciminib demonstrated a superior treatment effect compared with bosutinib: asciminib nearly doubled the MMR rate at week 24, meeting the primary objective of the study. According to 2020 ELN recommendations, achieving BCR-ABL1IS ≤1% at 24 weeks is a treatment goal for patients receiving first-line or 2L therapy.4 However, in ASCEMBL, a more stringent level of response was adopted to provide an early indication of optimal treatment benefit in these heavily pretreated patients. At week 24, more patients on asciminib (49.0%) than on bosutinib (23.7%) had BCR-ABL1IS ≤1%. Because a suboptimal response to 2 or more TKIs warrants prompt consideration of allogeneic stem cell transplant,4 an early indication of optimal disease control (as in ASCEMBL) is clinically meaningful.
MMR is a surrogate end point indicative of successful CML treatment because of its association with superior long-term outcomes, including survival and progression-free survival.27,28 Although most studies conducted in patients with CML with R/I have used MCyR as the primary end point, there is an increasing interest in assessing deep molecular responses even in later lines of treatment.12,14,29-31 Some studies have shown that, in early lines of therapy, deep molecular response rates are associated with lower risks of treatment failure, disease progression, and death32,33; this could also be considered in studies of more heavily pretreated patients. In ASCEMBL, among patients who achieved MMR before data cutoff, deeper levels of response were achieved with asciminib than with bosutinib.
The MMR rate for bosutinib reported here is consistent with that of previous results: in a phase 2 trial of bosutinib as 3L therapy, the cumulative MMR rate was 15% after a median duration of 8.3 months on bosutinib treatment.11
Response rates (CCyR and MMR) with bosutinib as ≥2L therapy were higher in the BYOND phase 4 study than in ASCEMBL and in the pivotal phase 2 bosutinib study.11,31 This could be due to the more heavily pretreated population in ASCEMBL, where bosutinib was the 3L or fourth, fifth, or sixth or later line of therapy in 39.5%, 38.2%, 13.2%, and 9.2% of patients, respectively, and 71.1% discontinued their last TKI because of lack of efficacy (Table 1). In BYOND, bosutinib was the 2L, 3L, or fourth-line treatment in 29.5%, 39.1%, and 31.4% of patients, respectively, and 53.2% of patients were resistant to ≥1 of their prior TKIs (not specifically to the last one).31 Flexibility in dose modifications and dose levels may have allowed more patients to continue therapy and decrease treatment discontinuations in BYOND. However, the rate of treatment discontinuation because of AEs in BYOND was relatively similar to that with bosutinib in ASCEMBL (25.0% vs 21.1%, respectively).31 In ASCEMBL, because discontinuations from AEs occurred mainly during the initial months of treatment (supplemental Table 2), rates may remain similar after a longer follow-up. The lowest dose of bosutinib in BYOND was 200 mg once daily.31 In ASCEMBL, according to dose recommendations in the bosutinib label,20,21 the lowest dose was 300 mg once daily. In real-life practice, some patients with R/I may start bosutinib at lower doses (eg, 400 mg daily), which could improve tolerability and allow better responses. In the first-line BFORE study, the dose of 400 mg once daily was better tolerated than the 500 mg once daily dose used in the first-line BELA study.34,35 Further studies are needed to address how alternative dosing schemes may improve outcomes in patients with R/I, but it would have been outside the realm of this study. In ASCEMBL we used the licensed bosutinib dose of 500 mg daily for patients with R/I.20,21 The dose modifications for toxicities in ASCEMBL were similar for both treatment arms and comparable to those in the bosutinib prescribing information.20,21
Subgroup analyses demonstrated a homogeneous and consistent treatment effect across most major demographic and prognostic factors (Figure 2). The benefit of asciminib over bosutinib, regardless of line of TKI therapy, confirmed the potential role for asciminib in this difficult-to-treat patient population. The results favored asciminib in patients with lack of efficacy on their prior TKI, and the benefit was less pronounced in patients with intolerance. Moreover, the benefit of asciminib over bosutinib was confirmed even after adjusting for relevant prognostic variables that were not balanced between treatment arms, such as number of prior lines of TKI therapy and reason for discontinuation of the last TKI.
Conclusions on the impact of mutations on efficacy cannot be made because of the heterogeneity of reported mutations and low patient numbers. Three patients harboring baseline E255K/V mutations were in MMR at week 24, whereas 4 harboring baseline F359C/V mutations on asciminib were not in MMR. These findings are consistent with those of a previous report.36
The superior efficacy of asciminib and its improved safety profile as compared with bosutinib led to a longer duration of treatment with asciminib, thus providing a longer duration for the possibility of AE reporting. Despite this, asciminib demonstrated favorable safety and tolerability. No new safety findings were identified for asciminib, and the safety profile is consistent with that observed in the phase 1 study.18 Gastrointestinal events (all-grade) and increased ALT and AST levels (all-grade and grade ≥3) were notably higher with bosutinib. The safety profile of bosutinib in ASCEMBL is consistent with that reported in the studies by Khoury et al and Cortes et al; after median treatment durations of 8.3 and 8.6 months, respectively, the most common nonhematologic AEs with bosutinib included diarrhea, nausea, and vomiting.11,12 Rates of dose interruptions/reductions from AEs with bosutinib in ASCEMBL are consistent with previous results.11,12 Overall, fewer grade 3/4 AEs and discontinuations and dose modifications from AEs occurred with asciminib than with bosutinib.
The rate of treatment discontinuation with bosutinib reported in ASCEMBL (71.1%) is similar to that reported by Khoury et al (71%) after a relatively similar median treatment duration (ASCEMBL, 6.7 months; Khoury et al, 8.3 months). As in ASCEMBL, the most frequent reasons for treatment discontinuation in patients randomized to bosutinib reported by Khoury et al were unsatisfactory response (or lack of efficacy; 21%) and AEs (20%).11
In the absence of specific recommendations for 3L therapy and beyond, the 2013 ELN response criteria for 2L therapy were applied in ASCEMBL to ensure that a consistent and homogeneous definition for lack of efficacy was applied to both arms. Patients in either treatment arm with documented lack of efficacy based on 2013 ELN recommendations were to discontinue study treatment.
Patients who experienced treatment failure on bosutinib had few remaining treatment options and were offered the option to switch to asciminib. However, only those who met failure criteria per 2013 ELN recommendations while on bosutinib treatment could do so. Patients who discontinued bosutinib treatment because of intolerance were not allowed to switch. Data collected after a patient met lack of efficacy criteria on either treatment arm were not considered in any efficacy analyses. Because failure criteria for patients who discontinued bosutinib for lack of efficacy are based on objective measures, the switch did not have an impact on the assessment of efficacy in ASCEMBL. Of the 24 patients who discontinued bosutinib for lack of efficacy, 22 switched to asciminib. The safety profile of asciminib in these 22 patients is comparable to that observed in patients receiving asciminib in the study’s randomized period and to that reported in the phase 1 study18 (supplemental Table 9).
Overall, the clinically meaningful and statistically significant, superior efficacy of asciminib compared with that of bosutinib, deeper molecular response rates, and favorable safety profile support the potential of asciminib to transform the CML treatment landscape, particularly for patients with R/I to ≥2 prior TKIs, who may benefit from a treatment with a novel mechanism of action. Our results demonstrate that asciminib, the first STAMP inhibitor, has potential to be the standard of care for 3L and later-line therapies.
Acknowledgments
The authors thank Rohini Roy and Karen Kaluza Smith (Nucleus Global) for medical editorial assistance with this manuscript. J.F.A. is grateful for the support of the Imperial National Institute for Health Research (NIHR) Oxford Biomedical Research Centre.
The ASCEMBL study and work presented here were sponsored and funded by Novartis Pharmaceuticals Corporation. Financial support for medical editorial assistance was provided by Novartis.
Authorship
Contribution: D.R., M.J.M., C.B., Y.M., E.L., S.V., A.T., D.-W.K., J.F.A., A. Abdo, L.M.F., D.D.H.K., P.l.C., S.S., M.A., T.P.H., N.C., K.S., L.C., V.G.-G., J.E.C., P.A., A. Allepuz, S.Q., V.B., and A.H. contributed to data acquisition and interpretation, writing and reviewing the manuscript, and reviewing and approving the final manuscript.
Conflict-of-interest disclosure: All authors received nonfinancial support (assistance with manuscript preparation) from Nucleus Global, which received funding from Novartis Pharmaceuticals Corporation. D.R. received personal fees from Novartis, Pfizer, and Incyte. M.J.M. received personal fees from Bristol Myers Squibb, Takeda, and Pfizer. C.B. received grants from Novartis during the conduct of the study. Y.M. received honoraria from Bristol Myers Squibb, Novartis, Pfizer, and Astellas. E.L. received grants from Novartis and personal fees and nonfinancial support from Novartis, Pfizer, and Bristol Myers Squibb. S.V. received personal fees and nonfinancial support from Novartis, AbbVie, Janssen, Sanofi, and Biocad; nonfinancial support from Pfizer; and personal fees from Takeda and Astra Zeneca. A.T. has no other relationships to disclose. D.-W.K. received grants from Novartis, Bristol Myers Squibb, Pfizer, ILYANG, and Takeda. J.F.A. received honoraria, grants, and personal fees from Novartis and grants and personal fees from Pfizer. A. Abdo received honoraria from Novartis and Takeda. L.M.F. has no other relationships to disclose. D.D.H.K. received grants, honoraria, and consulting fees from Novartis; grants from Bristol Myers Squibb; and honoraria from Pfizer and Paladin. P.l.C. received speaker’s honoraria from Novartis, Incyte, Pfizer, and Bristol Myers Squibb. S.S. received honoraria, grants, and personal fees from Novartis and grants and personal fees from Bristol Myers Squibb and Incyte. M.A. has no other relationships to disclose. T.P.H. received grants and honoraria for advisory boards and symposia from Novartis and grants from Bristol Myers Squibb. N.C. has no other relationships to disclose. K.S. received research funding and honoraria for advisory boards from Novartis. L.C. received honoraria for travel and meeting attendance from Novartis. V.G.-G. received grants, nonfinancial support, and honoraria from Novartis, Pfizer, Bristol Myers Squibb, and Incyte. J.E.C. received grants and consulting fees from Novartis, grants and consulting fees from Pfizer, and grants from Bristol Myers Squibb. P.A. is an employee of Novartis. A. Allepuz is an employee of Novartis. S.Q. is an employee of Novartis. V.B. is an employee of Novartis. A.H. received research support and personal fees from Novartis, Bristol Myers Squibb, and Pfizer.
Correspondence: Delphine Réa, DMU Hématologie, Hôpital Saint-Louis, 1 Ave Claude Vellefaux, 75010 Paris, France; e-mail: delphine.rea@aphp.fr.
Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel based on scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial consistent with applicable laws and regulations. This trial data availability is according to the criteria and process described on www.clinicalstudydatarequest.com.
There is a Blood Commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.