Abstract
Chronic inflammation with aging (“inflammaging”) plays a prominent role in the pathogenesis of myeloid malignancies. Aberrant inflammatory activity affects many different cells in the marrow, including normal blood and stromal marrow elements and leukemic cells, in unique and distinct ways. Inflammation can promote selective clonal expansion through differential immune-mediated suppression of normal hematopoietic cells and malignant clones. We review these complex roles, how they can be understood by separating cell-intrinsic from extrinsic effects, and how this informs future clinical trials.
Introduction
Aging and chronic inflammation are tightly linked, and the term “inflammaging” has been coined to describe these intertwined processes.1 Chronic exposure to exogenous pathogen associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), endogenous proteins released from damaged cells, produces an “immune biography” that installs an environment of sterile inflammation.2 The result is reduced adaptive immunity,3 enhanced proinflammatory reactions,4 and predisposition to clonal disorders and malignancies.5,6 Inflammaging also drives hematopoietic stem cell (HSC) dysfunction, causing a loss of HSC quiescence,7 reduced self-renewal capacity,8,9 and myeloid differentiation bias.10,11 Inflammation drives pathogenesis in myeloid malignancies across the disease spectrum, including myeloproliferative neoplasms (MPNs),12 myelodysplastic syndromes (MDS),13,14 and acute myeloid leukemia (AML).15
There is also evidence that clonal hematopoiesis (CH) causes inflammation, and inflammaging also likely provides a permissive environment for clonal expansion.16 In addition, chronic inflammatory conditions (eg, cardiovascular17 and rheumatologic18 diseases) are strongly associated with CH, though whether inflammation can initiate CH is less clear. Although these are distinct disease states (eg, MDS causes ineffective hematopoiesis, whereas AML is proliferative), there are commonalities in the pathways involved, and inflammation can drive disease progression.19 Beyond the marrow, systemic inflammatory disorders are also increasingly associated with myeloid malignancy (eg, vacuoles, E1-enzyme, X-linked, autoinflammatory, somatic [VEXAS] syndrome and MDS).20 Approaches targeting inflammation are being rapidly translated into clinical trials.9,21,22
In this article, we outline the complex role of inflammation in myeloid malignancies and, using examples from preclinical models and the framework of cell-intrinsic vs extrinsic effects, emphasize that inflammation is not a monolithic entity in myeloid malignancies, but rather has a cytokine, signaling pathway, and cell context-dependent nature that is dynamic throughout the disease course. Understanding these dependencies will be crucial to successfully modulating inflammation for clinical benefit in myeloid malignancies.
Inflammation in myeloid malignancies: context matters
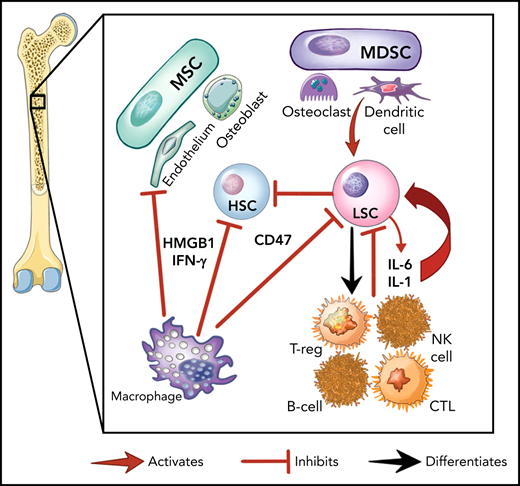
Here we provide an overview of the specific inflammatory pathways involved in myeloid malignancies, demonstrating how the malignant cells, the marrow stroma, and immune cells interact to establish the inflammatory environment (Figure 1A). This is nuanced by the fact that malignant hematopoietic stem and progenitor cells (HSPCs) can differentiate into mature immune cells (eg, natural killer [NK], myeloid derived suppressor cells [MDSCs] and others).23-25 Further, preleukemic mutations (eg, DNMT3A, TET2) that alter inflammatory signaling are transmitted to the entirety of myeloid and lymphoid cells in patients with MDS.23,26,27 Differentiated malignant cells can then elaborate inflammatory cytokines such as tumor necrosis factor (TNF), interleukin-6 (IL-6), and interferon-γ (IFN-γ).9,28

The role of inflammation in myeloid malignancies. (A) Intrinsic leukemic stem cell (LSC) signaling as well as interactions with the microenvironment result in inflammation and a permissive environment for further clonal expansion. Cytokines secreted from the LSC or their progeny may promote clonal expansion and/or suppress normal HSCs. Immune and stromal cells can also respond to the LSC by secreting cytokines, which can have differential effect on the HSC and LSC. (B) Activation of inflammatory pathways in LSCs can be divided into cell-intrinsic and cell-extrinsic. Cell-intrinsic alterations can drive increased intracellular signalling through activated intracellular pathways, causing proliferative signalling and cytokine secretion. Cell-extrinsic factors refer to cytokines or other factors secreted by LSCs, immune cells, or the stroma which can (i) suppress normal HSCs, (ii) suppress the LSCs, or (iii) activate the LSCs to different degrees after interacting with their cellular receptors. MDSC, myeloid-derived stromal cell; MSC, mesenchymal stromal cell. Illustration by Vicky Earle.
The role of inflammation in myeloid malignancies. (A) Intrinsic leukemic stem cell (LSC) signaling as well as interactions with the microenvironment result in inflammation and a permissive environment for further clonal expansion. Cytokines secreted from the LSC or their progeny may promote clonal expansion and/or suppress normal HSCs. Immune and stromal cells can also respond to the LSC by secreting cytokines, which can have differential effect on the HSC and LSC. (B) Activation of inflammatory pathways in LSCs can be divided into cell-intrinsic and cell-extrinsic. Cell-intrinsic alterations can drive increased intracellular signalling through activated intracellular pathways, causing proliferative signalling and cytokine secretion. Cell-extrinsic factors refer to cytokines or other factors secreted by LSCs, immune cells, or the stroma which can (i) suppress normal HSCs, (ii) suppress the LSCs, or (iii) activate the LSCs to different degrees after interacting with their cellular receptors. MDSC, myeloid-derived stromal cell; MSC, mesenchymal stromal cell. Illustration by Vicky Earle.
Innate immune signaling is frequently aberrantly activated through Toll-like receptor (TLR) signaling in malignant myeloid cells.29 TLRs, which recognize PAMPs and DAMPs, produce an inflammatory response upon activation in hematopoietic cells.29,30 In malignant myeloid cells, TLRs31 and downstream effectors (eg, MYD88,32IRAK1,33TRAF634) are often expressed at higher levels or in alternate isoforms (eg, IRAK4L35), and their intrinsic regulators (eg, miR-145, miR-146a36) are downregulated. This activated TLR axis results in the secretion of several cytokines (eg, IL-1,37 IL-6,36 IL-8,32 TNF,38 IFN-γ, granulocyte colony stimulating factor39) from malignant myeloid cells. IL-1 receptor signalling is linked with TLRs through the shared cytoplasmic Toll-IL-1-receptor domain and is also upregulated by TLR activation.40 Overexpression of the IL-1 receptor accessory protein on HSCs has also been shown to increase AML cell viability and clonal output in patients.41
Another key innate immune component in malignant myeloid cells are nucleotide-binding domain and leucine-rich repeat (NLR) receptors. NLRs associate with nucleotide-binding domains to form the inflammasome complex, producing proinflammatory, lytic cell death (pyroptosis) on activation by PAMPs or DAMPs.42 The alarmin S100A9, a DAMP secreted by stromal cells and found at high levels in MDS marrows,43 can bind NLRs and activate the NLRP3 inflammasome, which drives clonal expansion and pyroptosis by activating caspase-1, thereby generating mature IL-1β and IL-18 in patients with MDS.42 The CD33 receptor, expressed on malignant cells and MDSCs, senses S100A8/9 and can activate the inflammasome.44 Overactivity of TLRs and the inflammasome, with other factors, results in a heightened inflammatory milieu in the MDS/AML marrow microenvironment.24,45-47 This inflammatory signaling generally promotes differential fitness of the malignant MDS/AML LSC over normal HSPCs, although this effect varies depending on the disease context.47
Our understanding of the role for cellular immunity in myeloid malignancies continues to evolve. MDS/AML demonstrate relatively low rates of somatic mutations,48 and generating robust T-cell responses to malignant myeloid cells with low neoantigen burdens can be challenging.49,50 However, the curative graft-versus-leukemia effect from allogeneic hematopoietic stem cell transplant is mediated through T and NK cells,51,52 and inflammation may promote the graft-versus-leukemia effect.53 Other new dimensions in this area are the advent of cellular therapies54-56 and the recognition of the role for regulatory T cells (Tregs) in the MDS/AML marrow microenvironment.57,58 Tregs can facilitate or suppress CD8+ T-cell-mediated control of malignant clones.57,58 Tregs in lower risk MDS have more proinflammatory immune responses and effector-type cells,57,58 whereas higher risk MDS has expanded Treg and MDSC compartments.57,59 Higher Treg infiltrates are correlated with worse outcomes in MDS/AML, possibly because of suppression of T cell-mediated immune surveillance.57,59-61 It is also becoming evident that macrophage and NK cells are crucial to immune surveillance of malignant myeloid cells. Overexpression of the macrophage inhibitory molecule CD47 on LSCs has been shown to drive disease progression,62 whereas MDS clone-derived mesenchymal stromal cells can inhibit NK-cell function and promote malignant clonal expansion.17,22,26,63
Inflammation in myeloid malignancies can also occur outside the marrow space, with up to 7.4% of patients with MDS meeting criteria for a systemic autoinflammatory disease.64 Autoinflammatory features are associated with acquired transcription factor mutations and abnormal karyotype.64,65 Recently, a new systemic autoinflammatory disease associated with low-grade MDS was described and named vacuoles, E1-enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome, which arises from somatic mutations in the ubiquitin-activating enzyme UBA1.20
Cell-intrinsic vs cell-extrinsic effects
One way to conceptualize inflammation is by separating cell-intrinsic from cell-extrinsic effects, each of which may differentially influence the fate of individual malignant cells (Figure 1B). In the context of therapeutic targeting of inflammation, we refer to cell-intrinsic effects as those that originate and act principally within the malignant cell (eg, somatic mutations, cytogenetic, epigenetic changes).66 In contrast, we refer to cell-extrinsic effects as those that impinge on the malignant cell through an external signal such as cytokines. Extrinsic factors may arise from activated signals in the malignant cell, from normal hematopoietic cells, or cells in the marrow microenvironment. These signals may impinge on all these various cell types, thereby having a major impact on the cellular environment as well as the malignant cell. Thus, although therapeutic targeting of cell extrinsic factors, such as cytokines, may relieve cytopenias or dampen proliferative signals in malignant cells, the intrinsic drivers of myeloid malignancy would likely not be affected, and such an approach would not likely eliminate the malignant cell as a result. The corollary is that targeting tumor cell intrinsic drivers, although more likely to suppress the malignant clone, may also have wider toxicity issues unless the target is tumor specific, as these pathways have significant autonomous effects on the cell independent of activating cytokines. Though targeting cytokines may also have systemic effects, these are less likely to cause cellular toxicity.
Although from the standpoint of therapy separating intrinsic from extrinsic factor targeting provides a convenient conceptual framework, these effects are inextricably linked and interwoven. For example, spliceosome mutations in MDS induce innate immune signalling by altering splice isoform expression of intermediaries such as IRAK4,35MAP3K7, and CASP8.67 Somatic TET2 mutations, frequently seen in CH,68 also activate the NLRP3 inflammasome.69 Loss of the intrinsic TRAF6 regulator miR-146a alters stem cell activity and promotes myeloid malignancy in mice,9 though this has recently been shown to also act as a tumor suppressor through MYC in myeloid malignancy.70 However, all the intrinsic perturbations described previously result in the release of one or more cytokines such as TNF, IL-6, IL-1, or others. Articulating these mechanistic distinctions are important to establish specific goals for therapeutic approaches (eg, reducing clonal expansion vs relieving cytopenias) and avoiding unexpected adverse events (eg, by blocking cytokines that suppress some malignant clones). Thus, inflammation can be targeted at multiple levels including the cytokine, receptor, and downstream pathways, but depending on the target or cell type affected, the overall global effect may be different.
Differential effects: the pathways and cytokines matter
In myeloid malignancies, multiple inflammatory pathways can be simultaneously activated, producing a multitude of signals that affect marrow cells in a variety of context-dependent and cell-specific ways. For example, activation of one inflammatory pathway might drive malignant clonal expansion, whereas a different cytokine induces global marrow dysfunction by suppressing HSPCs and LSCs, and another is simply a bystander. The role these pathways play can also shift with disease progression, for example, by exacerbating cytopenias in MDS24 but regulating proliferation in AML.71 It is important to note that, although inflammation can drive clonal expansion, it is also part of the immune response that can suppress clonal expansion, and the multiple types of secreted cytokines can have differential effects on malignant and normal cells.
A diseased stroma and microenvironment, resulting from the influence of malignant cells or inflammaging, alters the cytokine milieu and promotes the relative fitness of malignant over normal cells.7,43 These cytokines can then differentially suppress normal hematopoiesis, facilitating the emergence of the malignant clone, and affect the marrow niche to different degrees.72,73 For normal HSPCs, chronic inflammation can permanently imprint HSPC differentiation programs, even after removal of the stimulus, suggesting that cell-intrinsic factors can perpetuate inflammatory phenotypes in the context of normal hematopoiesis.10,11 Cytokines can also simultaneously stimulate multiple pathways. In a mouse bone marrow failure model initiated by constitutive TIRAP expression, IFN-γ suppresses erythropoiesis and megakaryopoiesis possibly through direct interaction with the thrombopoietin receptor,28,74 but inhibits myelopoiesis by releasing high mobility group box 1 (HMGB1), which suppresses the endothelial niche.28 Extrinsic stimuli such as infection also can drive IFN-γ-induced CH proliferation in DNMT3A-deficient HSPCs.16
The discordant effects of inflammatory signaling is exemplified by IL-1, which expands myeloid progenitors in an AML xenograft, but suppresses normal progenitors.75 Heterogeneity can also emerge during malignant clonal evolution. Recent data show that expression of inflammatory signalling genes (BST2, IFITM1, IFITM3) can promote clonal expansion of TP53-mutant LSCs in patients with MPNs, and appears necessary for AML transformation while simultaneously suppressing antecedent TP53-wild type clones present before AML transformation.12 In contrast, secreted IL-10 and TGF-β from MDSCs suppress both malignant and normal hematopoiesis in the MDS context, producing global marrow dysfunction and cytopenias in mice.24 However, reducing TGF-β activity by blocking the central adaptor protein Disabled-2 accelerates leukemic progression by increasing stem cell activity at the expense of mature progenitors in a mouse AML xenograft model.71 A TGF-β superfamily ligand trap that reduces downstream SMAD signalling (luspatercept) improves anemia in patients with low-risk MDS,76 and no similar risk has been seen with this drug, though follow-up is short.71 The differences seen may also be due to the fact that Disabled-2 blockade also affects other intrinsic signaling pathways independent of TGF-β, with blockade at the receptor level being more specific.
In another MDS mouse model, IFN-γ depletion caused progression from marrow failure to a myeloproliferation.28 Interestingly IFN-γ is also expressed at higher levels in MDS compared with AML patient samples.28 In a mouse model of TRAF6-driven MDS, which results in marrow failure or acute leukemia, IL-6 deletion blocked marrow failure but not leukemic progression.36 In contrast, inhibiting IL-6 prolonged survival in an AML xenograft model,77 but had no effect in a mouse MPN model.78 It may be that IL-6 blockade can prevent AML-induced anemia, prolonging survival, but in the MDS or MPN context, suppression of IL-6 may repress an inhibitory signal resulting in myeloproliferation and disease progression. These findings emphasize that inflammation likely suppresses some malignant clones in addition to normal hematopoiesis,9 and relieving cytokine-induced suppression may permit expansion and evolution of independent preleukemic or leukemic clones.79
How can we target inflammation in myeloid malignancies?
Several agents are under investigation that target cell-intrinsic innate immune pathways, such as the TLR axis (Table 1). One example is the IRAK4-long isoform (IRAK4L). U2AF1 mutations in AML generate IRAK4L, which hyperactivates NF-ĸB, driving proliferation in a cell-intrinsic manner.35 An IRAK4L inhibitor (CA-4948) blocked leukemic proliferation in an AML model, and is in early-phase trials (NCT04278768), with early results showing tolerability.35,80,81 Other IRAK inhibitors (eg, IRAK1) are being developed.33,82 Inhibitors of UBE2N/Ubc13, an essential E2 ubiquitin-conjugating enzyme required for TRAF6 signalling, are under investigation.83 Blocking NLRP3 inflammasome signaling is another appealing cell-intrinsic approach, given its importance in MDS/AML pathogenesis. Many early-stage NLRP3 inhibitors have been developed targeting the NACHT ATPase domain (MCC-950),84 ATPase activity by cysteine modification (MSN),85 and ATPase activity by blocking NLRP3 activation (CY-09) and pyroptosis, though few clinical data are available about these drugs.86 Whether inhibition of pyroptosis is a targetable strategy in MDS is open to debate, as in another mouse model, genetic targeting of caspase-1 had no impact on marrow failure.28 The cyclic GMP-AMP synthase stimulator of interferon genes pathway is another overactive intrinsic pathway with inhibitors in early development.87 Targeting receptors may block cell-extrinsic or cell-intrinsic effects by blocking autocrine activation. Direct TLR2 inhibition (OPN-305, NCT03337451) has undergone a phase 1/2 trial, for which results are awaited.88 It has also been suggested that targeting CD33 as a ligand for S100A8/9 may be an approach to block the inflammasome. Although CD33 has been targeted previously in AML, the rationale of this approach was not to explicitly block the CD33 and S100A8/9 interaction, but rather to eradicate blast cells.89
Cell-extrinsic approaches are more mature in their development. Luspatercept, a ligand trap for TGF-β superfamily members, was approved after a phase 3 trial demonstrated reduced transfusion burden and good tolerance for lower risk MDS with ring sideroblasts or SF3B1 mutations.90 Although luspatercept attenuates anemia, it does not alter disease course or prevent progression, suggesting it primarily alleviates suppression of normal erythropoiesis or promotes maturation in the context of ineffective erythropoiesis. Recent evidence suggests other lineages are also improved, pointing toward a global cell-extrinsic and stroma-modulating activity of luspatercept.91,92 One early-phase trial attempted TNF-α blockade, though this has not entered later phase trials.93 Surprisingly, given its importance in MDS/AML pathogenesis, few approaches targeting IL-1 receptor accessory protein have been tried.41,91,92,94 A phase 2 trial of canakinumab, an IL-1β-blocking monoclonal antibody that is well tolerated in other inflammatory conditions, has recently opened (NCT04239157), and the results are highly anticipated. Interestingly, an exploratory analysis suggested that the presence of CH predicts for cardiovascular benefit with canakinumab.95 Another promising cytokine target is IL-6. Several studies have demonstrated IL-6 blockade might improve AML-induced anemia and possibly survival in mice,9,77 though accelerated AML progression has also been observed.36 Other cell-extrinsic approaches in early-stage investigation include targeting DAMPs such as S100A8/943,96 and HMGB1.97
Immune-mediated therapies in MDS/AML are also currently used. Immunosuppressive therapy with horse antithymocyte globulin and cyclosporine produced an overall response rate of 48.8% in patients with hypoplastic MDS,98,99 and alemtuzumab up to 60% in younger, less transfused HLA-DR-15+ patients.100 Although these approaches improve cytopenias and modulate the immunome, only hematopoietic stem cell transplant is disease-modifying.101-103 Immunotherapeutic approaches have been tried in MDS/AML (eg, checkpoint inhibitors), with disappointing overall response rates.104 However, targeting macrophage activity with an anti-CD47 antibody (magrolimab) and 5-azacitidine is promising, particularly in TP53-mutated MDS, and results are eagerly awaited.105
Future directions
Inflammation plays a dynamic and heterogeneous role in the pathogenesis of myeloid malignancies that is both context- and patient-dependent. It can simultaneously drive or suppress clonal expansion and alter global marrow function through cell-extrinsic and cell-intrinsic mechanisms. There remain many burning questions around which patients might benefit from these approaches and to what degree. For example, should we be targeting different inflammatory pathways in different disease states, or should these be patient-tailored? Eventually, a “precision medicine” approach might define patients who will benefit from specific immune-targeting therapies, based on disease features or overactive pathways. One other major conceptual question that remains unanswered is whether modulating inflammation can ever modify the disease course in myeloid malignancies. We suggest that blocking cell-extrinsic pathways primarily alleviates global marrow suppression, whereas targeting cell-intrinsic pathways is more likely to be disease-modifying, though possibly with more off-target effects. Further, is targeting one pathway enough, or do we need multitargeting approaches? To move the field forward, we must think beyond inflammation as a general concept and focus on both the inflammatory context and actions of specific pathways and cytokines.
Acknowledgments
R.J.S. received funding from the Leukemia Lymphoma Society of Canada, Canadian Institutes of Health Research (202002LFC-439884), and the Clinician Investigator Program of the University of British Columbia. Work in the laboratory of A.K. is funded by grants from the Canadian Institutes of Health Research, the Terry Fox Research Institute, the Canadian Cancer Society Research Institute, Genome BC, Genome Canada, the Leukemia and Lymphoma Society of Canada, and the BC Cancer Foundation through the Leukemia and Myeloma Program. A.K. is the recipient of the BC Cancer Foundation John Auston Clinical Scientist Award. U.P. received funding from the Jose Carreras Leukemia Fund, the German Ministry of Education and Health, and the Jackstädt Foundation.
Authorship
Contribution: R.J.S., U.P., and A.K. contributed equally to conceiving, constructing, reviewing, and approving the final manuscript.
Conflict-of-interest disclosure: U.P. has received honoraria from Novartis and Curis. A.K. has received a grant-in-aid from AstraZeneca and an honorarium from Jazz Pharmaceuticals. R.J.S. declares no competing financial interests.
Correspondence: Ryan J. Stubbins, BC Cancer Research Centre, 675 West 10th Ave, Vancouver, BC V5Z 1L3, Canada; e-mail: ryan.stubbins1@bccancer.bc.ca; Uwe Platzbecker, Medical Clinic and Policlinic I, Hematology, Cellular Therapy and Hemostaseology, Leipzig University Hospital Medical Center, Liebigstraße 22, Haus 7, Leipzig 04103, Germany; e-mail: uwe.platzbecker@medizin.uni-leipzig.de; and Aly Karsan, BC Cancer Research Centre, 675 West 10th Ave, Vancouver, BC V5Z 1L3, Canada; e-mail: akarsan@bcgsc.ca.