

Key Points
All 3 starting doses showed clinical benefit in patients with resistant CP-CML.
Optimal benefit/risk outcomes occurred with the 45 mg starting dose decreasing to 15 mg upon achievement of a response (≤1% BCR-ABL1IS).

Abstract
In PACE (Ponatinib Ph+ ALL and CML Evaluation), a phase 2 trial of ponatinib that included patients with chronic-phase chronic myeloid leukemia (CP-CML) resistant to multiple prior tyrosine kinase inhibitors (TKIs), ponatinib showed deep and durable responses, but arterial occlusive events (AOEs) emerged as notable adverse events. Post hoc analyses indicated that AOEs are dose dependent. We assessed the benefit/risk ratio across 3 ponatinib starting doses in the first prospective study to evaluate a novel, response-based, dose-reduction strategy for TKI treatment. Adults with CP-CML resistant to or intolerant of at least 2 prior BCR-ABL1 TKIs or with a BCR-ABL1 T315I mutation were randomly assigned 1:1:1 to 3 cohorts receiving ponatinib 45, 30, or 15 mg once daily. In patients who received 45 or 30 mg daily the dose was reduced to 15 mg upon response (BCR-ABL1IS transcript levels ≤1%). The primary end point was response at 12 months. From August 2015 through May 2019, 283 patients were randomly assigned to the cohorts: 282 (94 per dose group) received treatment (data cutoff, 31 May 2020). The primary end point (98.3% confidence interval) was achieved in 44.1% (31.7-57.0) in the 45-mg cohort, 29.0% (18.4-41.6) in the 30-mg cohort, and 23.1% (13.4-35.3) in the 15-mg cohort. Independently confirmed grade 3 or above treatment-emergent AOEs occurred in 5, 5, and 3 patients in the 45-, 30-, and 15-mg cohorts, respectively. All cohorts showed benefit in this highly resistant CP-CML population. Optimal benefit/risk outcomes occurred with the 45-mg starting dose, which was decreased to 15 mg upon achievement of a response. This trial is registered on www.clinicaltrials.gov as NCT02467270.
Introduction
Although most patients with chronic-phase chronic myeloid leukemia (CP-CML) have good long-term outcomes with tyrosine kinase inhibitors (TKIs),1 a significant percentage require a change in treatment for lack of efficacy or adverse events (AEs). Only a minority (≈21% to 35%)2-7 of patients with CP-CML who have disease resistant to a second-generation TKI have a complete cytogenetic response (CCyR) when treated with a different second-generation TKI, with 1 study reporting a 30-month overall survival (OS) of only 47%.4
Ponatinib is a third-generation TKI that inhibits BCR-ABL1, with or without kinase domain mutations, including T315I.8 The phase 2 PACE (Ponatinib Ph+ ALL and CML Evaluation) trial tested the efficacy and safety of ponatinib 45 mg once daily in patients with CML and Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia that is resistant to or intolerant of dasatinib or nilotinib or that has the T315I mutation.9,10 Ponatinib demonstrated robust clinical activity regardless of BCR-ABL1 mutation status, with rapid, deep, and durable responses, including favorable survival (5-year OS of 73% in CP-CML), although 80% of patients with CP-CML were resistant to prior therapy.10 However, arterial occlusive events (AOEs) were reported in 84 of 270 (31%) patients with CP-CML.10-12 A post hoc multivariate regression analysis of pooled data from 3 clinical trials of ponatinib in patients with Ph+ leukemia suggested a 33% reduction in the risk of an AOE for each 15-mg/d decrease in average ponatinib dose intensity.13 To further understand the impact of the ponatinib dose on safety and efficacy, we conducted the phase 2 OPTIC (Optimizing Ponatinib Treatment in CP-CML) trial, exploring a novel, response-based, dose-reduction strategy.
Methods
Study oversight
The protocol, amendments, and informed consent form were approved by the institutional review boards/ethics committees of the participating centers. A 5-member independent data-monitoring committee was responsible for providing safety recommendations. AOEs were prospectively reviewed by an independent committee of cardiologists, neurologists, and vascular experts (separate from the data monitoring committee) and blinded to dose. All authors contributed to and reviewed the data reported and vouch for the integrity of the analysis.
Patients
Eligible patients were adults (aged ≥18 years) with CP-CML resistant or intolerant to at least 2 prior TKIs or who had a T315I mutation. Patients with risk factors for AOEs were eligible, provided the comorbidities were under control at the time of enrollment. All patients provided written informed consent. BCR-ABL1 transcript levels had to measure >1% on the International Scale (BCR-ABL1IS) by quantitative real-time polymerase chain reaction at screening. Additional inclusion/exclusion criteria are listed in supplemental Table 1 (available on the Blood Web site).
Study design and treatment
Patients were enrolled at 61 sites in 19 countries, spanning North America, Europe, Latin America, and the Asia-Pacific region. Patients were allocated randomly in a 1:1:1 ratio to receive a once-daily starting dose of ponatinib 45 (45-mg cohort), 30 (30-mg cohort), or 15 mg (15-mg cohort). Randomization was stratified by age (≥60 vs <60 years) and history of arterial hypertension, diabetes mellitus, and/or hyperlipidemia (yes/no). Patients in the 45- and 30-mg cohorts were required to have the dose reduced to 15 mg once daily upon achievement of ≤1% BCR-ABL1IS (grossly equivalent to CCyR).14 Patients in the 15-mg cohort had no response-based dose changes.
Patients could escalate to their original starting dose for loss of response confirmed in 2 consecutive assessments in the absence of AE-driven dose modifications. Dose reduction caused by AEs, to a minimum of 10 mg, was permitted in all cohorts. Patients in the 45- and 30-mg cohorts could undergo dose reduction for AEs before having ≤1% BCR-ABL1IS. BCR-ABL1 mutations were assessed by Sanger sequencing at a central laboratory (MolecularMD, Portland, OR).
End points
The primary end point was ≤1% BCR-ABL1IS at 12 months. Secondary efficacy end points, described in detail in the supplemental Appendix, included rates of molecular, cytogenetic, and hematologic response, and survival outcomes. Safety evaluations included rates of AEs including AOEs, serious AEs, discontinuations for AEs, dose reduction, and dose interruption.
Efficacy assessments
Response assessments performed per standard criteria15 included cytogenetic response by bone marrow karyotyping at cycle 12 (partial cytogenetic response, CCyR or major cytogenetic response [MCyR]), molecular response by BCR-ABL1IS assessment (≤1% BCR-ABL1IS and major molecular response) in peripheral blood samples at 3-month intervals, and complete blood count for assessment of complete hematologic response (CHR) at 3-month intervals (additional details in supplemental Table 2).
Survival assessments
Progression-free survival (PFS) was defined as the interval between the first dose and disease progression (progression to accelerated-phase CML or blast-phase CML, loss of CHR or MCyR, or doubling of white blood cell count to >20 000 on 2 occasions at least 4 weeks apart in patients without CHR) or death from any cause. PFS-defining events are presented in supplemental Table 2. OS was defined as the interval between the first dose and death from any cause.
Safety assessments
Safety was assessed by physical and laboratory evaluations, electrocardiograms, echocardiograms, and AE monitoring throughout the study. AE severity was assessed according to National Cancer Institute Common Terminology Criteria for Adverse Events, v4.0. Investigator’s assessment of causality was provided for all AEs. The independent cardiovascular (CV) end point adjudication committee reviewed all documentation related to AOEs (details in the supplemental Appendix).
Statistical analysis
Each cohort was analyzed separately for efficacy and safety. Categorical data were summarized by number and percentage of patients and continuous data by descriptive statistics. The intent-to-treat population included all randomly assigned patients for whom BCR-ABL1IS could be measured, regardless of whether they received the assigned study drug, and the safety population included all patients who received at least 1 dose of study drug. Duration of treatment was defined as the time interval from the first to the last dose of study treatment. Dose intensity was calculated as the total cumulative dose divided by the duration of drug exposure. PFS and OS were estimated by the Kaplan-Meier method.
A sample size of 92 patients per cohort was chosen to distinguish a favorable ≤1% BCR-ABL1IS rate of 35% from a null or uninteresting response rate of 20% with a nominal 80% power and a 1-sided type 1 error rate of 0.0083 by exact binomial test. The study design targeted 35% as the interesting response rate to align with the poor response rates observed in resistant populations with ≥2 prior TKIs (22% to 26% for second-generation TKIs at any time3). The lower boundary of the 2-sided exact 98.3% confidence interval (CI) for the ≤1% BCR-ABL1IS rate was required to be >20% for the primary end point to be met. Missing and partial data were imputed according to conventions described in the statistical analysis plan (supplemental Appendix).
Results
Patient disposition and baseline characteristics
The data cutoff was 31 May 2020, and the median (range) duration of follow-up for the total population was 32 months (1-57). From August 2015 through May 2019, 283 patients were randomly assigned, of whom 282 received at least 1 dose of study drug (safety population). Of those, 134 (47.5%) were still receiving treatment and were included in the analyses in this study (supplemental Figure 1). The most common reason for treatment discontinuation was lack of efficacy (18.8%; 53 of 282); importantly, 77.4% of those patients were in the lower dose cohorts. Additional reasons for treatment discontinuation are listed in supplemental Table 3. All patients were evaluable for response.
Patient baseline characteristics are summarized in Table 1. Most patients (55%) had received at least 3 prior TKIs, and 99% were resistant to at least 1 prior TKI therapy. The best response to last prior therapy was CHR or worse for 61%. One-third (33%) had at least 1 CV risk factor at baseline. More than 40% of patients had at least 1 kinase domain mutation at study entry. A total of 204 of 282 patients (72.3%) had a duration of exposure >12 months, and 100 of 282 patients (35.5%) had a duration of exposure >24 months.
Efficacy
The rate of ≤1% BCR-ABL1IS (98.3% CI) at 12 months (the primary end point), was 44.1% (31.7-57.0) in the 45-mg cohort, 29.0% (18.4-41.6) in the 30-mg cohort, and 23.1% (13.4-35.3) in the 15-mg cohort (Figure 1A). The prespecified statistical end point was met in the 45-mg cohort (P < .017). The median (range) time to response was 6.0 (2.9-18.0), 3.0 (2.9-15.3), and 6.0 (2.9-31.9) months in the 45-, 30-, and 15-mg cohorts, respectively. Cumulative responses (by 6, 12, and 24 months) increased over time in all cohorts with the 45-mg cohort showing the most robust increase, despite the response-directed dose reduction over time in this cohort (Figure 1B; supplemental Figure 2).

Response to once-daily ponatinib. Twelve months (A) and median dose intensity and response over time (B). (A) ≤1 % BCR-ABL1IS at 12 months (98.3% CI). (B) Median dose intensity and ≤1% BCR-ABL1IS by 6, 12, and 24 months.
Response to once-daily ponatinib. Twelve months (A) and median dose intensity and response over time (B). (A) ≤1 % BCR-ABL1IS at 12 months (98.3% CI). (B) Median dose intensity and ≤1% BCR-ABL1IS by 6, 12, and 24 months.
We also calculated the cumulative rates of ≤1% BCR-ABL1IS by 12 months in the subgroups of patients (Table 2; supplemental Figure 3). Patients with or without the T315I mutation at baseline had high ≤1% BCR-ABL1IS rates by 12 months (60.0% and 48.5%, respectively) in the 45-mg cohort. In the lower dose cohorts, a lower proportion of patients with the T315I mutation had ≤1% BCR-ABL1IS rate compared with those without T315I. This difference was more notable in patients in the 15-mg cohort (10.5% vs 29.6%) than in those in the 30-mg cohort (25.0% vs 38.4%). Nine patients with the T315I mutation at baseline had additional mutations at baseline. Among them, 7 patients did not achieve ≤1% BCR-ABL1IS at any time. For the patients who had end-of-treatment mutation results available (n = 26), there were 5 who had the T315I mutation at baseline and acquired an additional BCR-ABL1 mutation during treatment. None of those 5 patients achieved ≤1% BCR-ABL1IS at any time. Response rates in patients with no mutations at baseline were 46.0%, 37.9%, and 28.3% in the 45-, 30-, and 15-mg cohorts, respectively. In an analysis based on best response to last prior therapy (CHR or worse vs better than CHR), a high ≤1% BCR-ABL1IS rate was seen for the 45-mg cohort in the 2 subgroups, whereas responses were higher in the “better than CHR” subgroup in the lower starting dose cohorts (Table 2). The overall major molecular response rates were 34.4%, 24.7%, and 23.1% in the 45-, 30-, and 15-mg cohorts, respectively; the overall MCyR rates were 50.5%, 33.3%, and 43.8%, respectively (supplemental Table 4).
Disease progression and survival
During the study, progression to accelerated-phase CML and blast-phase CML occurred in 11% to 12% and 1% to 3% of patients, respectively, across the 3 cohorts. Median PFS was not reached (NR) in the 45- and 30-mg cohorts and was 45.6 months in the 15-mg cohort (Figure 2A). The estimated probability of 24-month PFS was 80%, 76%, and 78% in the 45-, 30-, and 15-mg cohorts, respectively. Median (95% CI) OS was NR in all cohorts (Figure 2B); the estimated probability of 24-month OS was more than 90% in all 3 cohorts.

Safety and tolerability
Treatment-emergent adverse events (TEAEs) and dose modifications for TEAEs are summarized in Table 3. The most common nonhematologic TEAEs were arterial hypertension (28%), headache (18%), and lipase increase (17%) in all cohorts combined; the majority were grade 1 or 2. Overall, the most common hematologic TEAEs were thrombocytopenia (40%), neutropenia (26%), and anemia (19%). Nonhematologic and hematologic TEAE rates for each cohort are reported in supplemental Table 5. There were 4 deaths related to AEs (2 sudden deaths in the 45-mg cohort in patients who had CV risk factors at baseline and 2 deaths from pneumonia in the 15-mg cohort).
Rates of treatment-emergent AOEs (TE-AOEs) and dose modifications for TE-AOEs are summarized in Table 3. Overall, 17 of 282 patients (6%) experienced any TE-AOE (9 in the 45-mg cohort, 5 in the 30-mg cohort, and 3 in the 15-mg cohort). These events are summarized in supplemental Table 6. The exposure-adjusted TE-AOE rates were 5.6%, 3.6%, and 2.1% for the 45-, 30-, and 15-mg cohorts, respectively. Exposure-adjusted TE-AOEs decreased over time, as seen in the 45-mg cohort: 7.6 events per 100 person-years during the first year, 5.9 events per 100 person-years during the second year, and 0 events for years 3 to 5 (supplemental Table 7; supplemental Figure 4). Grade 3 to 5 TE-AOEs were reported in 13 patients (4.6%; 5, 5, and 3 in the 45-, 30-, and 15-mg cohorts respectively). Eight patients discontinued treatment because of TE-AOEs. One venous thromboembolism was reported in the study (grade ≤3 retinal vein occlusion in the 45-mg cohort). In the current analysis, a starting dose of 45 mg compared with 15 mg was associated with a 6.4 percentage-point increase in the AOE rate (9.6% to 3.2%), but with a 26.3 percentage-point improvement in the response rate (51.6% to 25.3%; Figure 3).
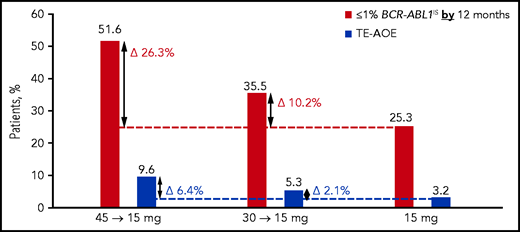
Overall safety and efficacy by starting dose. The analysis is a descriptive clinical summary of the data to illustrate the relationship between the efficacy and AOE rate.
Overall safety and efficacy by starting dose. The analysis is a descriptive clinical summary of the data to illustrate the relationship between the efficacy and AOE rate.
Dynamics of dosages
Overall median (range) dose intensity (in milligrams per day) was 27.7 (10.5-45.0) in the 45-mg cohort, 23.0 (5.1-30.0) in the 30-mg cohort, and 14.7 (6.0-15.0) in the 15-mg cohort, and dose reductions were reported in 75 (79.8%), 58 (61.7%), and 33 (35.1%) patients in the 45-, 30-, and 15-mg cohorts, respectively. Median (range) time to dose reduction was shortest in the 45-mg cohort, 3.4 (0.1-41.9) months, compared with the 30- and 15-mg cohorts, 7.1 (0.1-40.5) months and 11.4 (0.2-46.6) months, respectively.
In the 30- and 45-mg cohorts, 73 patients had a dose reduction to 15 mg after achieving ≤1% BCR-ABL1IS. Of those, 55 of 73 (75%) maintained a response for any period of time (48 maintained a response at a lower dose, and 7 had their doses reescalated without confirmation of loss of response at the second assessment); the 2 arms were similar in dose reductions and maintenance of response. Ninety-one percent (50 of 55) of those patients continued to maintain a response for 6 months or longer. Of the 18 patients who did not maintain the response, more than 70% lost the response within the first 6 months (supplemental Table 8), and the majority (61%; n = 11) had the T315I mutation at baseline. Because reescalation for loss of response was allowed for in the protocol, 13 and 5 patients reescalated to 45 and 30 mg, respectively; of those, 8 (61.5%) and 4 (80.0%) regained the response by the data cutoff (supplemental Table 9). Of the patients who achieved ≤1% BCR-ABL1IS, 37 of 45 (82%) patients in the 45-mg cohort and 21 of 28 (75%) patients in the 30-mg cohort remained on trial and were receiving ponatinib at the time of data cutoff.
AEs led to dose reductions in 42 (44.7%), 32 (34.0%), and 29 (30.9%) patients in the 45-, 30-, and 15-mg cohorts, respectively, with dose reduction to 10 mg (driven by dose modifications for safety) in 5.3%, 17.0%, and 35.1% of patients, respectively (supplemental Table 10).
Discussion
This global, multicenter, phase 2 study is the first to prospectively evaluate a response-based dose-reduction strategy to optimize the benefit/risk of a TKI in patients with CP-CML. The OPTIC study showed benefit with ponatinib in all 3 dosage regimens in a largely resistant (99%) population, in which the majority (61%) had not achieved a response better than a CHR on the immediate preceding therapy, and 55% had received 3 or more prior TKIs. Such patients typically have poor outcomes if treated with another second-generation TKI.2-7 Ponatinib acted in patients with resistant disease, both with and without a BCR-ABL1 mutation.
The maximum benefit was observed in the 45-mg cohort. Patients in 30- and 15-mg cohorts also experienced a benefit, especially those without the T315I mutation or who had better than a CHR to the last prior therapy. The data support a higher starting dose (45 mg) in patients with the T315I mutation. Patients with T315I at baseline have a higher probability of losing response after dose reduction. The benefit/risk of dose reduction in such patients should be carefully assessed and the patients closely followed after dose reduction. Although the median time to response was similar in the 45- and 15-mg cohorts, this result most likely occurred because of a higher rate of discontinuation for lack of efficacy in the 15-mg cohort compared with the 45-mg cohort. The responses were maintained in most of the patients after the response-based dose reduction from 45 or 30 to 15 mg (73.3% and 78.6%, respectively; supplemental Table 8; supplemental Figure 5). Most of the patients in the 45- and 30-mg cohorts in whom the response was lost when the dose was reduced had the T315I mutation at baseline. Most patients were able to regain a response after reescalation of the dose. The results in OPTIC also showed robust long-term survival in all 3 arms, with rates of 88.6% to 91.7% at 3 years, consistent with the results of the PACE trial,10 indicating that the dose-reduction strategy did not affect survival.
Patients with CML often have comorbidities that may increase the risk of CV AEs with TKI, and cardiac toxicity has been reported with second-generation TKIs (eg, in ENESTnd, CV events occurred in 16.5% and 23.5% in the 2 nilotinib arms, respectively, although the length of follow-up was considerably longer than in OPTIC16; in DASISION they occurred in 5% of patients treated with dasatinib),17 which can lead to discontinuation for intolerance at rates of 22% to 67%.5,16-20 The novel response-based ponatinib dosage regimens resulted in clinically manageable safety and AOE profiles. The rates of AOEs in OPTIC (6.0% overall and 9.6% in the 45-mg cohort) and exposure-adjusted AOEs appeared lower than those reported in PACE, where patients started at 45 mg ponatinib but response-based dose reduction was not part of the study design.10 Although differences in eligibility criteria preclude a direct comparison between the 2 studies, the CV exclusion criteria in OPTIC are consistent with those used in CP-CML studies of other TKIs.21-23 Thirty-three percent of the patients in OPTIC had at least 1 CV risk factor at baseline, and 4.6% had multiple risk factors. A previous post hoc analysis suggested that the incidence of AOEs and some other TEAEs correlated positively with ponatinib dose intensity.13 Exposure-adjusted AOEs decreased with time, most likely reflecting the combined effects of dose reductions for efficacy or safety and discontinuations because of safety concerns.
Although benefit/risk assessment is central to clinical decision making, it is often difficult to compare disparate end points such as response and toxicity. A descriptive analysis of data from this response-based study of dose-deescalation design suggests that a starting dose of 45 mg ponatinib daily may be associated with a modest (6.4 percentage point) increase in the AOE rate compared with the 15-mg starting dose (3.2% to 9.6%). In comparison, there appeared to be a 26.3 percentage-point improvement in the response rate (25.3% to 51.6%; Figure 3). Therefore, the absolute gain in efficacy is larger than the increase in AOEs, suggesting that the optimal benefit/risk may be achieved with a 45-mg starting dose followed by a reduction to 15 mg upon achievement of ≤1% BCR-ABL1IS. Although OPTIC was not powered to compare OS, CCyR (equivalent to BCR-ABL1 ≤1%) was the most robust predictor of OS, suggesting that this strategy may eventually translate into improved OS.
Benefit was also seen with starting doses of 30 and 15 mg in patients without a T315I mutation and in patients with less resistant disease, indicating that molecular characteristics may be useful in further refinement of risk-adapted therapy strategies. Despite limitations, such as open-label design and statistically inconclusive results of some subgroup analyses because of the small sample size, the results of the OPTIC trial support a novel ponatinib treatment regimen of a 45-mg starting dose reduced to 15 mg upon reaching ≤1% BCR-ABL1IS, which maximizes response while minimizing toxicity and provides a rationale to explore response-based dose-modification strategies for other BCR-ABL1 TKIs.
Acknowledgments
The authors thank the patients, their families, and their caregivers, the OPTIC Study investigators and their team members at each study site, and colleagues from Millennium Pharmaceuticals, Inc, Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Professional medical writing assistance was provided by Duprane Young of Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, and funded by Millennium Pharmaceuticals, Inc.
Authorship
Contribution: J.C., J.A., A.H., P.R., G.R., M.M., J.H.L., and M.D. designed the study; J.C., J.A., E.L., B.M., M.U.S., C.P., C.C., T.S., J.H.L., C.A.S., J.M., A.H., P.R., G.R., H.d.L, A.T., C.R., C.K.A., L.M., M.T., M.M., and M.D. performed data collection; S.S. served as the study monitor; T.H. served as the safety reviewer; and V.L. served as the study statistician; and all authors performed data analysis and interpretation, had full access to and verified all the data in the study, were involved in the drafting and critical revisions, and had were responsible for the decision to submit the article for publication.
Conflict-of-interest disclosure: J.C. has been a consultant to and received research funding from Bristol Myers Squibb, Daiichi Sankyo, Jazz Pharmaceuticals, Astellas, Novartis, Pfizer, Takeda, and BioPath Holdings; has received research funding from Sun Pharma, Telios, Arog, Merus, and Immunogen; has held membership on the board of directors or advisory committee of BioPath Holdings; and has been a consultant to Amphivena Therapeutics and BiolineRx. J.A. has received honoraria and research funding and is on the speakers bureaus of Incyte and Pfizer; and has received honoraria and served on the speakers bureaus of Bristol Myers Squibb and Novartis. E.L. has served on the speakers bureaus of Novartis and Pfizer; and has received travel and accommodation reimbursement from Novartis, Pfizer, and Bristol Myers Squibb. B.M. has served on the speakers bureaus of Novartis, Pfizer, and Takeda. M.U.S. has served on the advisory boards of AbbVie, Janssen, Novartis, Pfizer, and Roche and on the speakers bureaus of Janssen, Novartis, and Pfizer. C.C. has received honoraria from Novartis and Korea Otsuka Pharmaceutical; received honoraria and research funding from Bristol Myers Squibb; and received travel reimbursement and research funding from Pfizer. T.S. has been a consultant to, has received honoraria from, and has served on the speakers bureaus of Bristol Myers Squibb, Novartis, Pfizer, and Incyte; and has been a consultant to and received honoraria from Adamed. J.H.L. has been a consultant to and has received research funding from Bristol Myers Squibb, Ariad, Pfizer, and Novartis. C.A.S. has been a consultant to Bristol Myers Squibb and Novartis; and has received research funding from Takeda. A.H. has received honoraria and research funding from Bristol Myers Squibb, Novartis, and Pfizer; research funding from Incyte and Merck Sharp & Dohme; and honoraria from Takeda. P.R. has been a consultant to and has received funding from Incyte and Pfizer; and has been a consultant to Bristol Myers Squibb, Novartis, and Takeda. G.R. has received research funding from and served on the speakers bureau for Pfizer; and has served on the speakers bureaus of Bristol Myers Squibb, Incyte, and Novartis. H. de L. has received honoraria and research funding from Bristol Myers Squibb and Incyte, and honoraria from Pfizer and Novartis. C.R. has received personal fees from AstraZeneca, Roche, Novartis, and Janssen. M.T. holds membership on the board of directors or advisory committees of Bristol Myers Squibb and Constellation Pharmaceuticals; has received research funding from Takeda and Novartis; and has been a consultant to IMAGO. M.M. has been a consultant to and has received honoraria; reimbursement of travel, accommodation and expenses; and research funding from Bristol Myers Squibb, Novartis, Takeda, and Pfizer; and research funding from Sun Pharma/SPARC. T.H. and V.L. are employees of Millennium Pharmaceuticals. S.S. is an employee of Takeda. M.D. is a consultant to, holds a membership on the board of directors or advisory committees of; was part of a study management committee of; and received research funding from Blueprint Medicines Corporation; is a consultant to Fusion Pharma, Medscape, and DisperSol; is a consultant to, has held membership on the board of directors or advisory committees of, and has received research funding from Takeda; is a consultant to and holds membership on the board of directors or advisory committee of Sangamo; is a consultant to and received research funding from Novartis; is a consultant to and received honoraria and research funding from Incyte; and has received research funding from SPARC, DisperSol, and the Leukemia and Lymphoma Society. C.P., A.T., C.K.A., J. M., and L.M. declare no competing financial interests.
The current affiliation for J.C. is Georgia Cancer Center, Augusta University, Augusta, GA.
Correspondence: Jorge Cortes, Georgia Cancer Center, 1410 Laney Walker Rd, CN2222, Augusta, GA 30912; e-mail: jorge.cortes@augusta.edu.
Presented in oral form at the 2020 virtual meeting of the American Society of Clinical Oncology, 29 May 2020; presented in oral form at the 25th congress of the European Hematology Association virtual meeting, 12 June 2020 and presented in poster form at the 2020 virtual meeting of the Society of Hematologic Oncology, 9 September 2020.
The data sets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results of the completed study, will be made available after the publication of the final study results within three months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
There is a Blood Commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.


