Key Points
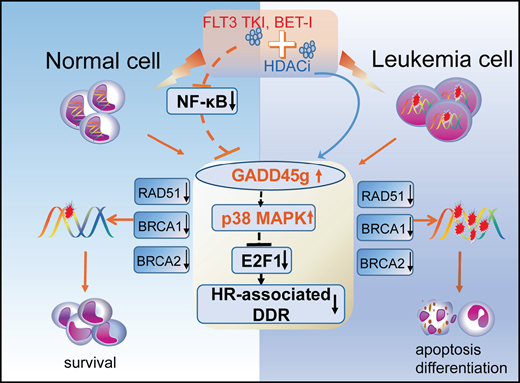
GADD45g is a novel tumor suppressor in AML and exerts selective and potent antileukemic activities.
GADD45g activation may be exploited therapeutically for treatment of FLT3-ITD+ and MLL-AF9+ AML by combining romidepsin with AC220 or JQ1.

Abstract
Acute myeloid leukemia (AML) is an aggressive hematopoietic malignancy for which there is an unmet need for novel treatment strategies. Here, we characterize the growth arrest and DNA damage-inducible gene gamma (GADD45g) as a novel tumor suppressor in AML. We show that GADD45g is preferentially silenced in AML, especially in AML with FMS-like tyrosine kinase 3–internal tandem duplication (FLT3-ITD) mutations and mixed-lineage leukemia (MLL)-rearrangements, and reduced expression of GADD45g is correlated with poor prognosis in patients with AML. Upregulation of GADD45g impairs homologous recombination DNA repair, leading to DNA damage accumulation, and dramatically induces apoptosis, differentiation, and growth arrest and increases sensitivity of AML cells to chemotherapeutic drugs, without affecting normal cells. In addition, GADD45g is epigenetically silenced by histone deacetylation in AML, and its expression is further downregulated by oncogenes FLT3-ITD and MLL-AF9 in patients carrying these genetic abnormalities. Combination of the histone deacetylase 1/2 inhibitor romidepsin with the FLT3 tyrosine kinase inhibitor AC220 or the bromodomain inhibitor JQ1 exerts synergistic antileukemic effects on FLT3-ITD+ and MLL-AF9+ AML, respectively, by dually activating GADD45g. These findings uncover hitherto unreported evidence for the selective antileukemic role of GADD45g and provide novel strategies for the treatment of FLT3-ITD+ and MLL-AF9+ AML.
Introduction
Acute myeloid leukemia (AML) is an aggressive hematologic neoplasm with distinct molecular and genetic features that is characterized by differentiation blockade and accumulation of immature myeloid cells.1,2 Among numerous genomic abnormalities in AML, FMS-like tyrosine kinase 3–internal tandem duplication (FLT3-ITD) and mixed-lineage leukemia (MLL) gene rearrangement are often associated with frequent relapses and unsatisfactory prognosis.3-5 Current treatments for AML are limited, and chemotherapy is still the standard therapeutic approach.6,7 A complete response to therapy is <40%, and the outcome is even worse for patients with poor-risk AML.8,9 It is imperative to identify new molecular targets and therapeutic approaches to improve treatment of AML, especially for patients with a poor prognosis.10
Cells use a tightly coordinated DNA damage response (DDR), which comprises a network of proteins to respond to DNA damage.11 After sensing DNA damage, cellular DDR either initiates signaling pathways that promote cell cycle checkpoint activation and repair the damage, or activates an apoptotic cell death program.12 The DNA repair system includes base-excision repair and mismatch repair for single-strand breaks, non-homologous end-joining pathways, and homologous recombination (HR)-mediated repair for DNA double-strand breaks (DSBs).13 DSBs are the most cytotoxic forms of DNA damage, and HR is the major repair pathway required for repairing DSBs.13,14 Numerous reports indicate that AML cells accumulate high levels of DNA lesions relative to normal cells,15 leading to their increased dependency on repair pathways and susceptibility to DDR inhibition than normal cells. Inhibition of DNA repair may create an opportunity to eradicate these cells while sparing normal cells.
The growth arrest and DNA damage–induced 45 (GADD45) family of genes, including GADD45a, GADD45b, and GADD45g, encode for small (∼18 kDa) evolutionarily conserved proteins. GADD45 family members are known as stress sensors, which can be rapidly induced by various physiological and environmental stresses, such as genotoxic agents and inflammatory cytokines.16,17 They have been implicated in regulating cell survival, apoptosis, senescence, cell cycle arrest, and genomic stability.16-19 Despite marked similarities, the GADD45 family exhibits functional diversity. Altered expression of the three GADD45 family members has been observed in a variety of human solid tumor types, acting as either a tumor promoter or a tumor suppressor, depending on cell type.17 Inactivation of the 3 genes is often associated with epigenetic dysregulation.20 In hematopoietic malignancies, studies have reported that GADD45a promoter methylation predicts poor overall survival (OS) in AML21 ; both Gadd45a and Gadd45b deficiency could accelerate BCR-ABL–driven chronic myeloid leukemia.22,23 To date, however, the role of GADD45g in hematopoietic malignancies remains completely unknown.
In the current study, we identify GADD45g as a tumor suppressor in AML and propose novel synergistic strategies for the treatment of AML, acting through dual activation of GADD45g.
Materials and methods
A complete Methods section is provided in the supplemental Material and methods (available on the Blood Web site.)
Cell culture
Molm-13, THP-1, U937, and HL-60 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS). MV4-11 cells were maintained in Iscove modified Dulbecco’s medium supplemented with 10% FBS. 293T cells were cultured in Dulbecco’s modified Eagle medium with 10% FBS. Cells were grown at 37°C in a humidified atmosphere containing 5% carbon dioxide.
Patient samples
Bone marrow specimens were collected from patients with newly diagnosed AML at the Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences, following receipt of informed consent. Human cord blood (CB) was obtained following receipt of informed consent from healthy postpartum women. All laboratory experiments with primary samples were performed under the ethical principles for medical research and were approved by the Ethics Committee of the Institute of Hematology and Blood Diseases Hospital.
AML xenograft model
Six- to eight-week-old NOD/SCID mice were purchased from the Institute of Laboratory Animals, Chinese Academy of Medical Sciences. All animal experiments were conducted in compliance with the animal care guidelines approved by the Institutional Animal Care and Use Committees of the State Key Laboratory of Experimental Hematology.
Transplanted mice were sublethally irradiated (200 cGy) 12 hours before injection. For experiments assessing the antileukemic activity of GADD45g in vivo, 8 × 105 Molm-13 cells expressing firefly luciferase (Molm13-luc2) were transfected with doxycycline (Dox)-inducible GADD45g lentiviral vector and engrafted into the irradiated NOD/SCID mice. Bioluminescence imaging was used to monitor and assess in vivo engraftment of human AML cells. After engraftment was confirmed 7 days following transplantation, Dox (1 mg/mL) supplemented with 7.5% sucrose was administered to the mice in their drinking water for 7 days. To evaluate the therapeutic potential of combination regimens in vivo, 8 × 105 Molm13-luc2 cells were engrafted into NOD/SCID mice. After engraftment was confirmed 5 days later, mice were treated intraperitoneally or by oral gavage with either vehicle (control), romidepsin (1.5 mg/kg), AC220 (10 mg/kg), JQ1 (50 mg/kg), or the indicated combination for 2 weeks. At the end of each experiment, bioluminescence images were taken, and the survival time was observed. Bioluminescent images were obtained by using Caliper IVIS Lumina II (Caliper Life Sciences).
Statistical analysis
Results are presented as mean ± standard deviation (SD) and were analyzed by using a paired or unpaired Student t test. Multiple groups were analyzed with one-way analysis of variance. Correlations between continuous variables were calculated by using the Pearson correlation. Survival was estimated by using the Kaplan-Meier method and compared by using the log-rank test. Values of P < .05 were considered significant in all experiments. Data were analyzed and plotted by using GraphPad Prism 8 software (GraphPad Software). Univariate and multivariate Cox proportional hazards models were established, and hazard ratios are presented with their 95% confidence intervals. The analyses were performed by using SPSS 20.0 (IBM SPSS Statistics, IBM Corporation).
Results
GADD45g is preferentially silenced in AML, and low GADD45g expression is correlated with poor prognosis of AML patients
To explore the potential role of GADD45g in leukemia, we first analyzed its expression in the Cancer Cell Line Encyclopedia data set (n = 947)24 and 3 large-cohort AML data sets (Microarray Innovations in Leukemia, GSE1159, and GSE37307).25-27 We found that GADD45g was expressed at a significantly lower level in AML cell lines and in bone marrow mononuclear cells (BMMNCs) from AML patients, compared with non-AML cell lines or BMMNCs and CD34+ BM cells from healthy donors, respectively (Figure 1A-B; supplemental Figure 1A-B). The results were validated when we examined the expression of GADD45g in 15 leukemia cell lines and primary BMMNCs from patients with AML (n = 85), and BMMNCs from healthy donors (n = 39) and MNCs from human CB (n = 5) as normal controls, at both messenger RNA (mRNA) and protein levels (Figure 1C-F). The clinical characteristics of the patients with AML, whose GADD45g protein levels are shown in Figure 1F, are presented in supplemental Table 1. These results show that GADD45g expression is markedly silenced in AML.
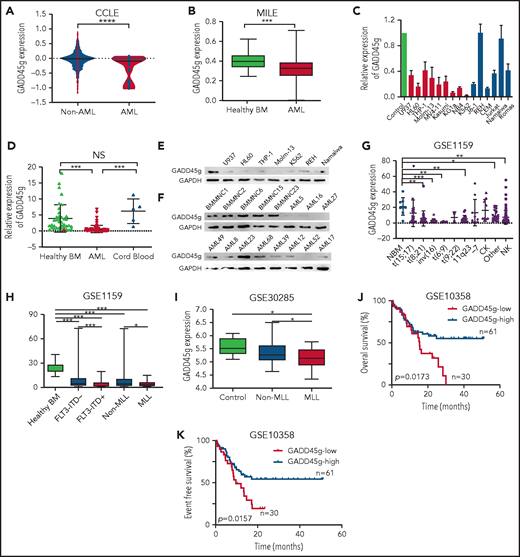
Significantly lower expression of GADD45g in AML cells and its correlation with poor prognosis. (A) Violin plot of GADD45g expression in the Cancer Cell Line Encyclopedia (CCLE) data set, comparing AML cell lines (AML; n = 34) vs non-AML cancer cell lines (Non-AML; n = 913). (B) Comparison of GADD45g expression between primary BMMNCs from patients with AML (n = 542) and normal controls from healthy volunteers (n = 74) in AML datasets Microarray Innovations in Leukemia (MILE) (GSE13204). (C) Relative mRNA expression of GADD45g in hematopoietic malignant cell lines quantified by qRT-PCR and normalized to β-actin. (D) GADD45g mRNA expression in primary BMMNCs from patients with AML (n = 85) and BMMNCs from healthy donors (n = 39) and MNCs from human CB (n = 5) as normal controls, as determined by qRT-PCR. Western blot analysis of GADD45g protein levels in hematopoietic malignant cell lines (E) and in primary BMMNCs from patients with AML (n = 11) and healthy donors (F) (n = 5). (G) The expression patterns of GADD45g in different disease subsets in the AML data set GSE1159 (n = 272). The correlation of GADD45g expression with MLL-rearrangements or FLT3-ITD mutations in AML data sets GSE1159 (H; n = 272) and GSE30285 (I; n = 93). OS (J) and event-free survival (K) of AML patient in AML data set GSE10358 stratified according to GADD45g expression. Survival curves were analyzed by using the Kaplan-Meier method, and P values were determined with the log-rank (Mantel-Cox) test. Data are presented as mean ± SD, and differences were compared by using the 2-tailed Student t test. Multiple groups were analyzed with the 1-way analysis of variance. *P < .05, **P < .01, ***P < .001, ****P < .0001. CK, complex karyotype; NBM, normal bone marrow; NK, normal karyotype; NS, not significant.
Significantly lower expression of GADD45g in AML cells and its correlation with poor prognosis. (A) Violin plot of GADD45g expression in the Cancer Cell Line Encyclopedia (CCLE) data set, comparing AML cell lines (AML; n = 34) vs non-AML cancer cell lines (Non-AML; n = 913). (B) Comparison of GADD45g expression between primary BMMNCs from patients with AML (n = 542) and normal controls from healthy volunteers (n = 74) in AML datasets Microarray Innovations in Leukemia (MILE) (GSE13204). (C) Relative mRNA expression of GADD45g in hematopoietic malignant cell lines quantified by qRT-PCR and normalized to β-actin. (D) GADD45g mRNA expression in primary BMMNCs from patients with AML (n = 85) and BMMNCs from healthy donors (n = 39) and MNCs from human CB (n = 5) as normal controls, as determined by qRT-PCR. Western blot analysis of GADD45g protein levels in hematopoietic malignant cell lines (E) and in primary BMMNCs from patients with AML (n = 11) and healthy donors (F) (n = 5). (G) The expression patterns of GADD45g in different disease subsets in the AML data set GSE1159 (n = 272). The correlation of GADD45g expression with MLL-rearrangements or FLT3-ITD mutations in AML data sets GSE1159 (H; n = 272) and GSE30285 (I; n = 93). OS (J) and event-free survival (K) of AML patient in AML data set GSE10358 stratified according to GADD45g expression. Survival curves were analyzed by using the Kaplan-Meier method, and P values were determined with the log-rank (Mantel-Cox) test. Data are presented as mean ± SD, and differences were compared by using the 2-tailed Student t test. Multiple groups were analyzed with the 1-way analysis of variance. *P < .05, **P < .01, ***P < .001, ****P < .0001. CK, complex karyotype; NBM, normal bone marrow; NK, normal karyotype; NS, not significant.
Despite the overall low expression of GADD45g in AML, we observed substantial heterogeneity among patients with AML (Figure 1G). This finding led us to investigate whether there was any correlation between the expression of GADD45g and specific genetic and chromosomal abnormalities in AML. By analyzing the gene expression profiles of GSE1159, GSE6891, and GSE30285, we found that GADD45g is aberrantly downregulated in AML subtypes that harbored MLL-rearrangements or FLT3-ITD mutations (Figure 1H-I; supplemental Figure 1C).
We subsequently investigated the correlation of GADD45g expression with outcome of patients with AML. We analyzed 2 independent data sets of AML patients (GSE10358 and GSE12417) whose gene expression and time-to-event data were available,28 and we found that patients with lower GADD45g expression exhibited strikingly inferior OS as well as event-free survival than those with higher GADD45g expression (Figure 1J-K; supplemental Figure 1D-E). Furthermore, univariate and multivariate analyses were performed in the GSE10358 data set. Ten variables were considered in univariate analysis for OS, and variables with a value of P ≤ .1 were included in a multivariate model. The variables GADD45g expression, age, and BM blast were entered into multivariate analysis with the Cox proportional hazards model. We observed that only age remained associated with statistically significantly inferior OS (hazard ratio, 3.201; 95% CI, 1.751-5.852; P = .000), whereas low GADD45g expression was not an independent predictor for OS (supplemental Tables 2-3).
Overall, these observations reveal that GADD45g expression is preferentially silenced in AML, especially in AML with MLL-rearrangements and FLT3-ITD mutations, and reduced expression of GADD45g is closely related to poor prognosis in patients with AML.
GADD45g induces apoptosis and differentiation, inhibits growth and colony-forming capacity, and enhances the chemosensitivity of AML cells
To investigate the role of GADD45g in AML, we generated a Dox-inducible Tet-on lentiviral system to induce GADD45g ectopic expression in AML cell lines. After Dox administration, the expression level of GADD45g was significantly upregulated in U937, HL60, THP-1, and Molm-13 cells as validated by quantitative reverse transcription polymerase chain reaction (qRT-PCR) and/or western blot analysis (Figure 2A). We found that overexpression of GADD45g in AML cells led to significant decreases in proliferation and colony formation and marked increases in cell apoptosis (Figure 2B-D; supplemental Figure 2A-C), whereas the cell cycle status was only marginally affected (supplemental Figure 2D). We then analyzed the expression of key genes associated with the aforementioned phenotypes and observed that forced expression of GADD45g resulted in a dramatically increased expression of genes promoting apoptosis (p73, BAD, BAK, BAX, BID, BIK, BIM, NOXA, and PUMA) and a sharply decreased expression of proto-oncogenes (Myc, BCL-2, BCL-XL, and TRIB2) in all 4 AML cell lines (supplemental Figure 2E). Furthermore, to verify the findings in cell lines, primary cells from AML patients and normal human CB were transduced with noninducible lentivirus vectors carrying GADD45g, and significantly upregulated GADD45g expression was validated by qRT-PCR (supplemental Figure 2F). Consistent with the findings in AML cell lines, enforced expression of GADD45g dramatically induced apoptosis and impaired the colony formation ability of primary leukemia cells from patients with AML (Figure 2E-F); its knockdown with small hairpin RNAs exerted the opposite effect on apoptosis (Figure 2G; supplemental Figure 2G). By contrast, GADD45g overexpression had little deleterious impact on normal human CB MNCs and CD34+ cells. These findings indicate that GADD45g exhibits a selective cell-killing effect on hematologic malignant cells.
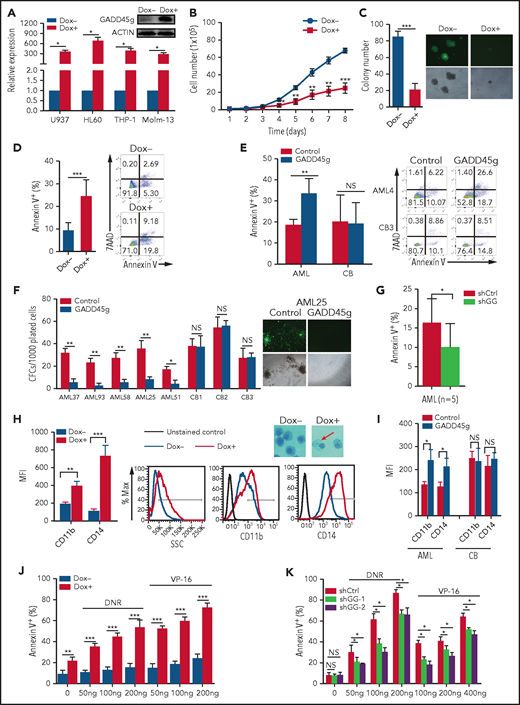
Human AML cells are sensitive to GADD45g overexpression. (A) qRT-PCR evaluation of GADD45g expression in AML cells with or without Dox-induced GADD45g overexpression and representative western blot in THP-1 cells. Effects of Dox-induced GADD45g expression on cell proliferation (B), colony formation (C), and apoptosis (D) in THP-1 cells as determined by cell counting, colony-forming assay, and flow cytometric analysis of Annexin V and 7AAD staining, respectively. (E) Primary BMMNCs from patients with AML (n = 5) and cord blood mononuclear cells from healthy donors (n = 5) were transfected with human GADD45g construct, and the percentage of apoptosis cells in green fluorescent protein (GFP)-positive cells were determined by fluorescence-activated cell sorting (FACS) analysis of Annexin V and 7AAD staining. (F) Colony formation of primary BM CD34+ from patients with AML (n = 5) and CB CD34+ cells from healthy donors (n = 3) after lentivirally transduced with GADD45g or empty vector control. (G) Primary BMMNCs from patients with AML (n = 5) were transfected with GADD45g small hairpin RNA (shGG) or scramble small hairpin RNA (shCtrl), and the percentage of apoptosis cells in GFP-positive cells were determined by FACS analysis of Annexin V and 7AAD staining. (H) Effects of Dox-induced GADD45g expression on cell differentiation of THP-1 cells, as determined by flow cytometric analysis of side-scatter profiles (SSC) and the expression of CD11b and CD14, and representative May-Grünwald-Giemsa staining in THP-1 cells with or without Dox treatment. Original magnification, ×400. (I) Effects of GADD45g overexpression on cell differentiation of primary BM CD34+ from patients with AML (n = 3) and CB CD34+ cells from healthy donors (n = 3), as determined by flow cytometric analysis of the expression of CD11b and CD14. THP-1 cells with GADD45g overexpression (J) or knockdown (K) were exposed to the indicated concentrations of daunorubicin (DNR) or VP-16 for 24 hours, and the percentage of apoptosis cells was determined by FACS analysis of Annexin V and 7AAD staining. Data are presented as mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. *P < .05, **P < .01, ***P < .001. MFI, mean fluorescence intensity; NS, not significant.
Human AML cells are sensitive to GADD45g overexpression. (A) qRT-PCR evaluation of GADD45g expression in AML cells with or without Dox-induced GADD45g overexpression and representative western blot in THP-1 cells. Effects of Dox-induced GADD45g expression on cell proliferation (B), colony formation (C), and apoptosis (D) in THP-1 cells as determined by cell counting, colony-forming assay, and flow cytometric analysis of Annexin V and 7AAD staining, respectively. (E) Primary BMMNCs from patients with AML (n = 5) and cord blood mononuclear cells from healthy donors (n = 5) were transfected with human GADD45g construct, and the percentage of apoptosis cells in green fluorescent protein (GFP)-positive cells were determined by fluorescence-activated cell sorting (FACS) analysis of Annexin V and 7AAD staining. (F) Colony formation of primary BM CD34+ from patients with AML (n = 5) and CB CD34+ cells from healthy donors (n = 3) after lentivirally transduced with GADD45g or empty vector control. (G) Primary BMMNCs from patients with AML (n = 5) were transfected with GADD45g small hairpin RNA (shGG) or scramble small hairpin RNA (shCtrl), and the percentage of apoptosis cells in GFP-positive cells were determined by FACS analysis of Annexin V and 7AAD staining. (H) Effects of Dox-induced GADD45g expression on cell differentiation of THP-1 cells, as determined by flow cytometric analysis of side-scatter profiles (SSC) and the expression of CD11b and CD14, and representative May-Grünwald-Giemsa staining in THP-1 cells with or without Dox treatment. Original magnification, ×400. (I) Effects of GADD45g overexpression on cell differentiation of primary BM CD34+ from patients with AML (n = 3) and CB CD34+ cells from healthy donors (n = 3), as determined by flow cytometric analysis of the expression of CD11b and CD14. THP-1 cells with GADD45g overexpression (J) or knockdown (K) were exposed to the indicated concentrations of daunorubicin (DNR) or VP-16 for 24 hours, and the percentage of apoptosis cells was determined by FACS analysis of Annexin V and 7AAD staining. Data are presented as mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. *P < .05, **P < .01, ***P < .001. MFI, mean fluorescence intensity; NS, not significant.
To investigate whether GADD45g is involved in cell differentiation in AML, we first analyzed the gene expression pattern of GADD45g in 2 independent data sets of AML cohorts (GSE63270 and GSE24006) and found that GADD45g was expressed at lower levels in AML stem/progenitor cells and upregulated in differentiated cells (supplemental Figure 3A-B). We also detected GADD45g expression levels in human CB cells at a different stage of differentiation by qRT-PCR but did not observe a similar tendency (supplemental Figure 3C). This observation was confirmed by analysis of a public data set of human BM-derived hematopoietic cells (supplemental Figure 3D). We then assessed the effect of GADD45g overexpression on the differentiation of U937, HL60, THP1, and Molm13 cell lines. As expected, enforced expression of GADD45g induced the differentiation of AML cells, as shown by the increased expression of cell-surface differentiation markers such as CD11b, CD14, and moderately altered side-scatter profiles, with cells becoming larger and more granulated in morphology (Figure 2H; supplemental Figure 3E). The expression of CEBPA, CEBPE, PU.1, and SCL, genes involved in myeloid cell differentiation, was markedly upregulated (supplemental Figure 3F). Evaluation of the effect of GADD45g on differentiation of CD34+ cells from AML patients and normal human CB revealed that GADD45g upregulation induced myeloid differentiation in AML cells but had no significant effect on CB cells (Figure 2I). These observations suggest that enforced expression of GADD45g induces myeloid differentiation of AML.
We next performed cell viability assay to evaluate the effect of GADD45g overexpression on the sensitivity of AML cells to chemotherapeutic drugs. As shown in Figure 2J and supplemental Figure 4A, daunorubicin or VP-16 treatment significantly increased the rate of apoptosis in GADD45g-overexpressing AML cells, compared with that in vector-transduced cells. When GADD45g was knocked down by small hairpin RNAs, as expected, the opposite effects were observed (Figure 2K; supplemental Figure 4B).
GADD45g induces DDR defect in human AML cells
To further explore the undiscovered role of GADD45g in AML and elucidate the mechanism underlying the function of GADD45g, we performed RNA sequencing (RNA-seq) analysis on Molm-13 cells, which harbor FLT3-ITD as well as MLL-rearrangement, with or without GADD45g overexpression. Overall, GADD45g overexpression resulted in dynamic alterations in transcription, with 1802 genes upregulated and 2095 genes downregulated (>1.5-fold change; P < .01) (Figure 3A). Consistent with the aforementioned phenotype analysis and gene expression changes, Gene Ontology analysis showed that categories related to “regulation of cell proliferation,” “growth,” “G1/S transition of mitotic cell cycle,” “myeloid leukocyte differentiation,” and “regulation of apoptotic process” were highly enriched (Figure 3B).
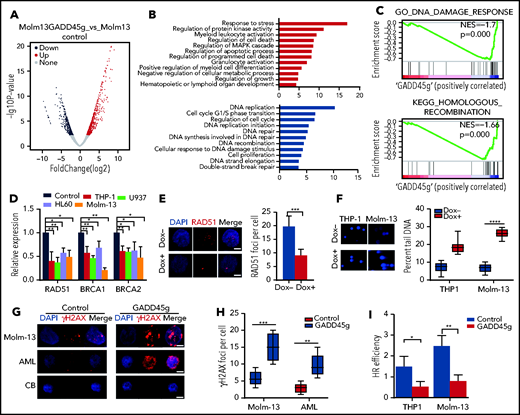
GADD45g overexpression represses HR DNA repair pathway. (A) Volcano plots of normalized gene expression in Molm-13 cells with or without Dox treatment as analyzed by RNA-seq. (B) Gene Ontology (GO) functional enrichment analysis of the RNA-seq data. Upregulated gene- enriched GO terms are represented by red, and downregulated gene-enriched GO terms are blue. (C) GSEA plot showing the enriched genes signature associated with HR in Molm-13 cells upon GADD45g overexpression. The normalized enrichment score (NES) and P values are shown. (D) qRT-PCR analysis of key HR genes including RAD51, BRCA1, and BRCA2 expression in THP-1, U937, HL60, and Molm-13 cells upon GADD45g overexpression. Data are presented as the fold change in gene expression relative to the negative control group, which is considered as 1. (E) Representative IF micrographs (left) of RAD51 foci in Molm-13 cells with or without Dox-induced GADD45g overexpression from 3 independent experiments, and quantification (right) of RAD51 foci (red) in nuclei (blue) per cell. Scale bar, 5 μm. (F) Representative micrographs (left) of the neutral comet assays for THP-1 and Molm-13 cells with or without Dox-induced GADD45g overexpression from 3 independent experiments, and quantification of tail-moments in comet assays (right). Representative IF micrographs showing γH2AX foci in Molm-13 cells, primary BMMNCs from patients with AML, and MNCs from human CB with or without GADD45g overexpression from 3 independent experiments (G), and quantification of the γH2AX foci per cell (H). Scale bar, 5 μm. (I) The efficiency of HR-mediated repair of I-SceI–induced DSB in THP-1 and Molm-13 cells transduced with pLV-GADD45g-mCherry (GADD45g) or lentiviral empty control vector (Control). Data are presented as the mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; NS, not significant.
GADD45g overexpression represses HR DNA repair pathway. (A) Volcano plots of normalized gene expression in Molm-13 cells with or without Dox treatment as analyzed by RNA-seq. (B) Gene Ontology (GO) functional enrichment analysis of the RNA-seq data. Upregulated gene- enriched GO terms are represented by red, and downregulated gene-enriched GO terms are blue. (C) GSEA plot showing the enriched genes signature associated with HR in Molm-13 cells upon GADD45g overexpression. The normalized enrichment score (NES) and P values are shown. (D) qRT-PCR analysis of key HR genes including RAD51, BRCA1, and BRCA2 expression in THP-1, U937, HL60, and Molm-13 cells upon GADD45g overexpression. Data are presented as the fold change in gene expression relative to the negative control group, which is considered as 1. (E) Representative IF micrographs (left) of RAD51 foci in Molm-13 cells with or without Dox-induced GADD45g overexpression from 3 independent experiments, and quantification (right) of RAD51 foci (red) in nuclei (blue) per cell. Scale bar, 5 μm. (F) Representative micrographs (left) of the neutral comet assays for THP-1 and Molm-13 cells with or without Dox-induced GADD45g overexpression from 3 independent experiments, and quantification of tail-moments in comet assays (right). Representative IF micrographs showing γH2AX foci in Molm-13 cells, primary BMMNCs from patients with AML, and MNCs from human CB with or without GADD45g overexpression from 3 independent experiments (G), and quantification of the γH2AX foci per cell (H). Scale bar, 5 μm. (I) The efficiency of HR-mediated repair of I-SceI–induced DSB in THP-1 and Molm-13 cells transduced with pLV-GADD45g-mCherry (GADD45g) or lentiviral empty control vector (Control). Data are presented as the mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; NS, not significant.
Noticeably, Gene Ontology and GSEA analysis also revealed that gene sets associated with DDR, especially HR, were significantly negatively enriched upon GADD45g overexpression (Figure 3C). qRT-PCR verified that the expression of genes involved in HR (RAD51, BRCA1, and BRCA2) was dramatically downregulated upon GADD45g overexpression (∼30%−80% reduction) (Figure 3D). Immunofluorescence (IF) assay also showed that GADD45g-overexpressing cells displayed significantly lower levels of RAD51 foci, indicative of defect in DSB repair pathways (Figure 3E; supplemental Figure 5A-B). To determine whether upregulation of GADD45g leads to accumulation of DNA damage, we performed a neutral comet assay and found that AML cells transduced with GADD45g exhibited significantly increased comet tail signal (Figure 3F). This increase in DSBs was also visualized as an elevated accumulation of Ser139 H2AX phosphorylation (γH2AX) by both IF and western blot assays (Figure 3G-H; supplemental Figure 5C-E). Next, to seek solid evidence that GADD45g contributes to impaired DNA repair activities, we performed an HR reporter assay and observed significant suppression of HR efficiency upon GADD45g upregulation (Figure 3I). Consistent with the results from the AML cell lines, primary AML cells also exhibited enhanced accumulation of γH2AX foci upon GADD45g overexpression, whereas GADD45g upregulation exerted a negligible effect on normal hematopoietic cells.
Together, these findings strongly suggest that enforced expression of GADD45g impairs HR DNA repair signaling and leads to accumulation of DNA damage, specifically in AML cells.
GADD45g inhibits leukemogenesis in xenograft model
To ascertain the antileukemic activity of GADD45g in vivo, we developed a xenograft model of human AML. Molm13-luc2 cells were transfected with Dox-inducible GADD45g lentiviral vector and engrafted into NOD/SCID mice by intravenous injection. The bioluminescence signal could be detected in vivo 7 days after transplantation. The mice were then treated with Dox for 1 week to induce GADD45g expression. The leukemia burden was significantly reduced, as determined by bioluminescence imaging (Figure 4A), and the survival time of these mice was remarkably prolonged compared with that of control mice (Figure 4B). Conversely, downregulation of GADD45g promoted AML growth and reduced the survival of tumor-bearing mice (Figure 4C-D). In contrast to the observation in the AML xenograft model, enforced GADD45g expression had no significant impact on the engraftment and differentiation (Figure 4E-G) of CB CD34+ cells in NOD/Shi-scid/IL2Rγnull (NOG) mice. These data suggest that GADD45g exerts a selective antileukemic effect in human AML cells in vivo.
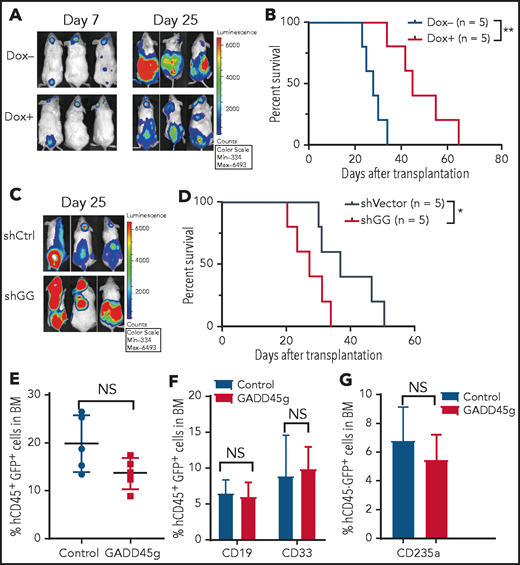
GADD45g inhibits progression of human AML cells in NOD/SCID mice. (A-B) Molm-13-luc2 cells were transfected with Dox-inducible GADD45g lentiviral vector and engrafted into NOD/SCID mice (8 × 105 cells per mouse). Dox 1 mg/mL was added to drinking water 7 days after transplantation for 1 week to induce GADD45g expression. (A) Bioluminescence imaging of representative mice from each group taken at day 7 and day 25 posttransplantation. (B) Kaplan-Meier survival curves are shown for 2 cohorts of transplanted mice. P values were determined by using the log-rank test (n = 5). NOD/SCID mice were inoculated via the tail vein with 5 × 105 Molm-13 cells with or without GADD45g knockdown. (C) Bioluminescence imaging of representative mice from each group taken at day 25 posttransplantation. (D) Kaplan-Meier survival curves are shown for 2 cohorts of transplanted mice. P values were determined by using the log-rank test (n = 5). (E) NOG mice were transplanted with 1 × 106 human CB CD34+ cells transduced with lentiviruses containing GADD45g or control vector. Percentages of hCD45+GFP+ cells in the BM of NOG mice at 16 weeks’ posttransplantation. Data are represented as the mean ± SD; n = 5 mice per group. (F) The percentage of CD19+ and CD33+ cells within the GFP+CD45+ population in the BM of NOG-recipient mice at 16 weeks’ posttransplantation. (G) The percentage of CD235a+ cells within the GFP+CD45– population in the BM of NOG-recipient mice at 16 weeks’ posttransplantation. Data are presented as the mean ± SD, and were analyzed by using the 2-tailed Student t test. *P < .05, **P < .01. NS, not significant; shCtrl, scramble small hairpin RNA; shGG, GADD45g small hairpin RNA.
GADD45g inhibits progression of human AML cells in NOD/SCID mice. (A-B) Molm-13-luc2 cells were transfected with Dox-inducible GADD45g lentiviral vector and engrafted into NOD/SCID mice (8 × 105 cells per mouse). Dox 1 mg/mL was added to drinking water 7 days after transplantation for 1 week to induce GADD45g expression. (A) Bioluminescence imaging of representative mice from each group taken at day 7 and day 25 posttransplantation. (B) Kaplan-Meier survival curves are shown for 2 cohorts of transplanted mice. P values were determined by using the log-rank test (n = 5). NOD/SCID mice were inoculated via the tail vein with 5 × 105 Molm-13 cells with or without GADD45g knockdown. (C) Bioluminescence imaging of representative mice from each group taken at day 25 posttransplantation. (D) Kaplan-Meier survival curves are shown for 2 cohorts of transplanted mice. P values were determined by using the log-rank test (n = 5). (E) NOG mice were transplanted with 1 × 106 human CB CD34+ cells transduced with lentiviruses containing GADD45g or control vector. Percentages of hCD45+GFP+ cells in the BM of NOG mice at 16 weeks’ posttransplantation. Data are represented as the mean ± SD; n = 5 mice per group. (F) The percentage of CD19+ and CD33+ cells within the GFP+CD45+ population in the BM of NOG-recipient mice at 16 weeks’ posttransplantation. (G) The percentage of CD235a+ cells within the GFP+CD45– population in the BM of NOG-recipient mice at 16 weeks’ posttransplantation. Data are presented as the mean ± SD, and were analyzed by using the 2-tailed Student t test. *P < .05, **P < .01. NS, not significant; shCtrl, scramble small hairpin RNA; shGG, GADD45g small hairpin RNA.
GADD45g exerts its antileukemic activities through inhibition of E2F1 via the p38 MAPK–dependent signaling pathway
To understand the molecular mechanism of the antileukemic activities of GADD45g, we further analyzed our RNA-seq data and found significantly positive enrichment in the p38 MAPK–signaling pathway and negative enrichment in the E2F1 signature (Figure 5A). E2F1, a well-known transcription factor, reportedly promotes cell proliferation, inhibits apoptosis and differentiation, and facilitates DNA repair by upregulating the expression of HR-associated genes.29-31 These roles seem to be just the opposite to GADD45g. We detected mRNA expression of both E2F1 and GADD45g by qRT-PCR in BMMNCs from patients with AML (n = 56) and found that the expression of E2F1 exhibited a significant negative correlation with GADD45g (r = –0.561; P < .001) (Figure 5B), suggesting that E2F1 may be a downstream target of GADD45g in AML cells. Numerous reports have revealed that GADD45g functions through activation of p38 MAPK.32-34 Intriguingly, it has been reported that p38 MAPK activation leads to E2F1 downregulation in solid tumor cells and differentiating epidermal keratinocytes.35,36 These clues tempted us to speculate that the enforced expression of GADD45g may lead to activation of p38 MAPK, which in turn represses the expression of E2F1 in AML cells.
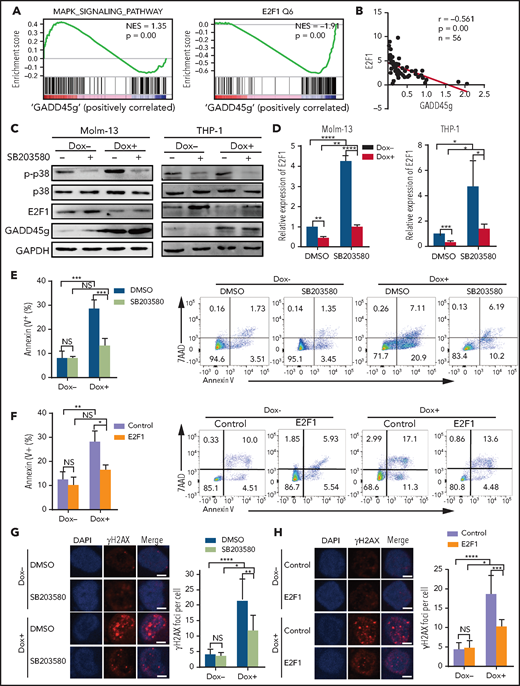
The antileukemic activities of GADD45g are mediated through inhibition of E2F1 via the p38 MAPK–dependent signaling pathway. (A) Significantly enriched GSEA signatures in the transcriptional profile of Molm-13 cells upon GADD45g overexpression. The normalized enrichment score (NES) and P values are shown. (B) Correlation of expression between GADD45g and E2F1 in BMMNCs from AML samples (n = 56). Correlation coefficient and P value of Spearman correlation test are shown. (C) Molm-13 cells and (left) and THP-1 cells (right) with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 for 16 hours and then subjected to western blot to detect the indicated proteins. (D) Molm-13 cells and THP-1 cells with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 for 16 hours. Relative mRNA expression of E2F1 was quantified by qRT-PCR. Molm-13 cells with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 (E) or transduced with lentiviral vectors expressing E2F1 (F), and the percentage of apoptosis cells was determined by fluorescence-activated cell sorting (FACS) analysis of Annexin V and 7AAD staining. Molm-13 cells with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 (G) or transduced with lentiviral vectors expressing E2F1 (H). Representative IF micrographs (left) showing γH2AX foci from 3 independent experiments and quantification (right) of γH2AX foci (red) in nuclei (blue) per cell. Scale bar, 5 μm. Data are presented as the mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. Correlations between continuous variables were calculated by using the Pearson correlation. *P < .05, **P < .01, ***P < .001, ****P < .0001. DMSO, dimethyl sulfoxide; NS, not significant.
The antileukemic activities of GADD45g are mediated through inhibition of E2F1 via the p38 MAPK–dependent signaling pathway. (A) Significantly enriched GSEA signatures in the transcriptional profile of Molm-13 cells upon GADD45g overexpression. The normalized enrichment score (NES) and P values are shown. (B) Correlation of expression between GADD45g and E2F1 in BMMNCs from AML samples (n = 56). Correlation coefficient and P value of Spearman correlation test are shown. (C) Molm-13 cells and (left) and THP-1 cells (right) with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 for 16 hours and then subjected to western blot to detect the indicated proteins. (D) Molm-13 cells and THP-1 cells with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 for 16 hours. Relative mRNA expression of E2F1 was quantified by qRT-PCR. Molm-13 cells with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 (E) or transduced with lentiviral vectors expressing E2F1 (F), and the percentage of apoptosis cells was determined by fluorescence-activated cell sorting (FACS) analysis of Annexin V and 7AAD staining. Molm-13 cells with or without Dox-induced GADD45g overexpression were treated with 1 μM SB203580 (G) or transduced with lentiviral vectors expressing E2F1 (H). Representative IF micrographs (left) showing γH2AX foci from 3 independent experiments and quantification (right) of γH2AX foci (red) in nuclei (blue) per cell. Scale bar, 5 μm. Data are presented as the mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. Correlations between continuous variables were calculated by using the Pearson correlation. *P < .05, **P < .01, ***P < .001, ****P < .0001. DMSO, dimethyl sulfoxide; NS, not significant.
To validate the hypothesis, we transduced Molm-13 and THP1 cells with Dox-inducible GADD45g lentiviral vector. As expected, GADD45g upregulation led to a remarkable increase in p38 MAPK activity and a significant reduction in E2F1 expression (Figure 5C-D) and corresponding alterations in the mRNA levels of E2F1 downstream target genes, including RAD51, BRCA1, BRCA2, TRIB2, and Myc (Figure 3D; supplemental Figure 2E). We then treated GADD45g-overexpressing AML cells with the p38 MAPK inhibitor SB203580, and found that SB203580 diminished the inhibitory effects of GADD45g on E2F1 and significantly induced E2F1 expression in Molm-13 and THP1 cells, suggesting that GADD45g-induced downregulation of E2F1 is mediated by the activation of p38 MAPK. Rescue experiments were performed to determine whether both p38 MAPK activation and inhibition of E2F1 are required for the antileukemic activities of GADD45g upregulation. The results showed that SB203580 markedly attenuated the cell apoptosis and DNA damage induced by GADD45g overexpression in Molm-13 and THP1 cells (Figure 5E,G; supplemental Figure 6A,C). Similar results were observed when the cells were transduced with E2F1 (Figure 5F,H; supplemental Figure 6B,D). These results suggest that GADD45g exerts an antileukemic effect via p38 MAPK–mediated E2F1 inhibition.
Silencing of GADD45g in AML is associated with epigenetic regulation and relevant leukemic oncogenes, and NF-κB mediates the activation of GADD45g by JQ1 and AC220
The finding that GADD45g upregulation exerted potent antileukemic activities in AML prompted us to explore the mechanisms underlying its silencing, which could help find effective GADD45g activating agents. Reduced expression of GADD45g gene in solid tumors is commonly associated with promoter hypermethylation or histone deacetylation.37,38 Thus, we first analyzed a genome-wide DNA methylation profiling of primary AML samples treated with the low-dose (100 nM) DNA demethylating agent decitabine (DAC) (GSE40871). The results revealed that GADD45g expression was modestly induced and the DNA methylation level was decreased in the second cytosine guanine dinucleotide island 200 bp upstream of the transcriptional start site of GADD45g (supplemental Figure 7A-B). Our methylation-specific PCR experiment also showed that methylated GADD45g alleles were remarkably decreased in AML cell lines after treatment with DAC (supplemental Figure 7C). The mRNA level of GADD45g was upregulated by fourfold in AML cell lines and by 1.85- to 4.03-fold in BMMNCs from AML patients, respectively, when treated with DAC (2 μM) for 96 hours (supplemental Figure 7D-E).
To address the contribution of histone modifications in GADD45g silencing in AML, we first treated AML cell lines and BMMNCs from patients with AML with suberoylanilide hydroxamic acid (SAHA), a pan-histone deacetylase (HDAC) inhibitor. The mRNA level of GADD45g was significantly increased following SAHA treatment (1 μM) for 48 hours (AML lines, fourfold to 20-fold; BMMNCs, threefold to eightfold) (supplemental Figure 7F-G). Notably, in 2 AML cohorts (GSE12417 and GSE10358), GADD45g exhibited a significant inverse correlation with HDAC1 and HDAC2 expression (supplemental Figure 7H), raising the possibility that selective inhibition of HDAC1,2 could be more efficacious in upregulating GADD45g expression. We screened various HDAC1, HDAC2, and HDAC1,2 inhibitors and compared their inductive effects on GADD45g expression with SAHA (data not shown), and found that the selective HDAC1,2 inhibitor romidepsin exhibited the strongest induction of GADD45g in AML cell lines and BMMNCs from patients with AML even at a low dose of 5 nM (AML lines, 8.8- to 90-fold; BMMNCs, fourfold to 16-fold) (Figure 6A-B). It has been reported that GADD45g transcriptional activation was associated with acetylation of histones H3 and H4 at the promoter.39 We therefore assessed the effect of romidepsin on the histone acetylation levels of GADD45g by chromatin immunoprecipitation (ChIP)-qPCR assay and observed increased levels of histones H3 and H4 acetylation at the GADD45g promoter (Figure 6C-D). These data suggest that GADD45g is silenced in AML by both promoter DNA methylation and histone deacetylation, and the HDAC1/2 inhibitor romidepsin is a powerful inducer of GADD45g.
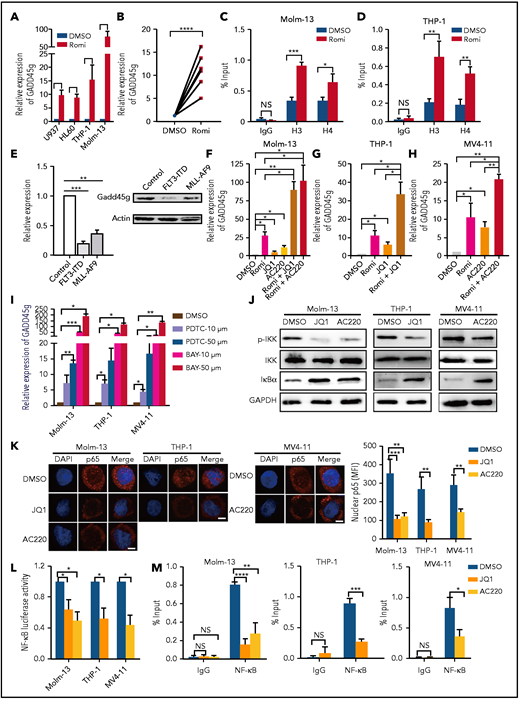
Epigenetic regulation and relevant leukemic oncogenes repress GADD45g expression in AML, and NF-κB mediates the activation of GADD45g by oncogene inhibitors JQ1 and AC220. qRT-PCR analysis of GADD45g expression in AML cell lines (A) or in BMMNCs from patients with AML (B) (n = 6) treated with 5 nM romidepsin (Romi) for 48 hours. Molm-13 cells (C) and THP-1 cells (D) were treated with or without 3 nM Romi for 48 hours. The cells were subjected to ChIP analysis by using antibodies against acetyl-histone H3 (Lys9) and anti–acetyl-histone H4 (Lys8). The enriched DNA that associated with the promoter region of GADD45g was quantified by using qPCR. (E) qRT-PCR and western blot analysis of Gadd45g expression in 32D cells transduced with FLT3-ITD, MLL-AF9, or control constructs. qRT-PCR analysis of GADD45g expression in Molm-13 cells (F), THP-1 cells (G), and MV4-11 cells (H) treated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combinations as indicated for 48 hours. (I) qRT-PCR analysis of GADD45g expression in AML cell lines treated with NF-κB inhibitors PDTC or BAY 11-70825 for 24 hours at the doses indicated. (J) The AML cell lines were treated with dimethyl sulfoxide (DMSO), JQ1 (100 nM), or AC220 (10 nM) for 24 hours and then subjected to western blot to detect the indicated proteins. (K) The AML cell lines were treated with DMSO, JQ1 (100 nM), or AC220 (10 nM) for 24 hours. Representative immunofluorescence (left) showing the localization of NF-κB p65 from 3 independent experiments, and quantification (right) of the mean fluorescence intensity in nuclear. Scale bars, 5 μm. (L) Relative activity of NF-κB–dependent luciferase reporter in the indicated AML lines treated with either DMSO, JQ1 (100 nM), or AC220 (10 nM) for 24 hours. (M) The AML cell lines were treated with DMSO, JQ1 (100 nM), or AC220 (10 nM) for 24 hours. The cells were subjected to ChIP analysis using antibodies against NF-κB p65. The enriched DNA that associated with the promoter region of GADD45g was quantified by using qPCR. Data are presented as the mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. Multiple groups were analyzed with the 1-way analysis of variance. *P < .05, **P < .01, ***P < .001, ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; p-IKK, phosphorylated IKK; NS, not significant; shCtrl, scramble small hairpin RNA; shGG, GADD45g small hairpin RNA.
Epigenetic regulation and relevant leukemic oncogenes repress GADD45g expression in AML, and NF-κB mediates the activation of GADD45g by oncogene inhibitors JQ1 and AC220. qRT-PCR analysis of GADD45g expression in AML cell lines (A) or in BMMNCs from patients with AML (B) (n = 6) treated with 5 nM romidepsin (Romi) for 48 hours. Molm-13 cells (C) and THP-1 cells (D) were treated with or without 3 nM Romi for 48 hours. The cells were subjected to ChIP analysis by using antibodies against acetyl-histone H3 (Lys9) and anti–acetyl-histone H4 (Lys8). The enriched DNA that associated with the promoter region of GADD45g was quantified by using qPCR. (E) qRT-PCR and western blot analysis of Gadd45g expression in 32D cells transduced with FLT3-ITD, MLL-AF9, or control constructs. qRT-PCR analysis of GADD45g expression in Molm-13 cells (F), THP-1 cells (G), and MV4-11 cells (H) treated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combinations as indicated for 48 hours. (I) qRT-PCR analysis of GADD45g expression in AML cell lines treated with NF-κB inhibitors PDTC or BAY 11-70825 for 24 hours at the doses indicated. (J) The AML cell lines were treated with dimethyl sulfoxide (DMSO), JQ1 (100 nM), or AC220 (10 nM) for 24 hours and then subjected to western blot to detect the indicated proteins. (K) The AML cell lines were treated with DMSO, JQ1 (100 nM), or AC220 (10 nM) for 24 hours. Representative immunofluorescence (left) showing the localization of NF-κB p65 from 3 independent experiments, and quantification (right) of the mean fluorescence intensity in nuclear. Scale bars, 5 μm. (L) Relative activity of NF-κB–dependent luciferase reporter in the indicated AML lines treated with either DMSO, JQ1 (100 nM), or AC220 (10 nM) for 24 hours. (M) The AML cell lines were treated with DMSO, JQ1 (100 nM), or AC220 (10 nM) for 24 hours. The cells were subjected to ChIP analysis using antibodies against NF-κB p65. The enriched DNA that associated with the promoter region of GADD45g was quantified by using qPCR. Data are presented as the mean ± SD of ≥3 independent experiments, and comparisons were evaluated by using the 2-tailed Student t test. Multiple groups were analyzed with the 1-way analysis of variance. *P < .05, **P < .01, ***P < .001, ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; p-IKK, phosphorylated IKK; NS, not significant; shCtrl, scramble small hairpin RNA; shGG, GADD45g small hairpin RNA.
Given that the expression level of GADD45g was particularly low in AMLs with MLL-rearrangements or FLT3-ITD mutations, we presumed that this expression pattern may be related to these oncogenes. We analyzed the published data sets and found that GADD45g exhibited a significant inverse correlation with FLT3 expression in 2 independent AML cohorts (supplemental Figure 7I). In addition, GADD45g expression is significantly increased when MLL-AF9 is inhibited in the MLL-AF9-Tet-off AML data set (supplemental Figure 7J). These findings led us to hypothesize that FLT3-ITD and MLL-AF9 mediate the downregulation of GADD45g. To validate this theory, we transduced the murine hematopoietic cell line 32D cells with MLL-AF9 or FLT3-ITD constructs and found that Gadd45g expression was significantly repressed by these 2 oncogenes to 20% to 35% at both mRNA and protein levels, compared with empty vector controls (Figure 6E). The observations that MLL-AF9 and FLT3-ITD repressed the expression of GADD45g suggest the possibility that inhibition of these oncogenes might lead to GADD45g activation. We first evaluated the current pharmacologic inhibitors targeting proteins critical for the development of MLL-AF9 leukemia. We found that among inhibitors for DOT1L, BRD4, β-catenin, and MENIN, only the BRD4 inhibitor JQ1 treatment could increase the expression of GADD45g in MLL-AF9+ AML cell lines (THP-1 and Molm-13) (Figure 6F-G), consistent with the published data GSE29799 (supplemental Figure 7K). As for FLT3-ITD, we found that the FLT3 inhibitor quizartinib (AC220) induced a significant increase in GADD45g expression in FLT3-ITD+ cells (MV4-11 and Molm-13) (Figure 6F,H). These results indicate that epigenetic regulation, as well as MLL-AF9 and FLT3-ITD oncogenes, contribute to the lower expression of GADD45g in patients with AML that have these abnormalities, and romidepsin, JQ1, and AC220 effectively activate GADD45g.
To elucidate the molecular mechanisms of the activation of GADD45g by JQ1 and AC220, we analyzed the published data sets of GSE29799 and GSE126933, in which AML cell lines were treated with JQ1 or AC220, respectively. The differentially expressed genes after JQ1 or AC220 treatment were analyzed by using the Metascape database (metascape.org) to identify the key transcription factors. Intriguingly, NF-κB is among the top 5 potential transcription factors predicted by both of the data sets (supplemental Figure 8), and it is the only one that has been reported to bind directly to GADD45g promoter and repress GADD45g transcription.40,41 In addition, previous studies have reported that JQ1 inhibits IκB kinase (IKK) activity, leading to accumulation of IκBα and reduced nuclear translocation of NF-κB in lymphoma cells and synoviocytes.42-44 We therefore speculated that JQ1 and AC220 activate GADD45g by inhibiting NF-κB–driven transcriptional repression of GADD45g. To verify it, we first treated THP-1, MV4-11, and Molm-13 cells with NF-κB inhibitors PDTC and BAY 11-7082 and observed that both inhibitors markedly unregulated GADD45g expression in a dose-dependent manner, suggesting that NF-κB represses GADD45g expression in AML cells (Figure 6I). We then treated MLL-AF9+ and FLT3-ITD+ AML cell lines with JQ1 and AC220, respectively, and found that the phosphorylated IKKβ was strongly decreased in these cells (Figure 6J), indicating inhibition of IKK activity. Consistent with decreased IKK signaling, accumulation of IκBα was observed in all 3 cell lines. As a consequence, immunofluorescence microscopy revealed a reduced nuclear translocation of NF-κB p65 (Figure 6K). Significantly decreased NF-κB activity was further confirmed by NF-κB–dependent luciferase reporter assay (Figure 6L). Moreover, ChIP assays showed that JQ1 and AC220 reduced the DNA-binding activity of NF-κB to the promoter of GADD45g (Figure 6M). Together, these results indicate that JQ1 and AC220 activate GADD45g by relieving the NF-κB–mediated transcriptional repression of GADD45g.
Combination of romidepsin with JQ1 or AC220 show synergistic antileukemic effects on MLL-AF9+ and FLT3-ITD+ AML, respectively
The findings that romidepsin, JQ1, and AC220 could effectively induce GADD45g expression led us to presume that combining romidepsin with JQ1 or AC220 may lead to greatly enhanced activation of GADD45g and exhibit potent antileukemic activities against MLL-AF9+ and FLT3-ITD+ AML, respectively. As expected, combination of romidepsin and JQ1 or AC220 dramatically increased GADD45g expression (Figure 6F-H) and resulted in synergistic induction of apoptosis in MLL-AF9+ and FLT3-ITD+ AML cell lines (Figure 7A-B). The combination index values were calculated as between 0.57 and 0.84 by using CompuSyn software (supplemental Figure 9A). In addition, the combination regimens also led to enhanced accumulation of γH2AX, as measured by both IF and western blot assays (Figure 7C-D), and a further reduction in expression of the key HR factors BRCA1, BRCA2, and RAD51 (supplemental Figure 9B). The synergistic antileukemic effects were confirmed in primary cells from patients with AML, whereas no significant toxic effect on normal cells could be observed (supplemental Figure 9C-D). In addition, GADD45g knockdown in MLL-AF9+ and FLT3-ITD+ AML cell lines significantly alleviated the cell apoptosis upon drug treatments, showing the contribution of GADD45g in these antileukemic activities (Figure 7E). To evaluate the therapeutic potential of these treatments in vivo, Molm13-luc2 were engrafted into NOD/SCID mice by intravenous inoculation followed by vehicle control or drug treatment. Bioluminescence imaging of luciferase revealed a significant delay in AML progression, which was associated with survival benefit in the romidepsin- and JQ1- or AC220-treated cohorts (Figure 7F-G).
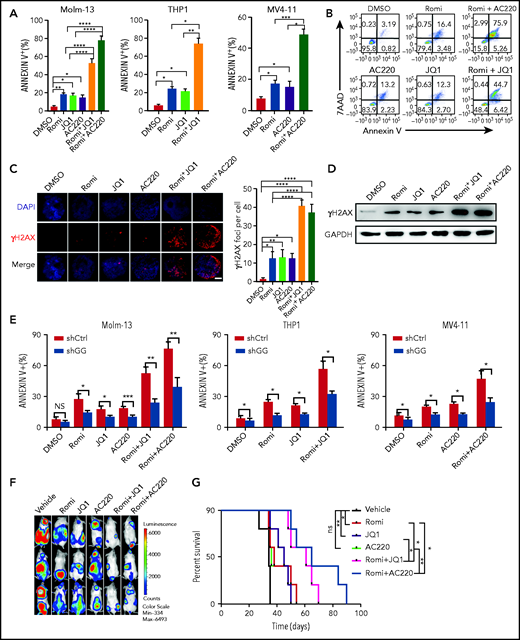
Combination of romidepsin (Romi) with AC220 or JQ1 exert synergistic antileukemic effects on FLT3-ITD+ and MLL-AF9+ AML, respectively. (A-B) Molm-13, THP-1, and MV4-11 cells were incubated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combination as indicated for 48 hours, and apoptosis was determined by flow cytometric analysis of Annexin V/7-AAD. Percentage of annexin V+ cells (A) and the representative fluorescence-activated cell sorting (FACS) plots of Molm-13 cells (B). (C) Representative IF micrographs (left) from 3 independent experiments showing γH2AX foci in Molm-13 cells treated with Romi (3 nM), AC220 (10 nM), JQ1(100 nM), or combination as indicated for 48 hours, and quantification (right) of γH2AX foci (red) in nuclei (blue) per cell. Scale bar, 5 μm. (D) Western blot analysis of γH2AX levels in Molm-13 cells treated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combination as indicated for 48 hours. (E) The AML cell lines with or without GADD45g knockdown were incubated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combination as indicated for 48 hours; the percentage of apoptosis cells was determined by flow cytometric analysis of Annexin V and 7AAD staining. (F-G) Molm13-luc2 cells were injected intravenously into sublethally irradiated NOD/SCID mice (8 × 105 cells per mouse). Five days later, mice were treated with vehicle or Romi (1.5 mg/kg), AC220 (10 mg/kg), JQ1 (50 mg/kg), or combination as indicated for 2 weeks. (F) Bioluminescence imaging of representative mice from each group taken at day 25 posttransplantation. (G) Survival curve of each group mice. P values were determined by using the log-rank test (n = 5). Data are presented as the mean ± SD of ≥3 independent experiments. Comparisons were evaluated by using the 2-tailed Student t test, and multiple groups were analyzed with the 1-way analysis of variance. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, not significant.
Combination of romidepsin (Romi) with AC220 or JQ1 exert synergistic antileukemic effects on FLT3-ITD+ and MLL-AF9+ AML, respectively. (A-B) Molm-13, THP-1, and MV4-11 cells were incubated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combination as indicated for 48 hours, and apoptosis was determined by flow cytometric analysis of Annexin V/7-AAD. Percentage of annexin V+ cells (A) and the representative fluorescence-activated cell sorting (FACS) plots of Molm-13 cells (B). (C) Representative IF micrographs (left) from 3 independent experiments showing γH2AX foci in Molm-13 cells treated with Romi (3 nM), AC220 (10 nM), JQ1(100 nM), or combination as indicated for 48 hours, and quantification (right) of γH2AX foci (red) in nuclei (blue) per cell. Scale bar, 5 μm. (D) Western blot analysis of γH2AX levels in Molm-13 cells treated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combination as indicated for 48 hours. (E) The AML cell lines with or without GADD45g knockdown were incubated with Romi (3 nM), AC220 (10 nM), JQ1 (100 nM), or combination as indicated for 48 hours; the percentage of apoptosis cells was determined by flow cytometric analysis of Annexin V and 7AAD staining. (F-G) Molm13-luc2 cells were injected intravenously into sublethally irradiated NOD/SCID mice (8 × 105 cells per mouse). Five days later, mice were treated with vehicle or Romi (1.5 mg/kg), AC220 (10 mg/kg), JQ1 (50 mg/kg), or combination as indicated for 2 weeks. (F) Bioluminescence imaging of representative mice from each group taken at day 25 posttransplantation. (G) Survival curve of each group mice. P values were determined by using the log-rank test (n = 5). Data are presented as the mean ± SD of ≥3 independent experiments. Comparisons were evaluated by using the 2-tailed Student t test, and multiple groups were analyzed with the 1-way analysis of variance. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, not significant.
Together, these results suggest that combination of the HDAC1/2 inhibitor romidepsin with AC220 or JQ1 may provide novel, effective, and selective therapeutic strategies for FLT3-ITD+ and MLL-AF9+ AML, respectively, by dual activation of GADD45g.
Discussion
GADD45g was generally considered as a DNA damage responsive gene in previous studies16,17,45 ; however, its precise role in DDR remains elusive. Here, of importance, our data show that upregulation of GADD45g inhibits DNA repair by suppressing the expression of key HR-associated genes in AML cells. Intriguingly, analysis of the AML gene expression data set revealed that GADD45g expression is higher in AML patients with AML1-ETO and PML-RARα translocations than those with MLL-rearrangements (Figure 1G), and Esposito et al14 reported that these 2 patient subsets exhibit suppressed expression of key HR genes and inherent DDR defects relative to those with MLL-rearrangements. Therefore, in patients with AML, high expression of GADD45g seems to be related to HR deficiency, consistent with our findings.
Malignant and normal cells are distinctly different in many aspects. Several studies indicate that AML cells exhibit higher levels of DNA lesions and genomic instability compared with normal hematopoietic cells, but they survive due to enhanced or altered activity of DNA repair systems.13 ,46-48 When the activity of the DNA repair system is inhibited, the DNA lesions become overwhelming and then surpass the ability of leukemia cells to repair, triggering the apoptosis pathway and eventually leading to cell death.49 Thus, the defects in DNA damage repair mechanisms caused by vigorous activation of GADD45g may account for its specific apoptosis induction effects in AML cells. Besides apoptosis, we also observed a selective induction of myeloid differentiation in leukemia cells. However, Thalheimer et al34 reported that GADD45g accelerates differentiation of murine hematopoietic stem cells. A possible explanation for this discrepancy may be that GADD45g plays different roles in human and mouse normal hematopoiesis. Analysis of gene expression profile of normal mice hematopoietic systems in the bloodspot database reveals that the expression of GADD45g increases with differentiation (supplemental Figure 10). However, analysis of GADD45g expression in human CB and BM cells did not reveal a similar tendency, providing indirect evidence for this explanation. In addition, the study by Santos et al50 found that excessive DNA damage induces myeloid differentiation and growth inhibition of leukemic blasts, which may account for the roles of GADD45g in promoting differentiation and suppressing proliferation in AML cells.
As a proof of principle, inhibition of poly (adenosine diphosphate [ADP]–ribose) polymerase (PARP) has been successfully applied for the treatment of BRCA1- and BRCA2-deficient breast, ovarian, and prostate tumors by targeting DNA repair mechanisms.51,52 A PARP inhibitor (PARPi) increases the frequency of DSBs and consequently the need for HR-dependent DSB repair, which is compromised in cancer cells carrying BRCA1/2 mutations, leading to their unique susceptibility to PARPi treatment.53,54 Intriguingly, although inactivating mutations of BRCA1/2 or other driver genetic mutations directly affecting DNA repair genes are infrequent in leukemias, the results of preclinical and clinical studies indicate the potential efficacy of PARPi treatment in selectively killing AML cells, suggesting a strong rationale for the use of targeting DNA repair mechanisms as a therapeutic approach in the treatment of AML.46 ,55-57 This strategy shows significant clinical benefit for patients compared with classical cytotoxic chemotherapy because of its selectivity against tumor cells. Nevertheless, adverse events to current PARPi treatment have been observed, and the presence of MLL-AF9 and FLT3-ITD in AML confers resistance to the PARPi, implying the necessity of new drug evaluation.14,58,59 Of note, our data indicate that GADD45g upregulation exerts potent antileukemic effects regardless of subtypes. Even in refractory AML carrying FLT3-ITD+ and MLL-AF9+, GADD45g upregulation could effectively eliminate leukemia cells. Thus, once activated, GADD45g may have a broader application in AML.
Epigenetic mechanisms are known to play an important role in silencing of tumor suppressor genes. After screening and comparing various HDAC inhibitors and DNA methyltransferase inhibitors, we identified the HDAC1,2 inhibitor romidepsin as the most efficacious activator of GADD45g. Interestingly, it has been reported that romidepsin treatment results in an increase in the expression of genes involved in apoptosis pathways and a decrease in DNA repair pathways,60 in concordance with the alterations upon GADD45g activation. In addition, we observed that knockdown of GADD45g significantly restored the romidepsin-induced apoptotic phenotype, suggesting that GADD45g may be an important target of this HDAC inhibitor. Romidepsin has received approval from the US Food and Drug Administration for the treatment of relapsed or refractory T-cell lymphoma.61 However, there are limited studies for its therapeutic potential for AML. Yan et al62 reported that romidepsin preferentially targets chemoresistant CD123+ AML cells and has a synergistic effect with all-trans retinoic acid in acute promyelocytic leukemia. Here, we show for the first time that romidepsin induces apoptosis in all 7 AML cell lines tested, suggesting that it has a broad antileukemic activity in AML (Figure 7A; supplemental Figure 9E).
The findings that GADD45g can also be upregulated by AC220 and JQ1 prompted us to evaluate the combined therapeutic efficacy of romidepsin with AC220 or JQ1 for FLT3-ITD+ and MLL-AF9+ AML, respectively. Our results show, for the first time, that the combination of romidepsin and AC220 or JQ1 exerts synergistic antileukemic activities against these subtypes of AML, both in vitro and in vivo. The efficacy of the pan-HDAC inhibitor panobinostat in combination with AC220 or JQ1 for the treatment of AML has been reported,63,64 although concerns have emerged regarding the severe adverse side effects of pan-HDAC inhibitors.65,66 Here, we show that romidepsin induces remarkable apoptosis in AML cells with an effective dose of 3 nM, which is 100 times less than SAHA (0.5 μM), suggesting that romidepsin monotherapy or combination therapy may be safer and more effective for the treatment of AML.
In summary, our studies identify GADD45g as a novel tumor suppressor in AML and show that its upregulation exerts selective and potent antileukemic effects. In addition, we provide novel therapeutic strategies for the treatment of FLT3-ITD+ and MLL-AF9+ AML by combination administration of romidepsin with JQ1 or AC220. Because GADD45g has been found to be silenced in a variety of cancer types, our findings may have important clinical implications for cancer therapy.
Acknowledgments
This work was supported by grants from The National Key Research and Development Program of China (2016YFA0100600 and 2020YFE0203000), the National Natural Science Foundation of China (82070113, 81890990, 81730006, and 81861148029), and the CAMS Innovation Fund for Medical Sciences (2016-I2M-1-017).
Authorship
Contribution: X.M., D.G., and Y.Z. designed the project and wrote the manuscript; D.G. and Y.Z. organized the experiments and performed statistical analysis of the data; D.G., Y.Z., N.Y., J.Y., P.Z., and W.Z. performed experiments and analyzed data; Q.R. provided reagents and materials; X.M. and T.C. supervised the experiments and analysis; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiaotong Ma, State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Institute of Hematology & Blood Diseases Hospital, 288 Nanjing Rd, Tianjin 300020, China; e-mail: maxt@ihcams.ac.cn; or Tao Cheng, State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Institute of Hematology & Blood Diseases Hospital, 288 Nanjing Rd, Tianjin 300020, China; e-mail: chengtao@ihcams.ac.cn.
The RNA-seq data reported in this article have been deposited in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo; accession number GSE152932).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.