Abstract
Epigenetic deregulation is now a well-recognized although not yet fully understood mechanism that contributes to the development and progression of myeloid malignancies. In the past 15 years, next-generation sequencing studies have revealed patterns of aberrant DNA methylation, altered chromatin states, and mutations in chromatin modifiers across the spectrum of myeloid malignancies. Studies into the mechanisms that drive these diseases through mouse modeling have helped identify new avenues for therapeutic interventions, from initial treatment to resistant or relapsed disease. This is particularly significant when chemotherapy with cytotoxic agents remains the general standard of care. In this review, we will discuss some of the recent findings of epigenetic mechanisms and how these are informing the development of more targeted strategies for therapeutic intervention in myeloid malignancies.
Introduction
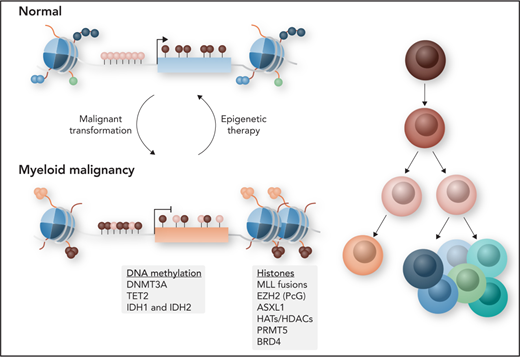
Spatiotemporal regulation of gene expression is controlled by chromatin status and regulatory components of the epigenome, which primarily consists of DNA methylation and histone modifications at the nucleosomal level. The combination of DNA methylation and histone modifications, which are re versible, promotes or inhibits transcriptional activity without changing the underlying DNA sequence, and disruption of these processes can lead to cancer and other disorders (Figure 1). Compared with cancers in other tissue types, myeloid malignancies have a relatively low frequency of somatic mutations. By contrast, epigenetic deregulation is now understood to be an important contributing mechanism to the development of myeloid malignancies. Within this landscape of an overall paucity of mutations, genes that regulate DNA methylation, histone modifications, and chromatin conformation are found to be among the most frequently mutated in hematologic malignancies.1 In normal hematopoiesis, these different layers of epigenetic information have been shown to be important for regulating transcriptional programs that instruct hematopoietic stem cell (HSC) self-renewal and lineage differentiation by regulating hematopoietic transcription factor access to regulatory elements such as enhancers and promoters.2 Any kind of imbalance can lead to abnormal hematopoiesis and the development of hematologic malignancies.

Schematic of regulatory components of the epigenome. The epigenome primarily consists of DNA methylation and histone modifications at the nucleosomal level. Most cytosine guanine dinucleotides (CpGs) in the mammalian genome are methylated in the form of 5-methylcytosine (5mC), except for those in CpG islands (CGIs), commonly found near promoter regions. The methylation is catalyzed by DNA methyltransferases (DNMTs). CpG shores, located within 2 kb of CGIs, as well as CpG canyons, which are large conserved regions of hypomethylation, can also be differentially methylated. Active removal of 5mC is catalyzed by the ten-eleven translocation (TET) family of dioxygenases through oxidation of 5mC to 5-hydroxymethylcytosine (5hmC). Isocitrate dehydrogenase 1 (IDH1) and IDH2 catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG), a metabolite required for TET catalytic function. Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of polycomb group (PcG) repressive complex 2 (PRC2), responsible for trimethylation of lysine 27 of histone H3 (H3K27me3), a repressive histone mark that is also regulated by additional sex combs–like 1 (ASXL1). The methylation of H3K4 by mixed-lineage leukemia (MLL), H3K79 by DOT1L, H3R2/R8 by protein arginine methyltransferase 5 (PRMT5), and histone acetylation by histone acetyltransferases (HATs) all promote active transcription. Lysine methylation and acetylation can be removed by demethylases such as lysine-specific demethylase 1 (LSD1) and histone deacetylases (HDACs). DOT1L, disruptor of telomeric silencing 1–like; me1, monomethylation; me2, dimethylation. Professional illustration by Somersault18:24.
Schematic of regulatory components of the epigenome. The epigenome primarily consists of DNA methylation and histone modifications at the nucleosomal level. Most cytosine guanine dinucleotides (CpGs) in the mammalian genome are methylated in the form of 5-methylcytosine (5mC), except for those in CpG islands (CGIs), commonly found near promoter regions. The methylation is catalyzed by DNA methyltransferases (DNMTs). CpG shores, located within 2 kb of CGIs, as well as CpG canyons, which are large conserved regions of hypomethylation, can also be differentially methylated. Active removal of 5mC is catalyzed by the ten-eleven translocation (TET) family of dioxygenases through oxidation of 5mC to 5-hydroxymethylcytosine (5hmC). Isocitrate dehydrogenase 1 (IDH1) and IDH2 catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG), a metabolite required for TET catalytic function. Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of polycomb group (PcG) repressive complex 2 (PRC2), responsible for trimethylation of lysine 27 of histone H3 (H3K27me3), a repressive histone mark that is also regulated by additional sex combs–like 1 (ASXL1). The methylation of H3K4 by mixed-lineage leukemia (MLL), H3K79 by DOT1L, H3R2/R8 by protein arginine methyltransferase 5 (PRMT5), and histone acetylation by histone acetyltransferases (HATs) all promote active transcription. Lysine methylation and acetylation can be removed by demethylases such as lysine-specific demethylase 1 (LSD1) and histone deacetylases (HDACs). DOT1L, disruptor of telomeric silencing 1–like; me1, monomethylation; me2, dimethylation. Professional illustration by Somersault18:24.
Myeloid malignancies comprise a group of biologically heterogeneous diseases, encompassing acute myeloid leukemia (AML), myelodysplastic syndromes (MDSs), myeloproliferative neoplasms (MPNs), and MDS/MPN overlap syndromes, all of which arise more frequently in the elderly. Despite their heterogeneity in clinical presentation and response to therapy, they have in common the presence of profound epigenetic abnormalities of varying types. This epigenetic deregulation remains a key factor that can be potentially exploited for the development of novel targeted therapies that can prevent relapse and improve long-term survival. In this review, we will discuss the current understanding of how deregulation of the different layers of the epigenome contributes to myeloid malignancies and how this is being exploited for the development of novel therapies.
DNA cytosine modifications
Aberrant DNA methylation is a hallmark of many cancers, and it has been extensively studied in different types of myeloid malignancies.3 DNA methylation in mammals occurs primarily at CpGs in the form of 5mC and is catalyzed by the family of DNMTs, including de novo methyltransferases DNMT3A and DNMT3B and maintenance methyltransferase DNMT1 (Figure 2). Under normal circumstances, most CpGs in the genome are methylated, except for those in CGIs, which are short clusters of CpG-rich sites ∼1000 bases long commonly found near promoter regions. CpGs located within 2 kb of CGIs, CpG shores, and large conserved regions of hypomethylation known as CpG canyons have also been identified as regions that can be differentially methylated.4,5 Although DNA methylation is most frequently associated with gene silencing, in reality, the precise impact on transcription depends on its genomic context. Promoter CGI methylation is indeed linked to gene silencing; however, 5mC across gene bodies is associated with active transcription.6 Seminal work by Tahiliani et al7 demonstrated for the first time in 2009 that 5mC can be actively removed through oxidation of 5mC to 5hmC, catalyzed by the TET family of dioxygenases. TET enzymes are oxygen-, α-KG–, and vitamin C–dependent dioxygenases that play a role in active DNA demethylation. Demethylation proceeds through the iterative oxidation of 5mC to 5hmC, 5-formylcytosine, and 5-carboxylcytosine, which are finally replaced by unmethylated cytosines through base excision repair (Figure 2).8,9 In addition to active demethylation, passive demethylation results from inadequate methylation maintenance by DNMT1, a situation which can arise through the increased number and rate of cell divisions of cancerous cells, because DNA maintenance is linked to DNA replication.10-12 The implication of this phenomenon in the context of cancer development is that aberrant methylation patterns may thus exist even in the absence of mutations in the DNA methylation machinery.

DNA methylation pathway. DNA methylation occurs on the C5 position of cytosine to form 5mC and is catalyzed by the family of DNA methyltransferases that includes DNMT3A. Active demethylation is catalyzed by TET enzymes, which are oxygen-, α-KG–, and vitamin C (VitC)–dependent dioxygenases. Demethylation proceeds through iterative oxidation of 5mC to 5hmC, 5-formylcytosine, and 5-carboxylcytosine. Final replacement with unmethylated cytosines is mediated by thymine DNA glycosylase (TDG) and the base excision repair (BER) pathway. Passive DNA demethylation occurs through inadequate methylation maintenance after DNA replication. Professional illustration by Somersault18:24.
DNA methylation pathway. DNA methylation occurs on the C5 position of cytosine to form 5mC and is catalyzed by the family of DNA methyltransferases that includes DNMT3A. Active demethylation is catalyzed by TET enzymes, which are oxygen-, α-KG–, and vitamin C (VitC)–dependent dioxygenases. Demethylation proceeds through iterative oxidation of 5mC to 5hmC, 5-formylcytosine, and 5-carboxylcytosine. Final replacement with unmethylated cytosines is mediated by thymine DNA glycosylase (TDG) and the base excision repair (BER) pathway. Passive DNA demethylation occurs through inadequate methylation maintenance after DNA replication. Professional illustration by Somersault18:24.
In cancer, there is a redistribution of DNA methylation, usually described as aberrant promoter methylation in the context of global hypomethylation. However, specific cancers and cancer subtypes may differ in the pattern of aberrant methylation observed.13 Aberrant CGI methylation has been linked to the silencing of tumor suppressor genes, whereas aberrant loss of methylation has been associated with the overexpression of oncogenes.14,15 However, changes in DNA methylation cannot always be easily linked to changes in gene expression, indicating that its role in transcription regulation is more complex than previously thought. In recent years, studies of different myeloid malignancies using deep sequencing have identified recurrent mutations in genes that regulate DNA methylation: DNMT3A, TET2, and IDH1/2.1,16-18
Mutations in DNMT3A
DNMT3A is mutated in ∼23% of AMLs, ∼10% of MDSs, ∼5% of MPNs, and ∼7% of chronic myelomonocytic leukemias (CMMLs); it is also the most frequently mutated gene in clonal hematopoiesis of indeterminate potential. The most common mutation in myeloid malignancies occurs at the R882 residue located within the catalytic domain.1,16-26 The heterozygous R882H mutation acts in a dominant-negative manner whereby the mutant DNMT3A prevents the wild-type enzyme from forming functional tetramers, thus reducing the methyltransferase activity by ∼80%. This leads to focal hypomethylation of CpGs at different types of genomic locations (eg, promoter, gene body, intergenic, CGI, or CpG shore or canyon regions).27 However, no clear correlation was found between differentially methylated regions (DMRs) and gene expression or other epigenetic modifications at nearby genes in the context of AML; therefore, how these DMRs contribute to leukemogenesis remains to be understood, although it is possible they may play a role in determining chromatin accessibility and chromosomal conformation.28 Other non-R882 mutations can increase or decrease the enzymatic activity of DNMT3A in vitro, but their impact on the methylome in the cellular context may not always be significant.29 In 1 study, mean methylation levels were not significantly different between non-R882 mutant and wild-type DNMT3A AMLs.27 How the different types of DNMT3A mutations contribute to pathogenesis, whether through catalytic or noncatalytic mechanisms, remains an area of active investigation.
Physiologically, loss of Dnmt3a in mice, where it is highly expressed in long-term HSCs, has been shown to bias HSCs toward self-renewal.30 Even though once again there was no clear correlation between DMRs and gene expression in mutant HSCs, there were multiple genes associated with HSC and leukemia function near hypomethylated DMRs, with some of the most significant regions found at enhancer regions and canyon edges, making them accessible to transcription factors such as FLI1, LMO2, RUNX1, and PU.1 that regulate hematopoietic cell fate.31,32 Mice transplanted with Dnmt3a knockout HSCs ultimately develop a range of hematologic malignancies as expansion of the HSC compartment coupled with aberrant DNA methylation increases the opportunity for secondary hits and subsequent leukemic transformation.33 Knockin of the murine mutant homolog Dnmt3aR878H, alone or in combination with cooperating oncogenes such as FMS-related tyrosine kinase 3–internal tandem duplication (Flt3ITD) and nucleophosmin 1 (Npm1c), also drives clonal expansion and leukemia development.34-36 These studies underscore the critical role that DNMT3A plays in normal hematopoiesis.
Mutations in TET2
TET2 and 5hmC have been found to have important biologic functions in clonal myeloid diseases. In their recent work, Rasmussen et al37 found that Tet2 binding in mouse embryonic stem cells is enriched at regions of open chromatin that overlap with enhancer regions marked by the presence of acetylation of H3K27 and monomethylation of H3K4, whereas it is depleted in promoters and CGIs. Notably, profiling of 5hmC in human AML identified better correlation between 5hmC and gene expression, regardless of genomic location, than 5mC at promoters. Nearly 90% of differential 5hmC regions are outside of CGIs, with approximately half of those located in enhancer regions. Indeed, binding sites for FLI1, RUNX1, and CEBPA/B were enriched in enhancer regions that are enriched for 5hmC.37,38 Tet2 deficiency in murine hematopoietic cells results in global loss of 5hmC, as expected, and changes in chromatin accessibility, leading to changes in transcription factor binding activity.37,39,40 Similar to Dnmt3a mutants, Tet2-deficient murine models exhibited increased self-renewal of hematopoietic stem and progenitor cells (HSPCs) while impairing differentiation, with hypermethylation of DMRs observed at distal enhancers.39-42 This resulted in a CMML-like phenotype found in human patients that includes splenomegaly, neutrophilia, and monocytosis.39,41
Loss-of-function TET2 mutations occur in ∼12% of AMLs, ∼22% of MDSs, ∼13% of MPNs, and ∼50% of CMMLs.1,16-18,21-23,43-45 Importantly, most TET2 mutations in leukemia patients are monoallelic, and recent work by Cimmino et al46 showed that restoring Tet2 activity in Tet2-deficient cells through the administration of vitamin C, a cofactor required for TET enzymatic activity, inhibits leukemogenesis. Treatment with vitamin C impaired self-renewal, promoted myeloid differentiation, and resulted in the reversal of DNA hypermethylation in murine hematopoietic cells and human AML cell lines.46
Mutations of IDH1 and IDH2
TET2 deficiency is also found in the context of IDH1 and IDH2 mutations. IDH1 and IDH2 belong to the family of IDH enzymes that catalyze the oxidative decarboxylation of isocitrate to α-KG. Mutations confer on these enzymes neomorphic activity to catalyze the reduction of α-KG into the oncometabolite 2-hydroxyglutarate (2-HG; Figure 3).47 High levels of 2-HG disrupt multiple cellular processes that include inhibition of α-KG–dependent dioxygenases like TET2, resulting in global hypermethylation and a block in differentiation.48,49 Likewise, histone demethylases like jumonji C domain–containing proteins are also affected by 2-HG, leading to an increase in histone methylation.50 Conditional knockin of IDH1R132H into mouse hematopoietic cells resulted in the expansion of HSPCs, with age, and DNA hypermethylation as observed in human AML.51 Mutations in IDH1 and IDH2 are found in ∼17% of AMLs (7% for IDH1 and 10% for IDH2), but they are rare in MDSs, MPNs, and MDS/MPN syndromes.1,16,18,21-23

Wild-type and mutant activities of IDH1/2. Mitochondrial metabolite α-KG is produced by reduced NAD phosphate (NADP; NAPDH)-dependent isocitrate dehydrogenases IDH1 and IDH2 in the mitochondrial matrix and in the cytosol by oxidative decarboxylation of isocitrate. Mutations (IDH1/2-mut) confer these enzymes with neomorphic activity to catalyze the reduction of α-KG into oncometabolite 2-HG. High levels of 2-HG disrupt multiple cellular processes that include inhibition of α-KG–dependent dioxygenases like TET2 and histone demethylases. Professional illustration by Somersault18:24.
Wild-type and mutant activities of IDH1/2. Mitochondrial metabolite α-KG is produced by reduced NAD phosphate (NADP; NAPDH)-dependent isocitrate dehydrogenases IDH1 and IDH2 in the mitochondrial matrix and in the cytosol by oxidative decarboxylation of isocitrate. Mutations (IDH1/2-mut) confer these enzymes with neomorphic activity to catalyze the reduction of α-KG into oncometabolite 2-HG. High levels of 2-HG disrupt multiple cellular processes that include inhibition of α-KG–dependent dioxygenases like TET2 and histone demethylases. Professional illustration by Somersault18:24.
Other mutations affecting DNA methylation
Much remains to be understood about how either global or focal DNA methylation or hydroxymethylation changes in hematopoietic cells drive malignant disease. However, it is clear that in addition to mutations of the epigenetic modifiers themselves, mutations in transcription factors may also affect the epigenome through the impaired recruitment of epigenetic modifiers to their normal target sites. Mutations in Wilms tumor 1, a zinc finger transcription factor that interacts with TET2, can impair either its binding to DNA or its recruitment of TET2 to the transcriptional complex. Both types of mutations lead to a phenocopy of TET2 deficiency.38,52
Another layer of complexity is added when combinations of different mutations are considered. On the basis of genomic classification studies in AML, a driver mutation can be identified in up to 99% of samples, with 86% of samples containing ≥2 driver mutations. Mutations of genes within the same molecular subgroup are usually mutually exclusive, such as transcription factor fusions, cohesin complex mutations, and kinase signaling. For cooccurring mutations, DNA methylation pathway mutations often cooccur with FLT3ITD mutations, which are found in ∼30% of AMLs.1,21 Modeling in mice has shown that Flt3ITD combined with Tet2 loss leads to leukemic transformation, and the number of DMRs is greatly increased in double mutants compared with single mutants, showing significant hypermethylation at promoters and CpG shores.53 Similarly, Dnmt3a deficiency in combination with Flt3ITD also leads to leukemic transformation, with leukemic cells showing significant hypomethylation at promoters and various CpG regions.32,54 The addition of an Npm1 mutation is sufficient to transform Dnmt3aR878H mutant clones into AML.36 Tet2−/−Flt3ITD AML cells were found to be more refractory to chemotherapy and FLT3 inhibition (quizartinib), and patients with the triple combination of DNMT3A, NPM1, and FLT3ITD mutations have a significantly worse prognosis than those with FLT3ITD plus either a DNMT3A or NPM1 mutation.21,53 Consequently, understanding how various combinations of mutations affect disease phenotype is equally important for identifying new strategies to improve therapeutic treatments of myeloid malignancies.
Malignant methylome
Aberrant DNA methylation profiles correlate strongly with molecular and cytogenetic subtypes as well as clinical outcomes in AML, irrespective of whether the leukemia carries mutations in the DNA methylation machinery.55 Each AML subtype is characterized by a distinct epigenetic profile, with different degrees of aberrant hyper- and hypomethylation when compared with normal CD34+ cells, indicating that there is no such thing as a uniquely characteristic malignant methylation profile. MLL-rearranged and DNMT3a mutant AMLs are characterized by predominant hypomethylation, whereas AMLs harboring silenced CEBPA, del(5q)/del(7q), or IDH1/2 mutations have a predominantly hypermethylated profile.13 Furthermore, specific DNA methylation profiles were sufficient to identify the existence of previously unrecognized AML subtypes, with distinct clinical outcomes, which could not be captured by gene expression profiling.55 Two of these 5 novel subtypes were shortly afterward identified as corresponding to IDH1/2 mutant cases, demonstrating that unlike gene expression profiling, DNA methylation could robustly segregate these distinct cases from other AML subtypes.48 Notably, although the cancer field focused on the role of aberrant promoter hypermethylation for many years, next-generation bisulfite sequencing studies of AML in recent years have revealed that enhancer methylation profiles play a much stronger role in determining the epigenetic identity of AMLs than promoter CGI profiles, stressing the important role that these regulatory elements play in malignant epigenomic regulation.13
Targeting DNA methylation
The first 2 epigenetic drugs approved for clinical treatment of myeloid malignancies were cytosine analogs 5-azacytidine (AZA) and 5-aza-2′-deoxycytidine (DAC), both of which function as DNA hypomethylating agents (HMAs; Figure 4). Both are currently used in the standard care of higher-risk MDS patients and as alternatives for AML patients ineligible for conventional chemotherapy. DAC is incorporated directly into the DNA, whereas AZA is predominantly incorporated into RNA, with a small percentage going into DNA.56 Incorporation of these analogs inhibits DNMT activity, resulting in DNA hypomethylation and upregulation of some silenced genes, although genome-wide studies have failed to report significant transcriptional changes.57,58 Seminal work by Chiappinelli et al59 and Roulois et al60 in 2015 demonstrated that HMAs lead to reexpression of endogenous retroviral elements capable of inducing an immune response through interferon signaling. However, ∼40% to ∼50% of MDS patients do not respond to HMA therapy even after 6 months of treatment, and there are still no clear clinical biomarkers that can predict response.61,62 Evidence for cell-cycle quiescence as a mechanism of resistance in mutant HSPCs to AZA has emerged, consistent with the need for DNA replication to occur for drug incorporation.63,64 Thus far, although some mutational profiling studies in MDS and CMML have shown a correlation between TET2 mutation status and response to HMAs, this did not correlate with overall survival.65-67 In mice, AZA treatment reduced the competitive advantage of Tet2-null hematopoietic cells.65 Given the strong correlation between DNA methylation profiles and clinical subtypes, it is possible that DNA methylation–based biomarkers may offer an alternative for outcome prediction in HMA-treated patients. In CMML, a 21-DMR biomarker predicts response to DAC with 87% accuracy.68

Targets of epigenetic therapies. Examples of epigenetic drugs developed to treat aberrant DNA methylation and chromatin in myeloid malignancies. Nucleoside analogs like AZA are incorporated into the DNA and inhibit DNMT activity, resulting in DNA hypomethylation. Mutant IDH1 and IDH2 (IDH1/2-mut) convert α-KG normally produced by wild-type enzymes into oncometabolite 2-HG, and the oncogenic effects caused by 2-HG can be reversed by IDH1 and IDH2 inhibitors ivosidenib and enasidenib, respectively. Chromatin complexes that drive leukemic gene expression or inhibit differentiation programs can also be targeted. Disruption of MLL fusion complexes includes inhibition of DOT1L methyltransferase by pinometostat and menin by menin inhibitors (MIs). Bromodomain and extra terminal (BET) inhibitors (BETi) target the bromodomains of BET proteins like bromodomain-containing protein 4 (BRD4) to prevent their binding to acetylated histones and interaction with transcription factors that promote leukemic gene expression programs. Conversely, repressed differentiation programs can be reversed with the use of LSD1 inhibitors (LSD1i). Disrupting the interaction between LSD1, GFI, and the CoREST repressive complex allows for upregulation of critical myeloid transcription factors and differentiation. me1, monomethylation; me2, dimethylation; NADP, NAD phosphate; NADPH, reduced NAD phosphate. Professional illustration by Somersault18:24.
Targets of epigenetic therapies. Examples of epigenetic drugs developed to treat aberrant DNA methylation and chromatin in myeloid malignancies. Nucleoside analogs like AZA are incorporated into the DNA and inhibit DNMT activity, resulting in DNA hypomethylation. Mutant IDH1 and IDH2 (IDH1/2-mut) convert α-KG normally produced by wild-type enzymes into oncometabolite 2-HG, and the oncogenic effects caused by 2-HG can be reversed by IDH1 and IDH2 inhibitors ivosidenib and enasidenib, respectively. Chromatin complexes that drive leukemic gene expression or inhibit differentiation programs can also be targeted. Disruption of MLL fusion complexes includes inhibition of DOT1L methyltransferase by pinometostat and menin by menin inhibitors (MIs). Bromodomain and extra terminal (BET) inhibitors (BETi) target the bromodomains of BET proteins like bromodomain-containing protein 4 (BRD4) to prevent their binding to acetylated histones and interaction with transcription factors that promote leukemic gene expression programs. Conversely, repressed differentiation programs can be reversed with the use of LSD1 inhibitors (LSD1i). Disrupting the interaction between LSD1, GFI, and the CoREST repressive complex allows for upregulation of critical myeloid transcription factors and differentiation. me1, monomethylation; me2, dimethylation; NADP, NAD phosphate; NADPH, reduced NAD phosphate. Professional illustration by Somersault18:24.
Clinical progress on the use of HMAs will depend on next-generation compounds or synergistic drug combinations. Next-generation HMAs like guadecitabine, which has an increased half-life, showed improved responses with upfront treatment, but whether they are superior remains to be determined.69 Various drug combinations with HMAs are being tested in clinical trials, from HDAC inhibitors to IDH inhibitors.70 One of the new promising combinations is with vitamin C; a recent study found it synergizes with DAC to significantly improve the complete remission (CR) rate and overall survival in AML patients.71
The oncogenic effects of 2-HG caused by IDH mutations can be reversed with the use of allosteric inhibitors ivosidenib and enasidenib, which target mutant IDH1 and IDH2, respectively (Figure 4). A recent phase 1 trial showed similar results for both inhibitors, achieving an overall response rate of ∼40% in patients with relapsed/refractory IDH mutant AML, reducing plasma levels of 2-HG in nearly all patients, and inducing blast maturation in responders.72,73 They are now being tested in frontline therapies, with initial results of ivosidenib treatment for newly diagnosed AML in patients age >75 years showing a CR rate of 30%.74 Moreover, combining the inhibitors with induction and consolidation therapy produced even more impressive results. Patients with newly diagnosed AML achieved CR in ∼55% of cases for ivosidenib and ∼47% for enasidenib.75 Despite this efficacy, primary and secondary mechanisms of resistance remain a problem. Recent results from studying patients with mutant IDH1 AML treated with ivosidenib showed resistance was associated with mutations in receptor tyrosine kinase pathway genes (NRAS, PTPN11), secondary mutations in IDH1 that reduce drug binding, and/or emergence of IDH2 mutant clones that restore 2-HG production.76 Thus, combinatorial treatment with kinase inhibitors or combined IDH1/2 treatment may improve responses and reduce relapse. The combination of ivosidenib and AZA also produced durable responses, with ∼61% of patients achieving CR, and of the responders, ∼71% showed clearance of mutant IDH1 clones.77
Posttranslational histone modifications
Another compartment of the epigenome that becomes deregulated in myeloid malignancies involves posttranslational modifications of nucleosomal histone proteins at their N-terminal tails. Histones can be modified by acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation, among others, and each type of modification alone or in combination is associated with particular patterns of gene activity and chromatin conformation, regulating accessibility of other factors to the DNA.78 Each type of modification is catalyzed and recognized by different classes of chromatin factors, which are frequently mutated or rearranged in myeloid malignancies.
MLL rearrangements
Rearrangements of MLL genes result in a class of aggressive leukemias. MLL genes are the mammalian homologs for the trithorax genes that catalyze the methylation of H3K4, a mark of active transcription, and are important for developmental homeotic (Hox) gene expression (Figure 1).79 Rearrangements involving the MLL1 gene on 11q23 create a chimeric gene that encodes the N terminus of MLL1 fused to the C terminus of at least 1 of the 70 known fusion partners.80 Although the methyltransferase domain of MLL is lost, some of the more common MLL fusion partners include members of the super elongation complex and DOT1L complex, both of which promote active transcription.79,81 The MLL fusion complexes induce leukemic transformation through transcriptional deregulation of target genes that activate or maintain a stem cell self-renewal gene expression program in HSPCs.82-84 Overexpression of HOXA cluster genes and MEIS1 is characteristic of MLL fusion leukemias, and recent work has shown that Hoxa9 overexpression actively drives leukemic transformation by increasing chromatin accessibility of enhancers and upregulating gene expression, including Msi1 and Bcl2, which have been previously implicated in myeloid leukemias.85
Given that MLL fusion complexes drive leukemic expression programs, they can be disrupted using small-molecule inhibitors. DOT1L is the only known H3K79 methyltransferase, and inhibition of its activity has been shown to be effective at targeting MLL-transformed leukemic cells (Figure 4). DOT1L inhibitors were the first histone methyltransferase inhibitors to enter human clinical trials. DOT1L inhibition reduces aberrant H3K79 dimethylation and gene expression driven by MLL fusions, consequently inhibiting proliferation and inducing apoptosis of MLL-transformed leukemic cells.86,87 A recent phase 1 study of the inhibitor pinometostat (EPZ-5676), however, showed moderate efficacy, with only 2 of 51 patients achieving CR.88
Menin, another cofactor of the MLL complex, is also required for MLL fusion–mediated transformation, and Borkin et al89 were the first to show in 2015 that menin inhibitors can also reverse the leukemic gene signature and block leukemia progression in preclinical models (Figure 4).90 Since then, 2 menin inhibitors have entered phase 1 clinical trials.91,92 Intriguing new findings suggest that the same targeting strategies can be applied to other leukemias that share an MLL dependency. NPM1c AMLs show similar HOXA/MEIS1 overexpression, and the use of menin inhibitors could eradicate preleukemic clones even in these non–MLL-rearranged leukemias.93,94 Other approaches include stabilizing wild-type MLL through IRAK inhibition.95
Mutations in members of the PcG complexes
The PcG proteins play an important role in gene silencing during embryonic development and tissue differentiation through H3K27me3 (Figure 1). Increased expression of PcG proteins and that of loss-of-function mutations have both been described for myeloid malignancies, indicating that balanced PRC2 activity is important for normal function.96 PcG proteins exert their activity through 2 main complexes: PRC1 and PRC2. Mutations in EZH2, the catalytic subunit of PRC2 responsible for H3K27me3, leads to loss of H3K27me3 and upregulation of HOX oncogenes as well as genes that confer chemoresistance, such as FHL1 and UBE2E1.97,98 ASXL1 is also frequently mutated in myeloid leukemias.1,21-23 Loss of ASXL1 leads to global loss of H3K27me3 and PRC2 activity and the development of an MDS-like phenotype.99-101 Mutations in EZH2 and other PRC2 components are relatively infrequent in myeloid malignancies.1,16-18,21-23 When present, they are loss-of-function mutations, but EZH2 deficiency is also found in SRSF2 splicing mutants.102 Patients with EZH2 or ASXL1 mutations consistently show worse prognosis in various malignancies.
Current approaches for EZH2 mutant malignancies are targeted at immediate downstream dependencies: targeting oncogenes derepressed by the loss of H3K27me3 such as HOX genes, proteasome inhibition (bortezomib) to restore EZH2 levels, inhibition of H3K27me3 demethylases like UTX/KDM6A, and dual inhibition with EZH1, which can compensate partially for EZH2.97,103,104 Recent work has shown that leukemia stem cells overexpress EZH1 and EZH2, and the reduction in H3K27me3 with dual inhibition allows for expression of cell-cycle genes and exit from quiescence.105
Deregulation of expression of epigenetic modifiers
In addition to mutations, other factors involved in regulating histone modifications have been shown to be abnormally expressed in myeloid malignancies and required for establishing or maintaining the leukemic state. Lysine methylation is targeted for removal by demethylases such as LSD1, which in complex with other proteins can demethylate mono- and dimethylation of H3K4 (Figure 1).106 LSD1 is overexpressed in myeloid malignancies, and its inhibition can induce differentiation of blasts in mouse models of MLL-rearranged leukemia.107,108 LSD1 inhibitors iadademstat and GSK-LSD1, even though they target the catalytic site, did not affect global histone methylation levels but rather disrupted LSD1's interaction with GFI and the CoREST repressive complex, leading to activation of enhancers and upregulation of critical myeloid transcription factors SPI1 and CEBPA to induce differentiation (Figure 4).109,110 Tranylcypromine and GSK2879552 were shown to have synergistic effects with all-trans retinoic acid to inhibit proliferation and induce differentiation and apoptosis in AML cell lines and in patients from a phase 1 study with relapsed/refractory AML and MDS.111,112 Multiple clinical trials studying the use of LSD1 inhibitors as mono- or combination therapy are ongoing. In vitro data in primary AML specimens have shown synergism between enhancer reprogramming through LSD1 inhibition and promoter reprogramming through HMAs, which was most effective in TET2 mutant AML.113
The family of PRMTs catalyzes histone arginine mono- and dimethylation, among many other cellular substrates, and regulates a wide variety of cellular processes, from gene transcription to RNA splicing and protein translation. Multiple family members have been found to be important for normal and malignant hematopoiesis.114 PRMT5-mediated methylation of histones is generally repressive, but recent work in AML cells has shown that PRMT5-mediated dimethylation of histone H3 on R2 and R8 antagonizes the deposition of H3K27me3 by PRC2 (Figure 1).115 PRMT5 is required for HSC self-renewal and is overexpressed in MPNs and lymphocytic leukemias.116-118 Although the mechanism of drug action remains to be elucidated, PRMT5 inhibitors activate the p53 pathway through alternative splicing of its target genes.118,119 Clinical trials are underway for AML, MDS, and CMML. Unlike PRMT5, coactivator-associated methyltransferase 1 is essential for leukemogenesis but not for normal hematopoiesis, providing an attractive therapeutic target for AML.114
Acetylation of histones is found in euchromatic regions and is dynamically regulated by the activity of histone lysine acetyltransferases and HDACs (Figure 1). Although they are not known to be mutated in myeloid malignancies, histone lysine acetyltransferases have been identified as leukemic fusion partners with other transcriptional activators such as the monocytic leukemia zinc finger and translation initiation factor MOZ-TIF2 fusion. Not only does expression of the fusion increase self-renewal and drive leukemic transformation of HSPCs in mice, its recruitment of another acetyltransferase, CREB binding protein, is essential for leukemogenesis.120,121 HDAC mutations in myeloid malignancies are likewise rare, but aberrant expression has been observed, as has aberrant recruitment by leukemia fusion proteins.1 The therapeutic effects of HDAC inhibitors, which target the catalytic site, range from restoration of gene expression programs leading to differentiation to induction of DNA damage and apoptosis.122,123 However, HDAC inihbitors (vorinostat, panobinostat) have not been effective clinically either as monotherapy or in combination with HMAs for the treatment of AML or MDS, even with newer agents like pracinostat.124-127 The broad effects of HDAC inhibitors on histone and nonhistone proteins make it challenging to identify signatures or somatic mutations that would confer sensitivity to treatment.
Acetylated lysine residues are recognized by BET domain–containing proteins. These proteins act as transcriptional coactivators and serve as scaffolds for the recruitment of other transcriptional regulators.128 Bromodomain-containing protein 4 (BRD4) colocalizes with hematopoietic transcription factors at gene promoters and enhancers such that inhibition by BET inhibitors, which target the conserved bromodomains, exerts strong antileukemic effects through bromodomain-containing protein 4 loss from enhancers of oncogenic drivers like MYC in AML (Figure 4).129,130 Given their efficacy, multiple BET inhibitors have been developed that are already in preclinical and clinical trials for several types of cancers, including advanced or refractory myeloid malignancies. However, reports from phase 1 trials completed in recent years have shown dose-limiting toxicities at doses insufficient to achieve clinical response.131-134 Furthermore, response to BET inhibitors is not uniform across subtypes. Notably, BET inhibitor–resistant AML cells have compensatory mechanisms to restore MYC expression after treatment through activation of a focal MYC enhancer located within PVT1, a long noncoding RNA.135,136 As a result, combinations of BET inhibitors with other therapies are being tested to overcome resistance and improve clinical response. In addition, although MPNs are mainly driven by mutations that activate tyrosine kinase signaling, recent work has shown that combination with BET inhibitors attenuates disease burden in a manner not seen with the single inhibitors because of the cooperation between BET proteins and NF-κB.137
Summary
Unlike genetic abnormalities, the cancer epigenome is amenable to reprogramming and therefore remains a target of interest for the development of novel therapies. Both somatic mutations and oncogenic fusions can hijack different chromatin regulators to enforce epigenomic reprogramming. This new oncogenic landscape results in the deregulation of critical regulatory elements such as promoters and enhancers and can promote oncogene expression and/or repress tumor suppressor programs. Despite recent advances in our understanding of the mechanisms involved in the development and progression of myeloid malignancies, epigenetic therapies thus far have not been curative in most cases, and resistance to these therapeutic agents involves a variety of mechanisms. The quiescent nature of HSPCs, tumor heterogeneity, and secondary mutations allows for the outgrowth of resistant clones and has proven to be a difficult hurdle to overcome. Although it remains to be seen how effective epigenetic therapies will be as a broad class, rational combinations are expected to provide the greatest benefit given the diverse landscape of myeloid malignancies.
Acknowledgment
This work was supported by Leukemia and Lymphoma Society Scholar Award 1357-19 (M.E.F.).
Authorship
Contribution: M.E.F. and H.-T.H. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Maria E. Figueroa, 1501 NW 10th Ave, BRB 709A, Miami, FL 33136; e-mail: mefigueroa@miami.edu.